
Abstract
Successful gene therapy of hemophilia A depends on the sustained expression of therapeutic levels of factor VIII (fVIII). Because of mRNA instability, interactions with resident endoplasmic reticulum (ER) chaperones, and the requirement for carbohydrate-facilitated transport from the ER to the Golgi apparatus, fVIII is expressed at much lower levels from mammalian cells than other proteins of similar size and complexity. A number of bioengineered forms of B domain-deleted (BDD) human fVIII have been generated and shown to have enhanced expression. Previously, we demonstrated that recombinant BDD porcine fVIII exhibits high-level expression due to specific sequence elements that increase biosynthesis via enhanced posttranslational transit through the secretory pathway. In the current study, high-expression recombinant fVIII constructs were compared directly in order to determine the relative expression of the various bioengineered fVIII transgenes. The data demonstrate that BDD porcine fVIII expression is superior to that of any of the human fVIII variant constructs tested. Mean fVIII expression of 18 units/106 cells/24 hr was observed from HEK-293 cells expressing a single copy of the porcine fVIII transgene, which was 36- to 225-fold greater than that of any human fVIII transgene tested. Furthermore, greater than 10-fold higher expression was observed in human cells transduced with BDD porcine fVIII versus BDD human fVIII-encoding lentiviral vectors, even at low proviral copy numbers, supporting its use over other human fVIII variants in future hemophilia A gene therapy clinical trials.
Introduction
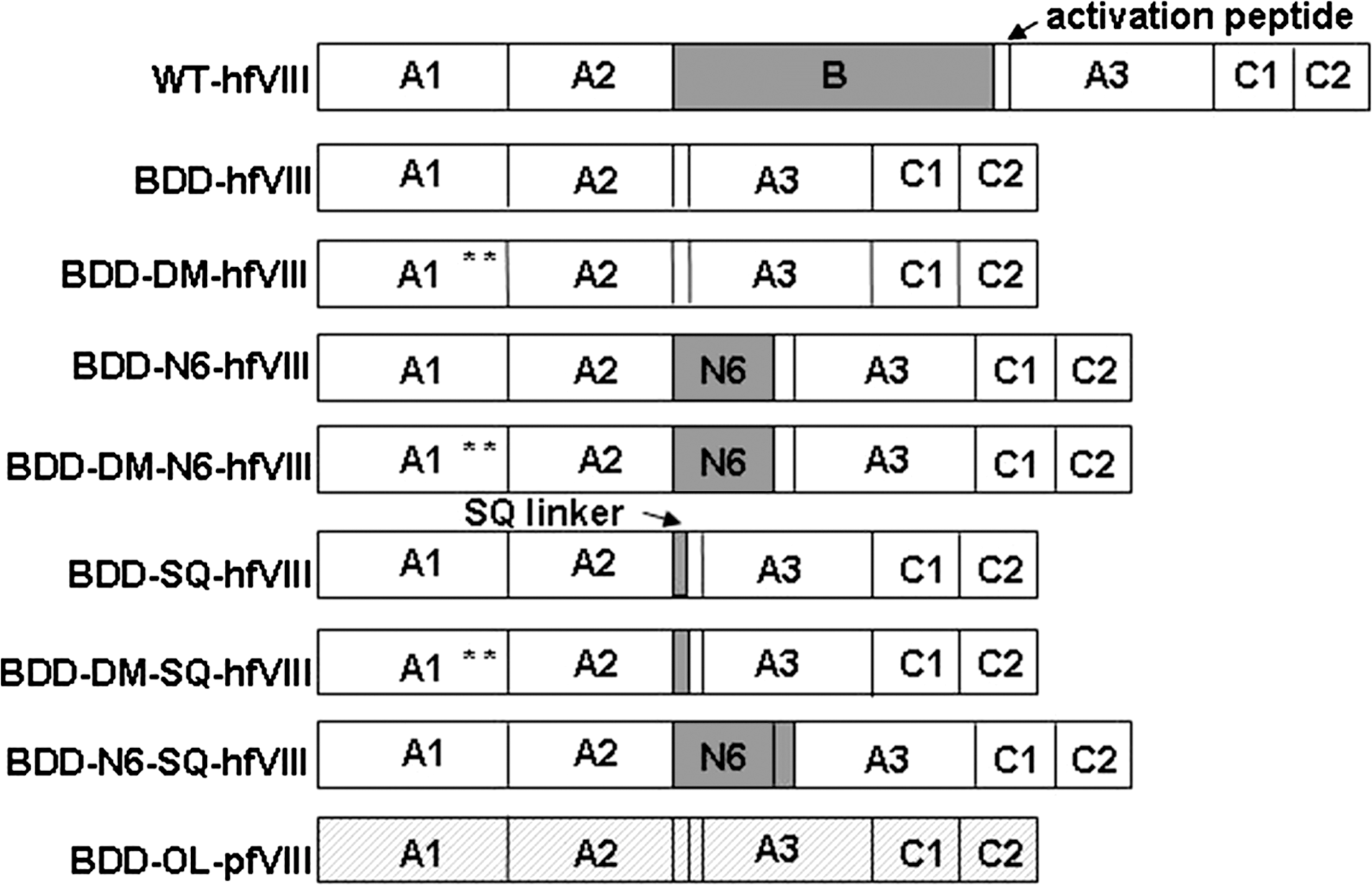
Schematic of fVIII constructs. Eight fVIII transgenes were generated for comparison of fVIII activity. Full-length wild-type fVIII was not used in these comparisons, but is shown here to illustrate the complete fVIII domain structure (A1-A2-B-ap-A3-C1-C2). Human fVIII domains are shown in white with the exception of the B domain, which is shown in gray. Porcine fVIII sequence is identified by hatching. The location of L303E/F309S in double-mutant (DM) constructs is depicted by asterisks.
Several bioengineered hfVIII constructs currently are being evaluated in a number of in vivo gene transfer systems. fVIII expression has been observed in mice after administration of transduced cells expressing BDD hfVIII (Dwarki et al., 1995; Moayeri et al., 2005), BDD hfVIII containing L303E/F309S (Moayeri et al., 2004; Kang et al., 2005), partial BDD hfVIII (Cerullo et al., 2007), and partial BDD hfVIII containing F309S (Sinn et al., 2007). We previously demonstrated that BDD porcine fVIII (pfVIII) is expressed at 10- to 14-fold greater levels than BDD hfVIII in vitro and that this increase in production is due to highly efficient secretion (Doering et al., 2002, 2004). We also demonstrated high-level expression of BDD pfVIII after ex vivo genetic modification and transplantation of hematopoietic stem and progenitor cells into genetically immunocompetent mice (Gangadharan et al., 2006). In these studies, moderate donor cell chimerism and sustained high-level expression of BDD pfVIII were achieved in mice after transplantation under myeloablative and reduced intensity conditioning (Ide et al., 2007). Furthermore, by employing a similar hematopoietic stem cell transplantation (HSCT) protocol, we demonstrated sustained BDD pfVIII expression even in the presence of preexisting fVIII immunity (Doering et al., 2007; Ide et al., 2007).
To achieve significant advances in gene therapy for hemophilia A, we hypothesize that it is necessary to combine an optimized fVIII transgene with an integrating retroviral vector to provide sustained high-level fVIII expression. We compared various high-expression fVIII constructs by transient transfection, analysis of stable clones, isogenic stable cell lines, and recombinant lentiviral gene transfer in an effort to determine the optimal fVIII transgene and to overcome the transient, low-level fVIII expression that hindered previous clinical gene therapy trials for hemophilia A. Direct comparison of bioengineered human and porcine fVIII transgenes demonstrated that BDD pfVIII is expressed at significantly greater levels than any other bioengineered hfVIII variant, supporting its future success in hemophilia A gene therapy applications.
