
Abstract
Deprivation of neurotrophic factors contributes to the pathogenesis of diabetic neuropathy. However, the role of glial cell-derived neurotrophic factor (GDNF) in the pathogenesis of diabetic neuropathy remains unclear. The present study evaluated the pathogenic role of GDNF deficiency and the therapeutic potential of GDNF gene transfer for diabetic neuropathy. After injection of streptozotocin (STZ) for 2 weeks, diabetic rats displayed significant alteration in electrophysiological parameters, which was associated with structural changes and defective myelination in the sciatic nerves. The early diabetic neuropathy was accompanied by attenuation of the GDNF/GFRα1/Akt signaling cascade and depletion of sensory neuropeptides in the peripheral nerves. After detection of neuropathy, intramuscular GDNF gene transfer reversed the deficiency of GDNF/Akt signaling in the sciatic nerve and improved the neurological functions of diabetic rats. Moreover, GDNF gene delivery alleviated the axonal demyelination and restored the sensory neuropeptide levels in the sciatic nerve of diabetic rats. In summary, peripheral GDNF gene delivery ameliorates the diabetes-induced downregulation of the GDNF signaling complex in the peripheral nervous system and holds promises for treatment of diabetic neuropathy.
Introduction
Peripheral neuropathy is one of the most devastating complications of diabetes, with estimates of prevalence between 50 and 90%. Diabetic neuropathy occurs as a complication of chronic hyperglycemia and affects peripheral nerve function and regeneration. Symptoms of diabetic neuropathy are variable, but most involve distal peripheral nerves and include abnormal touch, temperature, pain, and pressure sensation. Multiple factors contribute to the pathogenesis of diabetic neuropathy, including alterations in endoneural metabolism, defective neurotrophic factors, reduced nerve blood supply, and immune mechanisms (Siemionow and Demir, 2004). Structural changes in diabetic nerves include axonal loss, segmental demyelination, and impaired axonal regeneration (Vinik et al., 2000; Apfel, 2002). The mechanisms underlying diabetic neuropathy remain unclear; however, metabolic and/or vascular abnormalities are clearly involved.
Deficiency in neurotrophic support contributes to the development of diabetic neuropathy (Apfel, 1999; Pittenger and Vinik, 2003; Anand, 2004). Glial cell line-derived neurotrophic factor (GDNF), a member of the transforming growth factor β (TGF-β) superfamily, is perhaps the most potent trophic factor influencing the development, survival, and maintenance of neurons in the central and peripheral nervous systems (Lin et al., 1993; Sariola and Saarma, 2003; Paratcha and Ledda, 2008). The neurotrophic effects of GDNF are mediated by a multisubunit receptor system consisting of the glycosylphosphatidylinositol (GPI)-linked GDNF ligand-binding coreceptor, GFRα1, the transmembrane proto-oncogene-encoded Ret protein, and neuronal adhesion molecule (NCAM) (Airaksinen and Saarma, 2002; Paratcha and Ledda, 2008). GDNF has been applied in the treatment of various types of neurodegenerative diseases including Parkinson's disease (Gill et al., 2003) and amyotrophic lateral sclerosis (ALS) (Manabe et al., 2003).
Existing evidence indicates that the GDNF signaling complex may participate in the pathogenesis of diabetic neuropathy. For example, retrograde axonal transport of GDNF is decreased in experimentally induced diabetic mice (Akkina et al., 2001; Christianson et al., 2003). In addition, intrathecal administration of GDNF protein inhibits diabetes-induced changes in nerve fiber morphology and reductions in the sensory neuropeptides including substance P (SP) (Malcangio et al., 2000; Skoff et al., 2003) and calcitonin gene-related peptide (CGRP) (Ramer et al., 2003). Importantly, intrathecal GDNF injection potently stimulates axon growth and branching to improve nerve innervation deficits caused by diabetes (Christianson et al., 2003). Despite the previous studies, the exact role of the GDNF signaling pathway in the pathogenesis of diabetes-induced neurological deficits is far from delineated.