Materials and Methods
Materials
Dulbecco's modified Eagle's medium (DMEM), advanced DMEM–F12 medium, AIM V medium, Opti-MEM medium, heat-inactivated fetal bovine serum (FBS), penicillin–streptomycin solution, GlutaMax, and Dulbecco's phosphate-buffered saline (PBS) were purchased from Invitrogen (Carlsbad, CA). Zeocin and hygromycin B antibiotics for maintenance and selection of Flp-In 293 cells were purchased from Invitrogen. Cell transfections were performed with Lipofectamine 2000 from Invitrogen. Human fVIII-deficient plasma and normal pooled human plasma (FACT) were purchased from George King Bio-Medical (Overland Park, KS). Automated activated partial thromboplastin reagent was purchased from BioMérieux (Durham, NC). Clotting times were measured with an STart coagulation instrument (Diagnostica Stago, Asnieres, France). Genomic DNA and total RNA from cultured cells were isolated with a DNeasy blood and tissue kit and an RNeasy mini kit, respectively (Qiagen, Germantown, MD). Analysis of transgene integration events was performed by competitive polymerase chain reaction (PCR) analysis, using a Taq PCR master mix kit purchased from Qiagen. In vitro-transcribed fVIII RNA standards were generated with an mMessage mMachine kit (Ambion, Austin, TX). fVIII RNA quantitation was performed with SYBR green PCR master mix, TaqMan reverse transcription reagents, and an ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA). Oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). Chemicals were purchased from Fisher Scientific (Pittsburg, PA) unless otherwise specified. Southern and Northern blot analysis was performed with the digoxigenin (DIG) nonradioactive nucleic acid-labeling and detection system (Roche, Indianapolis, IN).
Construction of fVIII expression vectors
ReNeo constructs
BDD-SQ-hfVIII and BDD-OL-pfVIII transgenes in the ReNeo mammalian expression vector were generated as described previously (Doering et al., 2002). Both constructs contain the RHQR PACE (paired basic amino acid-cleaving enzyme)/furin recognition sequence responsible for increased protein processing and, it was previously published that expression data were similar for a BDD porcine fVIII transgene containing either the SQ linker or the OL linker (Doering et al., 2004). Human B domain-less fVIII (BDD-hfVIII) in the ReNeo vector was a gift from P. Lollar (Emory University, Atlanta, GA). L303E/F309S BDD-SQ-hfVIII (BDD-DM-SQ-hfVIII) and L303E/F309S BDD-hfVIII (BDD-DM-hfVIII) were created by PCR-mediated site-directed mutagenesis of BDD-SQ-hfVIII and BDD-hfVIII template DNA, respectively, using a Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA) with forward primer 5′-GAAGAGATATGACAAGACAGTAGAAA-3′ and reverse primer 5′-CTCTTGATGGACGAAGGACAGTTTCT-3′. BDD-N6-SQ-hfVIII was created by XhoI/AvrII digest of full-length, wild-type fVIII and ReNeo/BDD-SQ-hfVIII plasmid DNA followed by gel purification and ligation of the resulting 2282- and 10,163-bp fragments, respectively. BDD-N6-hfVIII was similarly created by subcloning of the 2282-bp XhoI/AvrII fragment into the BDD-hfVIII construct. BDD-DM-N6-hfVIII was created by subcloning the 2282-bp XhoI/AvrII fragment into the BDD-DM-hfVIII construct. All ReNeo constructs drive transgene expression from an adenovirus type 2 major late promoter.
pcDNA5/FRT constructs
BDD-SQ-hfVIII and BDD-OL-pfVIII in the pcDNA5/FRT vector (pcDNA5/FRT/BDD-OL-pfVIII and pcDNA5/FRT/BDD-SQ-hfVIII) were gifts from P. Lollar. pcDNA5/FRT/BDD-hfVIII was created by SpeI/XbaI digest of ReNeo/BDD-hfVIII and pcDNA5/FRT/BDD-SQ-hfVIII plasmid DNA followed by gel purification and ligation of the resulting 3636- and 5718-bp fragments, respectively. pcDNA5/FRT/BDD-DM-SQ-hfVIII was created by SpeI/MluI digest of ReNeo/BDD-DM-SQ-hfVIII and pcDNA5/FRT/BDD-SQ-hfVIII plasmid DNA followed by gel purification and ligation of the resulting 784- and 8612-bp fragments, respectively. pcDNA5/FRT/BDD-DM-hfVIII was similarly created by subcloning of the 784-bp SpeI/MluI fragment into the pcDNA5/FRT/BDDhfVIII construct. pcDNA5/FRT/BDD-N6-SQ-hfVIII was created by SpeI/XbaI digest of ReNeo/BDD-N6-SQ-hfVIII and pcDNA5/FRT/BDD-SQ-hfVIII plasmid DNA followed by gel purification and ligation of the resulting 4371- and 5718-bp fragments, respectively.
pcDNA5/FRT/BDD-N6-hfVIII was created by SpeI/XbaI digest of ReNeo/BDD-N6-hfVIII and pcDNA5/FRT/BDD-hfVIII plasmid DNA followed by gel purification and ligation of the resulting 4329- and 5718-bp fragments, respectively. pcDNA5/FRT/BDD-DM-N6-hfVIII was created by MluI/XbaI digest of ReNeo/BDD-N6-hfVIII and pcDNA5/FRT/BDD-hfVIII plasmid DNA followed by gel purification and ligation of the resulting 3545- and 6502-bp fragments, respectively.
Lentiviral vector production
Lentiviral vectors containing fVIII transgenes were manufactured by Lentigen (Gaithersburg, MD). Briefly, BDD-SQ-hfVIII and BDD-OL-pfVIII were subcloned into the LentiMax expression vector, a highly optimized HIV-based lentiviral vector. Transgene expression was driven by the human elongation factor-1α (EF-1α) promoter and research-grade lentiviral vector (LV) particles were manufactured with the LentiMax production system. Titers were obtained from the manufacturer by quantitative PCR (qPCR) for the gag region of the LV vector on extracted genomic DNA from transduced 293FT cells. LV particles were subsequently titered by performing competitive PCR analysis for fVIII transgene (as described later) on extracted genomic DNA from transduced human embryonic kidney (HEK)-293T cells. Lenti-GFP, a LentiMax LV expressing the enhanced green fluorescent reporter protein (eGFP), was used in transduction experiments as a negative control.
Cell culture, transfection, transduction, and clonal selection
Transient transfection of fVIII ReNeo vectors
Baby hamster kidney-derived (BHK-M) cells were grown to 90% confluency in 24-well plates containing 1 ml of Advanced DMEM–F12 supplemented with 10% FBS. Cells were transfected with 0.8 μg of plasmid DNA from each ReNeo construct and 2 μl of Lipofectamine 2000 per well in 0.5 ml of medium. Medium was changed to fresh Advanced DMEM–F12 with 10% FBS the next morning. Twenty-four hours later, medium was replaced with 0.5 ml of serum-free AIM V medium. Cells were cultured another 24 hr before fVIII activity was measured by one-stage coagulation assay.
Stable transfection of fVIII ReNeo vectors
BHK-M cells were grown to 90% confluency in 6-well plates containing 2 ml of Advanced DMEM–F12 medium supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml). Cells were transfected with 4 μg of plasmid DNA from each ReNeo construct and 2 μl of Lipofectamine 2000 per well in 2 ml of Opti-MEM medium. Twenty-four hours after transfection, cells were split into 100-mm cell culture plates and cultured in Advanced DMEM–F12 supplemented with 10% FBS, penicillin (100 units/ml), streptomycin (100 μg/ml), and G-418 (0.5 mg/ml) for 10–14 days. Visible colonies were picked with a pipette tip and transferred to a 96-well plate. Cells were then expanded onto a 24-well plate and grown to 90% confluency, before changing medium to serum-free AIM V medium. fVIII activity was measured by one-stage coagulation assay 24 hr later.
Stable transfection of fVIII pcDNA5/FRT vectors
Three million Flp-In HEK-293 cells were plated in 10 ml of DMEM containing
Lentiviral transduction
HEK-293T cells were maintained in complete Advanced DMEM–F12 supplemented with 10% FBS, 1% penicillin–streptomycin solution (100 units/ml each), and 2 mM GlutaMax at 37°C with 5% CO2. Transductions of LentiMax LV particles at various multiplicities of infection (MOIs) were performed by incubating approximately 100,000 cells per well plated onto collagen-1-coated 24-well plates (BioCoat; BD Biosciences, San Jose, CA) with virus in a final volume of 500 μl of complete Advanced DMEM–F12 supplemented with Polybrene (8 μg/ml; Specialty Media, Phillipsburg, NJ). Twenty-four hours posttransduction, virus-containing medium was replaced with fresh complete Advanced DMEM–F12 medium and transduced cells were cultured until reaching approximately 70–90% confluence, at which point each sample was transferred equally onto three wells of a 6-well plate for measurement of fVIII activity, integration events, and transcript expression as described later. Clonal isolation was performed by single-cell cloning by limiting dilution onto 96-well plates and visual examination of wells for single cells. After several weeks in culture, single-cell clones were expanded in sequence from a 96-well plate onto 48-well, 24-well, 12-well, 6-well, and 100-mm dishes for further testing.
Measurement of fVIII activity
For all in vitro studies, fVIII activity was measured with the activated partial thromboplastin reagent-based one-stage coagulation assay in an STart coagulation instrument (Diagnostica Stago) using human fVIII-deficient plasma as the substrate as previously described (Doering et al., 2002). All coagulation assays were carried out on cells cultured in low serum-containing Advanced DMEM–F12 medium (2% FBS) or serum-free (AIM V) medium for 24 hr before assaying fVIII activity in conditioned medium. Cells were counted at the time of assay in order to express fVIII levels as units per 106 cells per 24 hr. For in vivo fVIII measurement, blood was collected from mice through the retro-orbital plexus and fVIII was measured in a chromogenic substrate assay (Coatest SP FVIII; DiaPharma, West Chester, OH) as described previously (Doering et al., 2007).
Measurement of fVIII transgene copy number
Total DNA from transduced cells was isolated with the DNeasy blood and tissue kit in accordance with the manufacturer's protocol for cells. DNA was quantitated spectrophotometrically by absorbance at 260 nm (A 260) in distilled H2O. To determine transgene copy number, a competitive PCR protocol was developed in which a known amount of fVIII copies amplified from the pUA2 plasmid was added to the PCR reaction tube containing unknown total DNA. The derived product from the pUA2 plasmid comprises an fVIII DNA fragment containing a 70-bp deletion that can be distinguished from the integrated fVIII transgene. During the PCR, the pUA2 template competes with the integrated fVIII transgene in the genomic DNA and the point of equivalent amplification is used to determine the number of fVIII transgenes in the unknown sample. The PCRs were carried out in a 25-μl total volume containing 1× Taq PCR master mix, 2 μM forward and reverse primers, and 100 ng of total DNA. PCR cycling was carried out in a Mastercycler gradient (Eppendorf, Westbury, NY) by performing one cycle at 95°C for 3 min followed by 30 cycles at 95°C for 1 min, 58°C for 1 min, and 72°C for 1 min, and a final extension at 72°C for 10 min. The oligonucleotide primers used were located within the A2 domain of the fVIII sequence at positions 1402–1420 for the forward primer (5′-TGGGACCTTTACTTTATGG-3′) and at positions 1923–1946 for the reverse primer (5′-AAAAACATAGCCATTGATGCTGTG-3′) of the human fVIII cDNA sequence. The PCR products (520 bp for fVIII transgene and 450 bp for competitor) were separated in a 1.5% agarose gel and an ethidium bromide image was obtained with an ultraviolet transilluminator (BioDoc-It system; UVP, Upland, CA). Densitometry of PCR products was obtained with ImageJ version 1.37 software (developed by W. Rasband, National Institutes of Health, Bethesda, MD). Plots were performed to determine the percent competitor density as a function of the copies per reaction and the point of equivalence determined by linear regression analysis.
Measurement of fVIII transcript expression
Total RNA was extracted from cell lines, using the RNeasy mini kit in accordance with the manufacturer's instructions, and QIAshredder homogenizers. RNA concentrations were determined by A 260 in 10 mM Tris-HCl (pH 7.0). Porcine fVIII RNA standard, used for absolute quantitation of fVIII transcripts by quantitative RT-PCR, was generated as described previously (Doering et al., 2002). PCRs were carried out in a 25-μl total volume containing 1× SYBR green PCR master mix, 300 μM forward and reverse primers, 12.5 units of MultiScribe reverse transcriptase (Applied Biosystems), 10 units of RNase inhibitor, and 5 ng of sample RNA. PCRs using the porcine fVIII RNA standard also included 5 ng of yeast tRNA to mimic the RNA environment of sample RNA, using serial dilutions (102–106 transcripts per reaction) as template RNA. The oligonucleotide primers used were located in the fVIII sequence encoding the A2 domain located at positions 2047–2067 for the forward primer (5′-ATGCACAGCATCAATGGCTAT-3′) and positions 2194–2213 for the reverse primer (5′-GTGAGTGTGTCTTCATAGAC-3′) of the human fVIII cDNA sequence. Primers recognize both human and porcine fVIII, as sequences in this region are identical between both templates. One-step real-time RT-PCR was performed by incubation at 48°C for 30 min for reverse transcription followed by one cycle at 95°C for 10 min, and 40 amplification cycles of 90°C for 15 sec and then at 60°C for 1 min. Postreaction dissociation analysis was performed to confirm single-product amplification. Absolute quantitation was performed as described previously (Doering et al., 2002).
Northern blot analysis
Total RNA was extracted from cell lines with the RNeasy mini kit in accordance with the manufacturer's instructions, using QIAshredder homogenizers. RNA concentrations were determined as described previously. A total of 2.5 or 5 μg of RNA sample was separated, transferred, hybridized, and detected according to a protocol involving denaturing conditions in a 1% agarose–formaldehyde–MOPS (4-morpholinepropanesulfonic acid) gel according to the technique outlined in the DIG Application Manual for Filter Hybridization (Roche). Digoxigenin-labeled RNA molecular weight marker I (Roche) was used as a marker. Hybridization was performed with a PCR DIG-labeled probe specific to either the porcine A1 domain (A1 probe) or the human A2 domain (A2 probe) generated with the PCR DIG probe synthesis kit (Roche). The oligonucleotide primers used to generate for the A1 probe were as follows: forward (5′-CGGCAAAGTGAACTCCTCCGTG-3′) and reverse (3′-CTGACGTGAGCCTCCATGCCACCA-5′) from the porcine fVIII cDNA sequence. The oligonucleotide primers used to generate for the A2 probe were as follows: forward (5′-ACATTGCTGCTGAAGAGGAGGACT-3′) and reverse (3′-ATGTTGGAGGCTTGGAACTCTGGA-5′) from the human fVIII cDNA sequence. Posthybridization wash and block for immunological detection of DIG-labeled probes was performed with the DIG wash and block buffer set (Roche) at a dilution of anti-DIG–AP of 1:25,000. CDP-Star (Roche) was used for chemiluminescent substrate detection. Imaging was performed with a luminescence image analyzer (LAS-3000; Fujifilm, Tokyo, Japan) followed by X-ray film exposure (Eastman Kodak, Rochester, NY).