What remains to be identified are the pivotal genes/pathways involved in the pathogenesis of diabetic neuropathy. It may take years for diabetic patients to develop the symptoms of neuropathy after initial diagnosis (Said, 2007). Moreover, the control of plasma glucose level also critically influences the onset of diabetic neuropathy. Thus, it is difficult to identify the early changes in gene expression that contribute to the deficits of the peripheral nervous system in diabetic patients. In this paper we used the streptozotocin (STZ)-induced diabetes rat model, monitoring changes in the electrophysiological parameters of diabetic rats after induction to investigate the kinetics of the development of neuropathic symptoms. Once the onset of diabetic neuropathy was identified, the expression profiles of the GDNF signaling complex in the sciatic nerve of diabetic rats were examined to study whether the GDNF signaling complex was altered in the early phase of diabetic neuropathy. Last, peripheral GDNF gene delivery was achieved in diabetic rats with established neuropathy to evaluate the therapeutic potential of GDNF gene therapy.
Materials and Methods
STZ-induced diabetic rats
The protocol to induce diabetes in rats was performed as previously described (Hsieh et al., 2003) and approved by the Animal Care and Use Committee of Kaohsiung Veterans General Hospital (Kaohsiung, Taiwan). Briefly, Wistar rats (female, 200–250 g; Animal Center of the National Science Council, Taipei, Taiwan) were fed with standard laboratory rodent chow and water ad libitum and housed individually. After overnight fasting, diabetes was induced in anesthetized rats by intraperitoneal injection of STZ (50 mg/kg in 0.9% sterile saline; Sigma-Aldrich, St. Louis, MO) and maintained in parallel with age-matched control animals. Subsequently, rats were returned to standard laboratory chow and to water ad libitum. Blood glucose was measured with glucose test strips (BM-Accutest; Roche Diagnostics, Basel, Switzerland). Rats with blood glucose levels less than 400 mg/dl were excluded.
Preparation of adenoviral vectors
Recombinant adenoviral vectors encoding GDNF (Ad-GDNF) or enhanced green fluorescent protein (Ad-GFP) were prepared as described previously (Tai et al., 2003). For Ad-GDNF, GDNF cDNA was subcloned into pCA13 to yield the transfer vector Ad5-GDNF, which was used to transfect 293 cells with pJM17, a plasmid carrying the entire adenovirus genome, to generate recombinant virus through homologous recombination by a calcium phosphate protocol as described previously (Berns and Giraud, 1995). The virus was amplified in 293 cells, purified by two rounds of cesium chloride gradient ultracentrifugation, and dialyzed against buffer containing 10 mM Tris (pH 7.5), 1 mM MgCl2, 10% glycerol at 4°C. The titer of the virus solution was determined by measuring optical density at a wavelength of 260 nm and by plaque-forming assay in 293 cells before storage at −80°C.
Intramuscular injection
The experimental scheme of this study is depicted in Supplementary Fig. 1 (see
Electrophysiological analysis
Nerve conduction velocity (NCV) and the H-reflex of diabetic rats were measured with a Keypoint signal processor (Dantec Dynamics, Copenhagen, Denmark) according to a previously described protocol (Lai et al., 1997).
Electron microscopy analysis
Scanning electron microscopy (SEM) analysis was performed as described (Su et al., 2006). Briefly, formalin-fixed samples were postfixed in 0.1 M saline phosphate-buffered 1% osmium tetroxide, dehydrated in a graded series of acetone, dried with a critical point drying unit, and sputter coated with gold/palladium, and then examined by SEM (E101; Hitachi, Tokyo, Japan). Transmission electron microscopy (TEM) analysis was performed as previously described (Hoke et al., 2003). Briefly, formalin-fixed samples were postfixed in 1% osmium tetroxide for 30 min, dehydrated in a graded series of alcohol, and embedded in Spurr's resin. Thin sections (80 nm) were cut with a glass knife, stained with uranyl acetate and lead citrate, and then viewed by TEM (JEOL-1230; JEOL, Tokyo, Japan) operated at 80 keV.