Southern blot analysis
To confirm that pcDNA/FRT clones contained a single integration event, total genomic DNA was isolated from transfected cells with the DNeasy blood and tissue kit (Qiagen) and analyzed by Southern blot. Eight micrograms of genomic DNA from each stable clone was digested with ScaI and fragments were separated on a 1% agarose gel. DNA was transferred by capillary action, using 20 × SSC (saline–sodium citrate) transfer buffer to a positively charged nylon membrane and hybridized overnight with a probe complementary to either the porcine A1 domain or the human A2 domain. Probes were created by PCR-mediated digoxigenin labeling, using the DIG nonradioactive nucleic acid labeling and detection system (Roche). Stable transfectants with the correct single integration were confirmed by a single band at approximately 12,460 bp for BDD-OL-pfVIII clones, 12,430 bp for BDD-SQ-hfVIII and BDD-DMSQ-hfVIII clones, 13,120 bp for BDD-N6-SQ-hfVIII clones, 12,390 bp for BDD-hfVIII and BDDDM-hfVIII clones, and 13,080 bp for BDD-N6-hfVIII and BDD-DM-N6-hfVIII clones. All clones also produced a band at 8.6 and 5.5 kb when probed with the human A2 probe because of homology with two portions of the endogenous fVIII sequence when digested with ScaI. Transduced HEK-293T clones were also analyzed by Southern blot in order to determine the number of integration events using 5 μg of genomic DNA digested with AvrII.
Hematopoietic stem cell isolation, transduction, and transplant
Bone marrow was harvested from femurs and tibias of wild-type C57BL/6 mice and enriched for Sca-1+ cells using positive immunomagnetic bead selection, resulting in a 70–90% Sca-1+ cell population. Cells were cultured in serum-free medium and stimulated for 2 days with murine stem cell factor (100 ng/ml), murine interleukin-3 (20 ng/ml), human interleukin-11 (100 ng/ml), and human Flt3 ligand (100 ng/ml). On days 3 and 4, cells were transduced consecutively with 2.5 functional viral particles (FVP)/cell, using either SIV-BDD-SQ-hfVIII or SIV-BDD-OL-pfVIII. Plasmids necessary for generating simian immunodeficiency viruses (SIVs) were a kind gift from A. Nienhuis (Hanawa et al., 2004). Transduced cells were transplanted into exon 16-disrupted hemophilia A mice by tail vein injection at a dose of 7.5 × 105 cells per mouse. Mice were 8–12 weeks old at the time of transplantation and were lethally irradiated (11 Gy, split dose) before transplantation with a Gammacell 40 Exactor (Nordion, Ottawa, ON, Canada).
Statistical analyses
Analysis of variance (ANOVA) was used for all comparisons in which more than two groups were compared. When only two groups were compared, the Student t test or Mann–Whitney U test was employed.
Results
Expression of bioengineered fVIII constructs in heterologous mammalian cells
To determine the highest expressing fVIII transgene and overcome the barrier of low transgene expression associated with hemophilia A gene therapy applications, we compared the expression of several previously described bioengineered fVIII transgenes. The following fVIII variants were generated: BDD human fVIII (Toole et al., 1986) (herein referred to as BDD-hfVIII); BDD-hfVIII containing the L303E/F309S double mutation (Swaroop et al., 1997) (herein referred to as BDD-DM-hfVIII); BDD-hfVIII containing six N-linked glycosylation sites (Miao et al., 2004) (herein referred to as BDD-N6-hfVIII); BDD-hfVIII containing the double mutation and six N-linked glycosylation sites (herein referred to as BDD-DM-N6-hfVIII); BDD-hfVIII containing a 14-amino acid SQ linker between the A2 domain and the activation peptide, which includes the RHQR recognition sequence to increase secretion via PACE/furin processing (Seidah and Chretien, 1997; Doering et al., 2002) (herein referred to as BDD-SQ-hfVIII); BDD-SQ-hfVIII containing L303E/F309S (herein referred to as BDD-DM-SQ-hfVIII); BDD-SQ-hfVIII containing six N-linked glycosylation sites (herein referred to as BDD-N6-SQ-hfVIII); and BDD porcine fVIII containing a 24-amino acid linker (designated OL) between the A2 domain and the activation peptide, described previously (Doering et al., 2002) (herein referred to as BDD-OL-pfVIII) (Fig. 1). All fVIII cDNAs were cloned into the ReNeo mammalian expression vector. Transgene sequences were confirmed by automated DNA sequencing.
To compare fVIII expression, BHK-M cells were transfected with plasmids encoding each transgene and one-stage coagulation assays were performed after incubation for 24 hr in serum-free medium. No significant differences were observed among the human fVIII transgenes. However, transient expression of BDD-OL-pfVIII was approximately 15-fold higher than any of the human fVIII variants (p < 0.001, ANOVA) (Fig. 2A). Stable clones (n = 17–42) were selected by antibiotic resistance, and expanded in 6-well plates. Cells were incubated for 24 hr in serum-free medium before assaying for fVIII activity by one-stage coagulation assay. Again, no significant differences were observed among the human fVIII transgenes. However, mean expression of BDD-OL-pfVIII was 10 units/106 cells/24 hr, which was 5- to 16-fold higher than for any of the human fVIII variants (p < 0.001, ANOVA) (Fig. 2B).
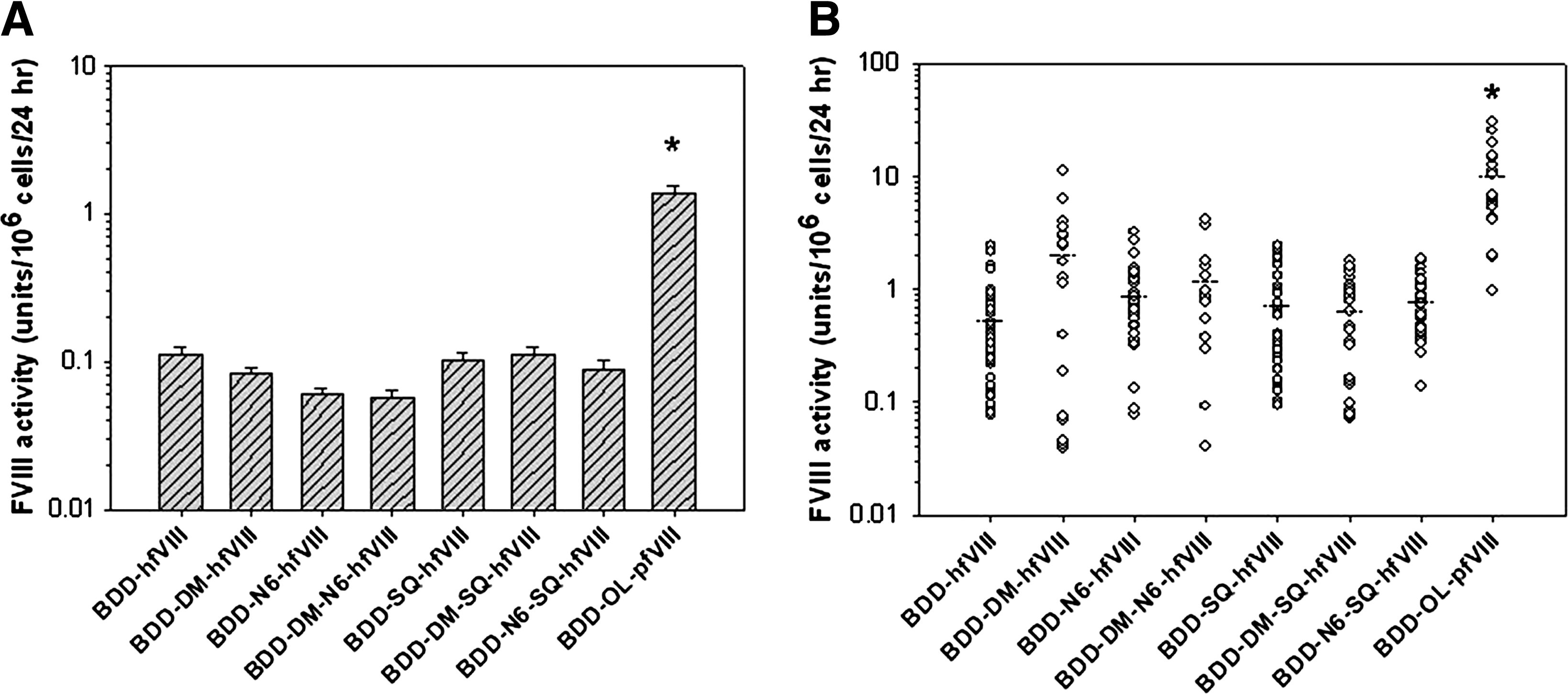
Heterologous expression of fVIII transgenes. BHK-M cells were transfected with fVIII transgenes in the ReNeo mammalian expression vector. (
Comparison of fVIII expression using targeted single transgene integration
Because of the large variances in fVIII expression observed for the human fVIII transgenes in transient and stable transfection experiments using BHK-M cells, we performed an additional, potentially more direct comparison of transgene expression. The Flp-In system is designed to allow expression of a single transgene from a specific location in the genome, thus eliminating the effects of random integration and position effect variegation that may have led to the high variability in fVIII expression observed among the BHK-M clones. We used Flp-In HEK-293 cells that were engineered to contain a single FLP recombination target (FRT) site. The FRT site serves as the binding and cleavage site for the recombinase and is located at the 5′ end of the open reading frame of a β-galactosidase/Zeocin fusion gene (lacZeo). This cell line is Zeocin resistant, lacZ positive, and hygromycin sensitive before transfection with the pcDNA5/FRT expression vector. As shown in Fig. 3A, FLP recombinase mediates site-specific recombination between the FRT site in the host cell line and the FRT site in the pcDNA5/FRT/fVIII expression vector, allowing transgene integration into the same transcriptionally active genomic location in every cell. After recombination, the fVIII transgene is expressed under the control of a cytomegalovirus (CMV) promoter and resistance to hygromycin B is driven by a simian virus 40 (SV40) promoter to allow selection of genetically modified cells. The efficiency of FLP-mediated, targeted recombination and ease in generating isogenic cell lines makes this an effective and potentially more predictive in vitro system for the comparison of relative transgene expression properties (Wirth and Hauser, 2004; Liu et al., 2006).
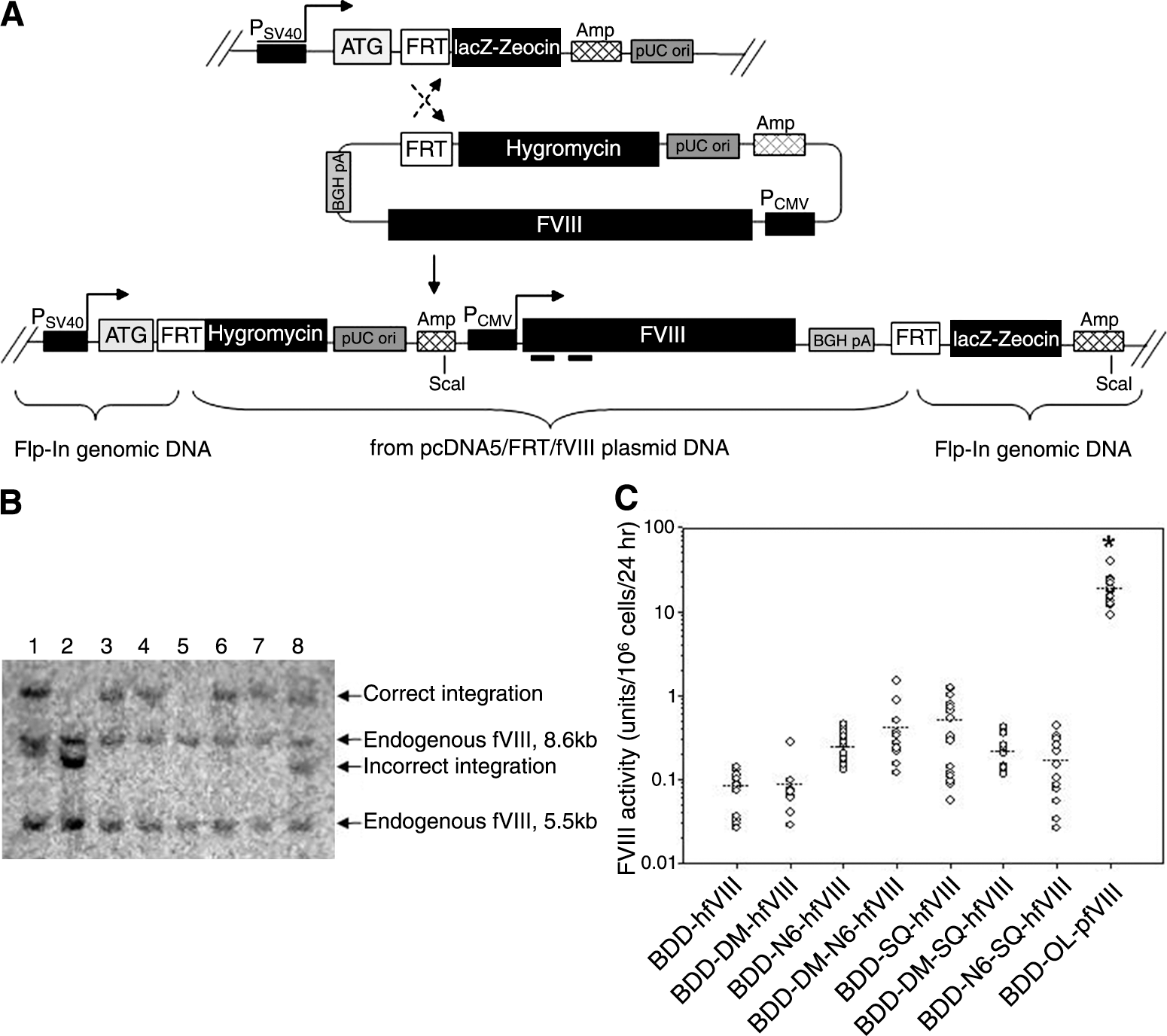
fVIII expression using the Flp-In system. (
After selection of 20–40 clones per fVIII transgene, the clonal cell lines were expanded and cultured for 24 hr in serum-free medium before fVIII activity was measured by one-stage coagulation assay. Cell viability and growth rates were also determined and found to be similar among all fVIII-expressing clones (data not shown). Genomic DNA was harvested from the clones with fVIII activity and screened by Southern blot to identify clones with multiple and/or incorrect integrations. A representative Southern blot analysis of genomic DNA from clonal cell lines expressing various correctly and incorrectly integrated human fVIII transgenes is shown in Fig. 3B. All clones exhibited bands at 8.6 and 5.5 kb when probed with the human A1 probe. These bands represent endogenous fVIII intron-containing sequences present in Flp-In HEK-293 cells. Transgene integration into the correct genomic location only is shown in lanes 3, 4, 6, and 7. Lanes 1 and 8 depict transgene integration into the correct location as well as an additional random integration potentially resulting in expression of two copies of the fVIII transgene. Lane 2 shows an incorrect integration event and lane 5 represents a clone with no probe-specific fVIII transgene sequence in the genome. Figure 3C shows the relative fVIII activity from Flp-In HEK-293 clones expressing a single correctly targeted fVIII transgene. Mean BDD-OL-pfVIII activity was 18 units/106 cells/24 hr, which was 36- to 225-fold higher than for any of the human transgenes (p < 0.001, ANOVA). Although no significant differences were found among human transgenes, mean BDD-SQ-hfVIII expression was the greatest at 0.5 unit/106 cells/24 hr, and only slightly higher than BDD-DM-N6-hfVIII with a mean fVIII expression of 0.4 unit/106 cells/24 hr. A 5.5-fold increase in fVIII expression was observed with the addition of the N6 segment to BDD-hfVIII (0.25 and 0.08 unit/106 cells/24 hr, respectively), which is similar to a previously published report (Miao et al., 2004). These data further demonstrate that BDD-OL-pfVIII is expressed at significantly higher levels than are previously characterized human fVIII transgenes.