Quantitative reverse transcription-polymerase chain reaction
RNA isolation and quantitative reverse transcription-polymerase chain reaction (qRT-PCR) were performed as described (Liu et al., 2006). Primer sequences for GDNF were as follows: forward primer, 5′-TTCCAGAGGGAAAGGTCGC-3′; reverse primer, 5′-CATTGTCTCGGCCGCTCCA-3′, which amplified a 160-bp GDNF cDNA fragment. The β-actin mRNA level was determined using forward primer 5′-TCACCCACACTGTGCCCATCTACGA-3′ and reverse primer 5′-CAGCGGAACCGCTCATTGCCAATGG-3′, which amplified a 295-bp β-actin cDNA fragment.
Immunoblot analysis
Protein extract was isolated from tissues with buffer containing 150 mM NaCl, 50 mM HEPES (pH 7), 1% Triton X-100, 10% glycerol, 1.5 mM MgCl2, 1 mM EGTA, and complete protease inhibitors (Roche Applied Science, Indianapolis, IN). After separation in sodium dodecyl sulfate (SDS)–12.5% polyacrylamide gels, proteins were transferred onto a polyvinylidene difluoride membrane, using a blotting apparatus. The membrane was blocked with 5% milk in TBS-T for 1 hr and then incubated with GDNF (1:500 dilution; R&D Systems, Minneapolis, MN), GFRα1 (1:500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), Ret (1:500 dilution; Santa Cruz Biotechnology), Akt (1:500 dilution; Cell Signaling Technology, Danvers, MA), and pAkt antibodies (1:500 dilution; Cell Signaling Technology) for 1 hr at room temperature. After incubation with secondary antibody conjugated with horseradish peroxidase (HRP) (1:5000 dilution in 5% milk) for 30 min, signals on the membrane were detected with ECL-plus luminol solution (GE Healthcare Life Sciences, Piscataway, NJ) and exposed to X-ray film for autoradiography.
Enzyme-linked immunoassay
GDNF, CGRP, and SP content in nerve tissue, plasma, and cell cultures was determined with an enzyme-linked immunosorbent assay (ELISA) kit for GDNF (GDNF Emax immunoassay system kit; Promega, Madison, WI), CGRP (R&D Systems), and SP (Cayman Chemical, Ann Arbor, MI), respectively, according to the manufacturers' instructions. Absorbance at a wavelength of 450 nm was determined with an ELISA reader (Dynatech Laboratories, Chantilly, VA). The level of GDNF, CGRP, and SP in samples was obtained by calculation from a six-point standard curve and expressed as picograms per milligram of tissue. The protein content in cell or tissue extracts was determined by Bradford assay for normalization.
Bioluminescence imaging
Animals were anesthetized with a cocktail of ketamine–xylazine (4:1 in PBS) and then injected intraperitoneally with 1 ml of d-luciferin solution (20 mg/ml in PBS; Promega). The animals were placed on a 37°C platform in a bioluminescence imaging system (NightOWL; Berthold Technologies, Bad Wildbad, Germany) for imaging. A gray-scale body surface image was obtained in the chamber under dim illumination followed by a 5-min acquisition and overlay of the pseudocolor images. The spatial distribution and quantity of photon counts emitted by cells producing luciferase within the animal were represented by colored pixels produced by the computer.