Characterization of fVIII production rates
It was shown previously that BDD-OL-pfVIII and BDD-SQ-hfVIII have similar specific activities and similar steady state levels of fVIII mRNA in transfected cell lines, suggesting that the increase in fVIII expression from BDD-OL-pfVIII is due to increased secretion resulting from more efficient translational and/or posttranslational modifications (Doering et al., 2002). To better understand the kinetics of fVIII in a cell culture system, we monitored the relative production and decay of BDD-OL-pfVIII and BDD-SQ-hfVIII in vitro. fVIII decay rates were assessed by adding 1–100 units of purified BDD-OL-pfVIII or BDD-SQ-hfVIII to a 6-well plate containing a nearly confluent layer of control Flp-In HEK-293 cells, and fVIII activity was measured by one-stage coagulation assay at various time points. The resulting time course analysis confirmed that BDD-OL-pfVIII and BDD-SQ-hfVIII exhibit similar decay rates in vitro (data not shown), suggesting that the higher fVIII activity levels observed for BDD-OL-pfVIII are attributable to increased biosynthesis and not to increased stability in conditioned medium. To confirm the increased rate of protein production for BDD-OL-pfVIII, cells were plated from BDD-OL-pfVIII and BDD-SQ-hfVIII Flp-In HEK-293 clones expressing a correctly targeted single transgene and fVIII activity was measured over time in serum-free medium by one-stage coagulation assay. Specific clones were selected whose 24-hr fVIII activity measurement was within 1 standard deviation of the mean expression level for that transgene. The rate of BDD-OL-pfVIII production was 18-fold greater than the rate of BDD-SQ-hfVIII production with a BDD-OL-pfVIII production rate of 0.92 unit/106 cells/hr and a BDD-SQ-hfVIII production rate at 0.05 unit/106 cells/hr (Fig. 4). The BDD-OL-pfVIII clone produced 75 fVIII molecules per cell per second, whereas the BDD-SQ-hfVIII clone produced 5 fVIII molecules per cell per second. These data confirm that BDD-OL-pfVIII is produced at a faster rate than BDD-SQ-hfVIII in vitro. Quantitative RT-PCR performed on RNA isolated from each clone confirmed that the BDD-OL-pfVIII and BDD-SQ-hfVIII clones expressed a similar number of transcripts (data not shown), thus confirming that the increase in fVIII expression from BDD-OL-pfVIII is due to more efficient secretion.
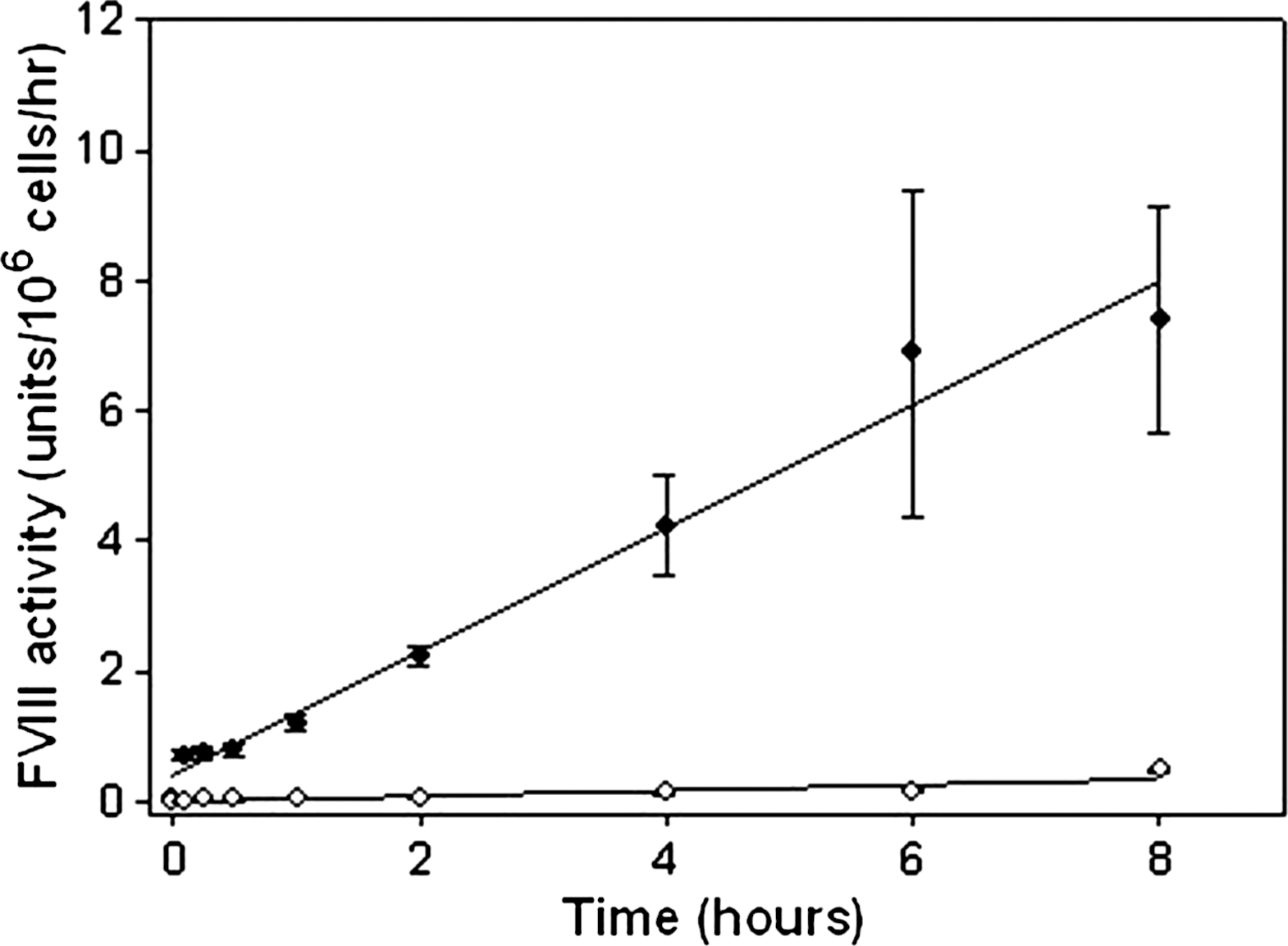
Kinetics of fVIII production. Two million cells were plated in 6-well plates and cultured for 48 hr before washing with PBS and replacing medium with serum-free medium. fVIII activity was measured by one-stage coagulation assay at time = 0, 5, 15, and 30 min, and then at 1, 2, 4, 6, and 8 hr in order to determine the rates of fVIII production from each clone. The BDD-OL-pfVIII clone is represented by the solid diamonds and the BDD-SQ-hfVIII clone is represented by the open diamonds. Each circle represents the mean value of three independent experiments ± sample standard deviation at each time point.