Immunofluorescence analysis
Transverse frozen sections (5 μm) of sciatic nerves and spinal cords were dried and incubated in blocking buffer containing 1.5% normal goat serum and 0.2% Triton X-100 in PBS. The slides were washed twice with PBS and incubated with primary antibodies including GDNF (1:100 dilution; R&D Systems), Akt (1:100 dilution; Cell Signaling Technology), pAkt (1:100 dilution; Cell Signaling Technology), P0 (1:100 dilution; Santa Cruz Biotechnology), and neurofilament 200 antibody (1:200 dilution; Dako, Carpinteria, CA) at 4°C overnight followed by repeated washing with PBS, and replaced with secondary antibodies conjugated with Alexa 488 or Alexa 588 (1:1000 dilution; Invitrogen, Carlsbad, CA) for 1 hr at room temperature. The immunostained slides were observed and recorded under a fluorescence microscope (Olympus America, Center Valley, PA).
Morphometric analysis of myelinated axons
Measurement of the size distribution of myelinated axons was performed by toluidine blue staining as described (Mazzer et al., 2008). Rats were anesthetized with an intraperitoneal injection of sodium pentobarbital (40 mg/kg) and perfused through the left cardiac ventricle with PBS, followed by 4% paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.2. Both sciatic nerves were removed and a 5-mm section of each nerve (10 mm distal to the injury site) was processed for semithin (1 mm) cross-sections. All transverse sections of the sciatic nerve were stained with 1% toluidine blue and their images were projected by camera lucida directly onto a digitizing tablet, where the myelinated axons were quantified. Data were expressed as the average percentage of myelinated axons of various sizes.
Statistical analysis
Within-group comparisons were made by one-way analysis of variance (ANOVA). Comparisons across groups were accomplished by two-way ANOVA and, if significant, discrete comparisons were accomplished by post hoc tests. A p value less than 0.05 was considered statistically significant. Data were expressed as means ± SEM. Results
Neuropathic symptoms and structural changes in sciatic nerve of diabetic rats after streptozotocin injection for 2 weeks
The STZ-injected rat model was employed to investigate the onset of diabetic neuropathy in rats without insulin supplementation. The plasma glucose level and body weight of diabetic rats were also assessed periodically. After STZ injection, rats developed typical signs of diabetes including polyuria and polydipsia in 48 hr. Two weeks after STZ injection, diabetic rats had a 4-fold increase in blood glucose levels and gained less weight compared with nondiabetic control rats (Table 1).
Characteristics of Control and Streptozotocin-Induced Diabetic Ratsa
Blood glucose and body weight of control and STZ-induced diabetic rats were compared on day 1 and 2 weeks after STZ injection. Data represent means ± SEM (n = 8 per group).
P < 0.01 versus control.
Whereas hyperglycemia and weight loss were promptly noted, altered electrophysiological functions of diabetic rats were not detected until day 14 after STZ injection, when the diabetic rats exhibited a significant reduction in nerve conductance velocity (NCV) (36.6 ± 1.8 vs. 45.2 ± 5.5 m/sec [control]; p < 0.05) with a concomitant delay in H-reflex (0.455 ± 0.044 vs. 0.378 ± 0.016 [control]; p < 0.05) (Fig. 1A). To confirm these neurological deficits, the sciatic nerves of diabetic rats were examined by electron microscope analysis, which revealed distorted ultrastructural organization and defective myelination in the sciatic nerve of diabetic rats 14 days after STZ injection (Fig. 1B and C). These studies indicate that neuropathic symptoms occur as early as 14 days after hyperglycemia begins in diabetic rats.
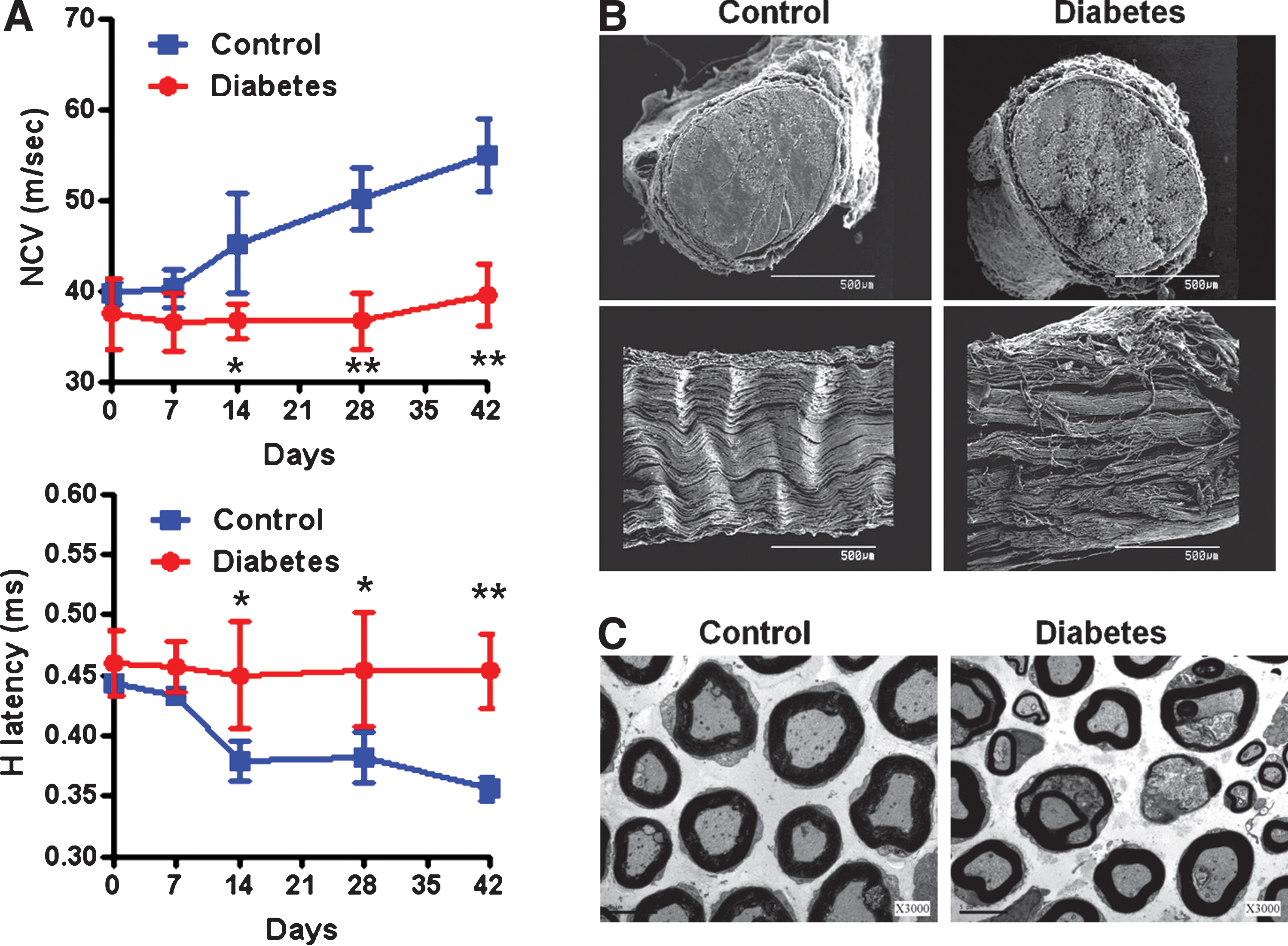
Effects of hyperglycemia on the neuropathic symptoms and ultrastructure of sciatic nerves in streptozotocin (STZ)-induced diabetic rats. (
Deficiency of GDNF signaling complex in sciatic nerve of diabetic rats during early stage of diabetic neuropathy
To investigate whether the GDNF signaling pathway participated in the emergence of diabetic neuropathy, the GDNF level in the sciatic nerve and lumbar spinal cord (L4–L5) was analyzed by ELISA on days 7 and 14 after STZ injection. No difference in GDNF level was found in either sciatic nerve or spinal cord on day 7 (data not shown). On day 14, when abnormalities in electrophysiological parameters were initially detected, the GDNF protein level in the sciatic nerve, but not in spinal cord, was significantly decreased in diabetic rats compared with control (Fig. 2A). Quantitative RT-PCR analysis further confirmed that the GDNF mRNA level in the sciatic nerve of diabetic rats was significantly decreased by 50% compared with control (p < 0.05; Fig. 2B), indicating that GDNF deprivation in the sciatic nerve occurred at the transcriptional level. Immunofluorescence analysis revealed that GDNF was expressed