High-level expression of BDD-OL-pfVIII using lentiviral gene transfer
Lentiviral vectors are an attractive tool for transgene delivery as they have the potential to transfer proviral DNA sequences into the genome of dividing and nondividing cells, thereby generating a source of long-term transgene expression. To compare human and porcine fVIII expression in cells after lentivirus-mediated gene transfer, self-inactivating HIV-1-based lentiviral vectors encoding either BDD-OL-pfVIII or BDD-SQ-hfVIII were generated (Fig. 5A). HEK-293T cells were transduced at low multiplicities of infection (MOIs) equal to 0.3, 0.9, and 2.7, in order to achieve low-level integration of proviral DNA. At 7 days posttransduction, BDD-OL-pfVIII expression levels were on average 7- to 8-fold higher compared with BDD-SQ-hfVIII (Fig. 5B). At the highest MOI, BDD-OL-pfVIII expression was 38 units/106cells/24 hr compared with 4.5 units/106 cells/24 hr for BDD-SQ-hfVIII. To ensure that the BDDhfVIII expression was not toxic to the cells, cell viability and growth rates were determined and found to be similar among GFP control and hfVIII-expressing cells (data not shown). Quantitative RT-PCR analysis performed on total RNA isolated from transduced HEK-293T cells indicated that the increased BDD-OL-pfVIII expression was not the result of disproportionately high steady state mRNA levels. In the populations tested, BDD-SQ-hfVIII-expressing cells had higher steady state mRNA levels than did BDD-OL-pfVIII-expressing cells at each MOI examined (Fig. 5C). When fVIII activity was normalized to transcript expression, BDD-OL-pfVIII expression was 25-fold higher than that of BDD-SQ-hfVIII. Northern blot analysis performed on the same samples, using either a human- or porcine-specific probe, showed expression of a single transcript of approximately 6300 nucleotides (Fig. 5D). To further characterize expression of BDD-SQ-hfVIII and BDD-OL-pfVIII from transduced HEK-293T cells, clones were isolated by limiting serial dilution 6 weeks posttransduction. Clonal BDD-OL-pfVIII expression was greater than that of BDD-SQ-hfVIII, ranging from 0.05 to 57 units/106 cells/24 hr and from 0.02 to 4.6 for BDD-OL-pfVIII and BDD-SQ-hfVIII, respectively (Fig. 6A). A significant difference in the median expression level of BDD-OL-pfVIII or BDD-SQ-hfVIII was observed from clones containing a single integration event (p < 0.001, Mann–Whitney U test) as well as from clones containing multiple integrants (p = 0.008, Mann–Whitney U test). Of note, BDD-OL-pfVIII clones derived from the population transduced at an MOI of 2.7 exhibited a lower number of integrations than did BDD-SQ-hfVIII clones at the same MOI. Although the viral titer of BDD-OL-pfVIII is slightly lower than that of BDD-SQ-hfVIII, cells were transduced at equivalent MOIs in order to prevent differences in the amount of virus introduced into the cells. Therefore, the difference in number of integrations cannot be due to differences in viral titer. Southern blot analysis was used to quantify integration of proviral transgene DNA within all clones and demonstrated a mean proviral copy number per clone for BDD-SQ-hfVIII of 0.9 and 2.2, and for BDD-OL-pfVIII of 0.9 and 1.5, at MOIs of 0.9 and 2.7, respectively. Band sizes were above the minimum expected after genomic DNA digestion with AvrII, a restriction enzyme that recognizes a single site within the transgene cassette, indicating successful integration (Fig. 6B). Steady state fVIII mRNA levels for all stable clones were measured by quantitative RT-PCR (Fig. 6C). Linear regression analysis revealed a significant correlation between fVIII activity and fVIII transcripts for both BDD-SQ-hfVIII and BDD-OL-pfVIII (p = 0.005 and p < 0.0001, respectively; two-tailed Student t test). Several clones that exhibited transcript levels below the detection limit and fVIII activity close to or below the baseline (0.03 units/106 cells/24 hr) were excluded from the analysis. These data demonstrate that the high-level BDD-OL-pfVIII expression achieved after transfection of heterologous mammalian cells is reproducible in cells modified by lentivirus-mediated gene transfer.

Expression of BDD-SQ-hfVIII and BDD-OL-pfVIII after lentiviral transduction of HEK-293T cells. (

Expression of BDD-SQ-hfVIII and BDD-OL-pfVIII from individual HEK-293T transduced clones. (
In vivo comparison of hfVIII and pfVIII expression
To compare lentiviral vector expression of pfVIII and hfVIII, Sca-1+ cells were isolated from donor mice and transduced with a self-inactivating lentivirus expressing either BDD-SQ-hfVIII or BDD-OL-pfVIII. SIV-transduced cells were transplanted into hemophilia A mice via tail vein injection and plasma fVIII levels were measured over time (Fig. 7). Plasma from mice transplanted with SIV-BDD-SQ-hfVIII-modified cells did not contain detectable fVIII activity at any time point, whereas mice transplanted with SIV-BDD-OL-pfVIII-modified cells showed sustained fVIII activity for at least 1 year.

In vivo expression of fVIII from genetically modified hematopoietic stem cells. Plasma fVIII levels were determined in hemophilia A mice after transplantation of Sca-1+ cells modified ex vivo with a lentivirus expressing either BDD-SQ-hfVIII (solid circles) or BDD-OL-pfVIII (open circles). Mice (n = 6 per group) were transplanted via tail vein injection with 7.5 × 105 cells transduced at an MOI of 2.5 twice. By week 26, three mice from each group had died from unknown causes.
Discussion
Low-level fVIII expression has been a limiting factor for both the commercial production of recombinant fVIII as well as the success of hemophilia A gene therapy. With respect to gene therapy, a number of preclinical studies have demonstrated therapeutic levels of human fVIII and phenotypic correction of bleeding in mice, using oncoretroviral vectors (VandenDriessche et al., 1999; Moayeri et al., 2004, 2005), adeno-associated vectors (Chao et al., 2000), and adenoviral vectors in mice (Balague et al., 2000; Reddy et al., 2002; Cerullo et al., 2007) and dogs (Chuah et al., 2003; McCormack et al., 2006). In a clinical setting, however, hemophilia A gene therapy has been unsuccessful. To date, three clinical trials for hemophilia A have been initiated. The first two trials used BDD-hfVIII and the third used full-length hfVIII; none was able to achieve sustained fVIII levels greater than 1% without treatment-related adverse effects (Chuah et al., 2004). On the basis of these clinical and preclinical studies, it is clear that improving fVIII expression is critical for future success.
Recombinant DNA technology has led to the development of fVIII molecules with increased half-life (Pipe and Kaufman, 1997), decreased immunogenicity (Lubin et al., 1997; Barrow et al., 2000; Saenko et al., 2003b), and increased rates of processing and secretion (Swaroop et al., 1997; Doering et al., 2002, 2004; Miao et al., 2004). Significant advancements also are being made in vector development including the use of stronger promoters or enhancer elements to drive expression of transgenes, although this may lead to increased risk of insertional mutagenesis when employed in retroviral vectors (Papadakis et al., 2004; West and Fraser, 2005). The focus of the current study was to compare the expression levels of the various bioengineered fVIII transgenes in order to determine the optimal one for use in clinical gene transfer. A number of bioengineered high-expression human fVIII constructs and a porcine fVIII construct were compared in order to determine which is expressed at the greatest level. Among the human fVIII transgenes, we evaluated differences in expression due to specific modifications previously shown to decrease interaction with the resident ER protein chaperone BiP and to increase carbohydrate-mediated transport from the ER to the Golgi (Dorner et al., 1987; Marquette et al., 1995). Our results show that BDD-OL-pfVIII is the optimal fVIII transgene with respect to expression levels, as it is expressed at significantly greater levels than any human fVIII variant when tested both in vitro and in vivo.
On transfection of each transgene into BHK-M cells, no significant differences in expression were observed among the human fVIII constructs. Although in each case BDD-OL-pfVIII expression was significantly greater, no conclusions could be made regarding differences in expression from the human transgenes. To decrease the interclonal expression variability, we expressed each transgene in the Flp-In system and compared expression from the resulting isogenic cell lines. The advantage of this system is that the integration of the gene of interest occurs in the exact same genomic location in each cell. Southern blot analysis can be used to screen for multiple and/or random integrations. Expected transgene integration has been shown to occur with high frequency and phenotypic characterization of expression to remain consistent as long as the transfected cells remain under selective pressure (Liu et al., 2006). Using the Flp-In system, BDD-OL-pfVIII was expressed at significantly greater levels than any human transgene whereas again no significant differences were detected among the human transgenes. BDD-OL-pfVIII was expressed at a 36-fold greater level than BDD-SQ-hfVIII and at a 225-fold greater level than BDD-hfVIII. Increased expression was not due to a disproportionately higher number of transcripts per cell in BDD-OL-pfVIII clones compared with BDD-SQ-hfVIII clones. This is consistent with our previous finding of enhanced secretion resulting in a faster protein production rate (Doering et al., 2002, 2004). At present, recombinant BDD-OL-pfVIII is being evaluated in a phase 2 clinical trial with patients with congenital hemophilia A possessing anti-human fVIII inhibitory antibody titers (Mahlangu et al., 2007). In this trial, BDD-OL-pfVIII was successful in controlling all bleeds, even in the presence of fVIII antibodies. Before the development of recombinant BDD-OL-pfVIII, plasma-derived porcine fVIII was used to treat patients harboring fVIII inhibitor antibodies.
After gene transfer, we previously showed that BDD-OL-pfVIII is expressed at significantly greater levels than BDD-SQ-hfVIII in bone morrow-derived mesenchymal stem cells (Gangadharan et al., 2006). Importantly, BDD-OL-pfVIII expression was sustained at levels greater than 1 unit/ml after HSCT without subsequent immune responses even when reduced intensity, radiation, or chemotherapeutic-based conditioning was used (Ide et al., 2007). Preimmunized hemophilia A mice, harboring clinically significant anti-fVIII titers, treated with the same HSCT and reduced intensity conditioning protocol also exhibited sustained BDD-OL-pfVIII expression, demonstrating the potential for using a porcine fVIII transgene to treat patients harboring inhibitory antibodies to human fVIII (Doering et al., 2007). These previous studies were performed with murine stem cell virus (MSCV) to drive genetic modification and expression of BDD-OL-pfVIII. In the current study, we aimed to compare human and porcine transgene expression in a more clinically relevant lentivirus-mediated gene transfer system. However, in order to ensure complete engraftment of transplanted cells lethal irradiation was used, which also prevents the production of inhibitory antibody formation. Therefore, these studies do not rule out the possibility that the porcine transgene may be immunogenic when translated to human gene therapy applications.
Lentiviruses are emerging as the vector of choice for stable gene delivery because of several attractive features that include (1) efficient transduction of dividing and nondividing cells, (2) the capacity to accommodate large genetic payloads, and (3) stable long-term transgene expression (Cockrell and Kafri, 2007). An increasing body of evidence suggests that lentiviruses are inherently safer than γ-oncoretroviruses, possibly because of differences in integration site preference (Schroder et al., 2002; Wu et al., 2003). To date, no evidence exists to link HIV to cancer. However, lentiviruses possess 3′ and 5′ LTRs, each containing a strong promoter and enhancer, which are capable inducing long-distance gene expression effects. To decrease this risk, self-inactivating (SIN) vectors have been developed in which the U3 regions of the LTRs have been deleted. Previous studies have demonstrated expression of therapeutic levels of fVIII expression in hemophilia A mice after systemic lentiviral gene delivery (Kootstra et al., 2003; Park, 2003; Kang et al., 2005), transplantation of transduced bone marrow cells (Kootstra et al., 2003), and transplantation of bone marrow cells with targeted platelet-specific expression (Ohmori et al., 2006; Shi et al., 2007). In the majority of these studies, only transient levels of fVIII expression where achieved because of the formation of neutralizing antibodies to fVIII, but platelet-specific fVIII expression from either the glycoprotein IIb (GPIIb) or GPIbα promoter has resulted in correction of bleeding in transplanted hemophilia A mice without inhibitor development (Ohmori et al., 2006; Shi et al., 2007). Using an alternative approach, Matsui and colleagues (2007) demonstrated in vivo fVIII expression from implanted lentivirally transduced blood outgrowth endothelial cells (BOECs) in hemophilia A mice, but an anti-fVIII humoral immune response develops unless tolerization or immunosuppression is employed.
Using an HIV-1-based, SIN lentiviral vector, we tested transduction efficiency and expression levels of BDD-OL-pfVIII compared with BDD-SQ-hfVIII in human cells. HEK-293T cells were efficiently transduced by the lentiviral vectors and expressed either BDD-SQ-hfVIII or BDD-OL-pfVIII, demonstrating a positive correlation between fVIII expression and MOI. Similar to our previous observations, BDD-OL-pfVIII exhibited enhanced expression compared with BDD-SQ-hfVIII, with a 7- to 8-fold differential in expression at each MOI tested. Analysis of steady state RNA levels indicated that higher BDD-OL-pfVIII expression was not due to increased transcript levels, which is consistent with our previous findings that the high-expression elements observed in porcine fVIII act through posttranscriptional differences that lead to enhanced secretion (Doering et al., 2004). fVIII biosynthesis from individual BDD-SQ-hfVIII- or BDD-OL-pfVIII-expressing stable clones correlated with steady state RNA levels and integration events. BDD-OL-pfVIII expression from individual clones was significantly greater than BDD-SQ-hfVIII expression when normalized to DNA copy number. Southern blot analysis revealed a number of clones with integration events but no detectable levels of fVIII expression, possibly because of transcriptional silencing or insertion site-dependent positional effects. Transgene expression driven by lentiviral vectors can be subject to epigenetic silencing by a number of pathways including DNA methylation and chromatin modification (Hamaguchi et al., 2000; Yao et al., 2004; Hong et al., 2007). Genomic silencing mechanisms described to date for lentiviral vectors are not well established and may be dependent on cell type, differentiation state, and the specific promoter used. One study of SIN lentivirus-mediated factor IX expression in bone marrow chimeras showed decreasing transgene expression within weeks of transplantation using the EF-1α promoter, resulting in barely detectable factor IX expression by week 30. However, stable transgene expression was maintained over time when driven by the β-globin locus control region (Chang et al., 2006). Southern blot analysis of BDD-SQ-hfVIII and BDD-OL-pfVIII clones also indicated efficient transduction at the MOIs tested, resulting in the expected range of one to three proviral integrants per cell. High-level BDD-OL-pfVIII expression was observed at low DNA copy numbers, suggesting a decreased risk of insertional mutagenesis because of the requirement for fewer insertional events to achieve therapeutic levels of fVIII expression. Interestingly, although efficient transduction was achieved in human cells, it was observed that our HIV-based lentiviral vectors did not efficiently transduce murine Sca-1+ hematopoietic stem and progenitor cells, resulting in undetectable plasma fVIII activity in vivo (our unpublished observation). However, when the fVIII transgenes were expressed in an SIV vector, efficient Sca-1+ cell transduction was achieved, resulting in sustained fVIII expression in mice transplanted with SIV-BDD-OL-pfVIII-transduced Sca-1+ cells. In summary, we have demonstrated significantly enhanced expression of BDD-OL-pfVIII compared with bioengineered human fVIII constructs, using both viral and nonviral methods. These data support the use of lentiviral vector-mediated gene transfer and expression of BDD-OL-pfVIII in clinical gene therapy for hemophilia A.
Footnotes
Acknowledgments
This work was supported by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL083531and HL084132) to C.B.D., Children's Healthcare of Atlanta, and the Georgia Research Alliance to H.T.S. Dr. Nienhuis kindly provided the plasmids necessary for generating SIV-based lentiviruses.
Author Disclosure Statement
The authors declare no conflict of interest.