mainly in Schwann cells surrounding the axons in the sciatic nerves (Fig. 2C). To further delineate whether the expression of GDNF receptors (including GFRα1, Ret, and NCAM) was influenced by hyperglycemia, immunoblot analysis showed that GFRα1 expression in the sciatic nerve was significantly reduced in diabetic rats (Fig. 2D). In contrast, Ret and NCAM protein levels in the peripheral nerves were not affected at this time. Importantly, the expression and phosphorylation of Akt, a downstream effector of the GDNF signaling pathway, was also significantly decreased in the sciatic nerve of diabetic rats (Fig. 2E) on day 14. Together, these results suggest that attenuation of the GDNF/GFRα1/Akt signaling pathway in the sciatic nerve is an early event during the pathogenesis of diabetic neuropathy.
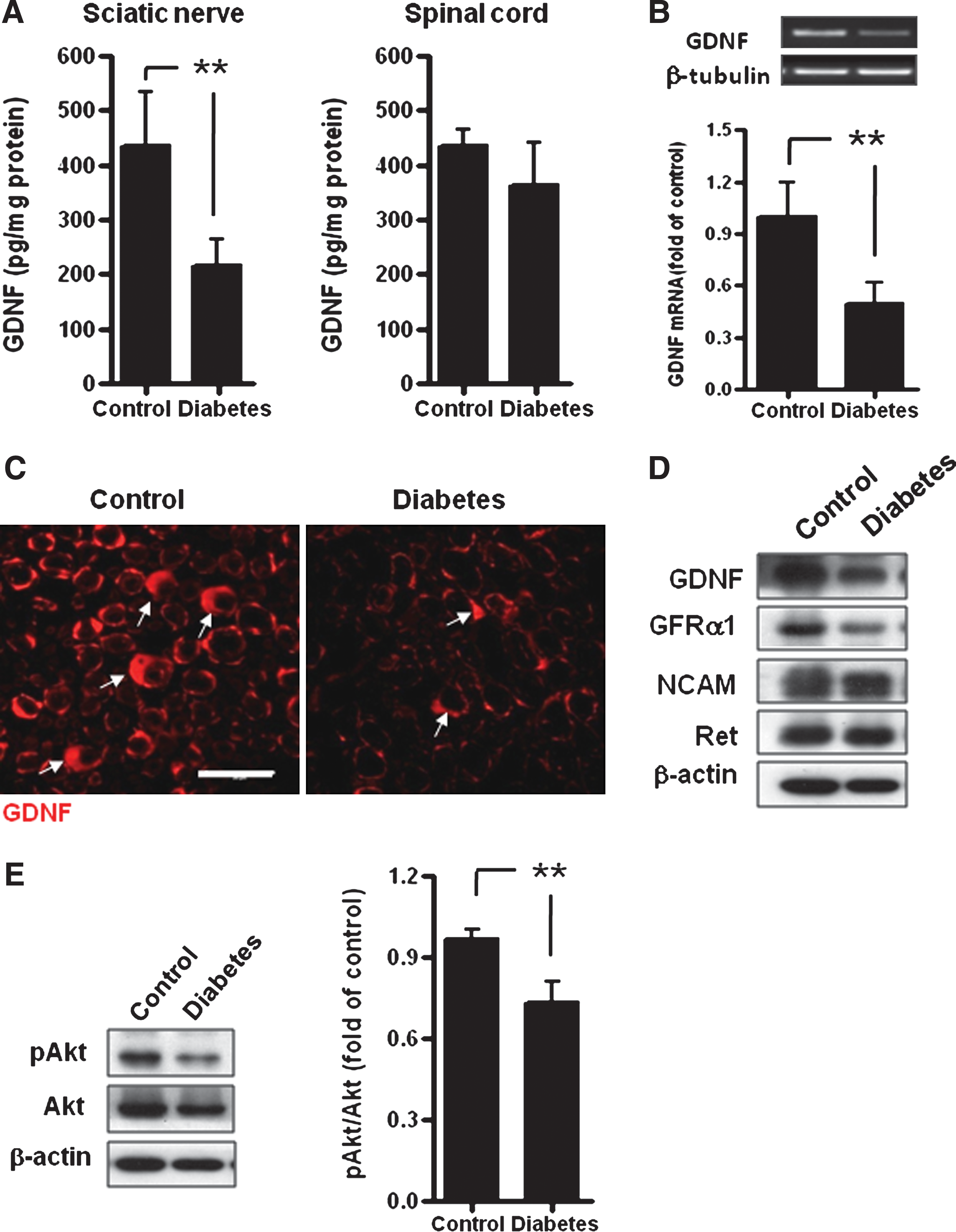
Effect of hyperglycemia on the expression of the glial cell-derived neurotrophic factor (GDNF) signaling complex in the sciatic nerve of diabetic rats. Fourteen days after STZ injection, GDNF expression in the sciatic nerve and spinal cord from diabetic (DM) and nondiabetic (control) rats was analyzed by (
Reduced sensory peptide expression in sciatic nerve of diabetic rats
To delineate the probable consequence of deficiency in GDNF signaling complex during early diabetes, the content of sensory neuropeptides, including CGRP and SP, in peripheral nervous tissue was determined. ELISA revealed that the CGRP content in the sciatic nerve of diabetic rats (280.3 ± 129.4 pg/mg) was significantly lower than that of controls (514.3 ± 158.4 pg/mg; p < 0.05; Fig. 3A). Similarly, the SP level in the sciatic nerve of diabetic rats (129.4 ± 58.8 pg/mg) was also significantly downregulated compared with that of controls (337.4 ± 211.6 pg/mg; p < 0.05; Fig. 3B). However, no difference in spinal CGRP and SP levels was found between control and diabetic rats (see Supplementary Fig. 2 at
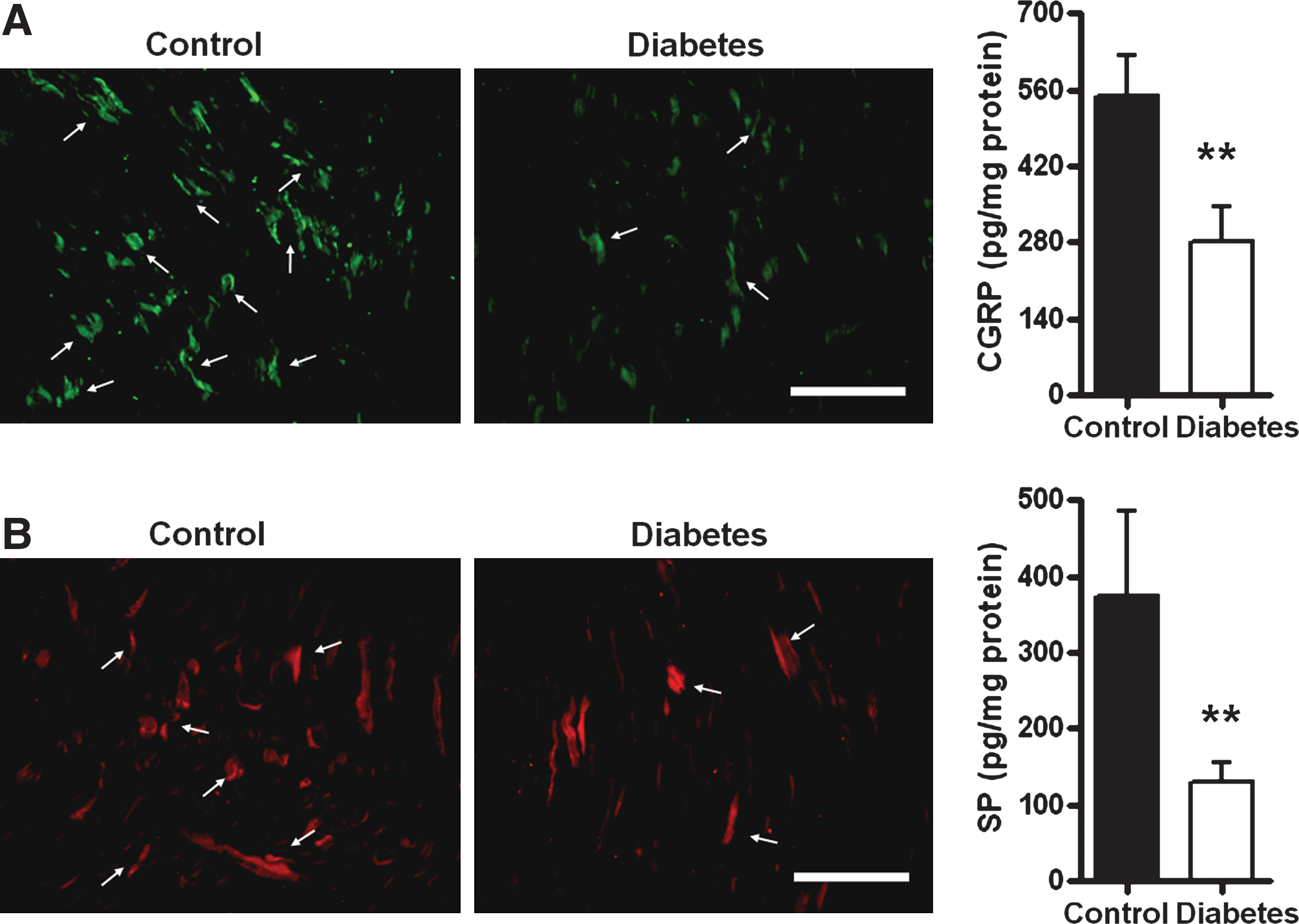
Reduction of calcitonin gene-related peptide (CGRP) and substance P (SP) contents in the sciatic nerve of diabetic rats. Fourteen days after STZ injection, the (
Intramuscular GDNF gene delivery restores GDNF level in sciatic nerve of diabetic rats
To correct GDNF depletion in the sciatic nerves of diabetic rats, intramuscular gene delivery by adenoviral vectors was employed to use the muscle cells as a reservoir for GDNF production, which could be delivered to the sciatic nerve via retrograde transport. To test such a hypothesis, adenovirus encoding luciferase (Ad-luc) was injected into the muscles of diabetic rats to monitor viral tropism and transgene expression after adenovirus gene delivery. By bioluminescence analysis, it was found that the luminescence signals of luciferase were localized mainly in muscles around the injection site (Fig. 4A). GDNF expression in muscles was determined by immunofluorescence and Western blot analysis, which showed elevated GDNF expression only in Ad-GDNF-injected muscles (Fig. 4B). Because GDNF-transduced muscle cells may release GDNF into the circulation, the circulating GDNF level was determined by ELISA at various time intervals after intramuscular administration of adenoviral vectors. An elevated plasma GDNF level was observed only in Ad-GDNF-injected rats and the enhanced increment in circulating GDNF reached the maximal level between 7 and 14 days after injection (Fig. 4C), yet remained significantly higher than that of control rats for at least 28 days. Thus, intramuscular GDNF gene delivery increases GDNF production in muscle as well as in the circulation.
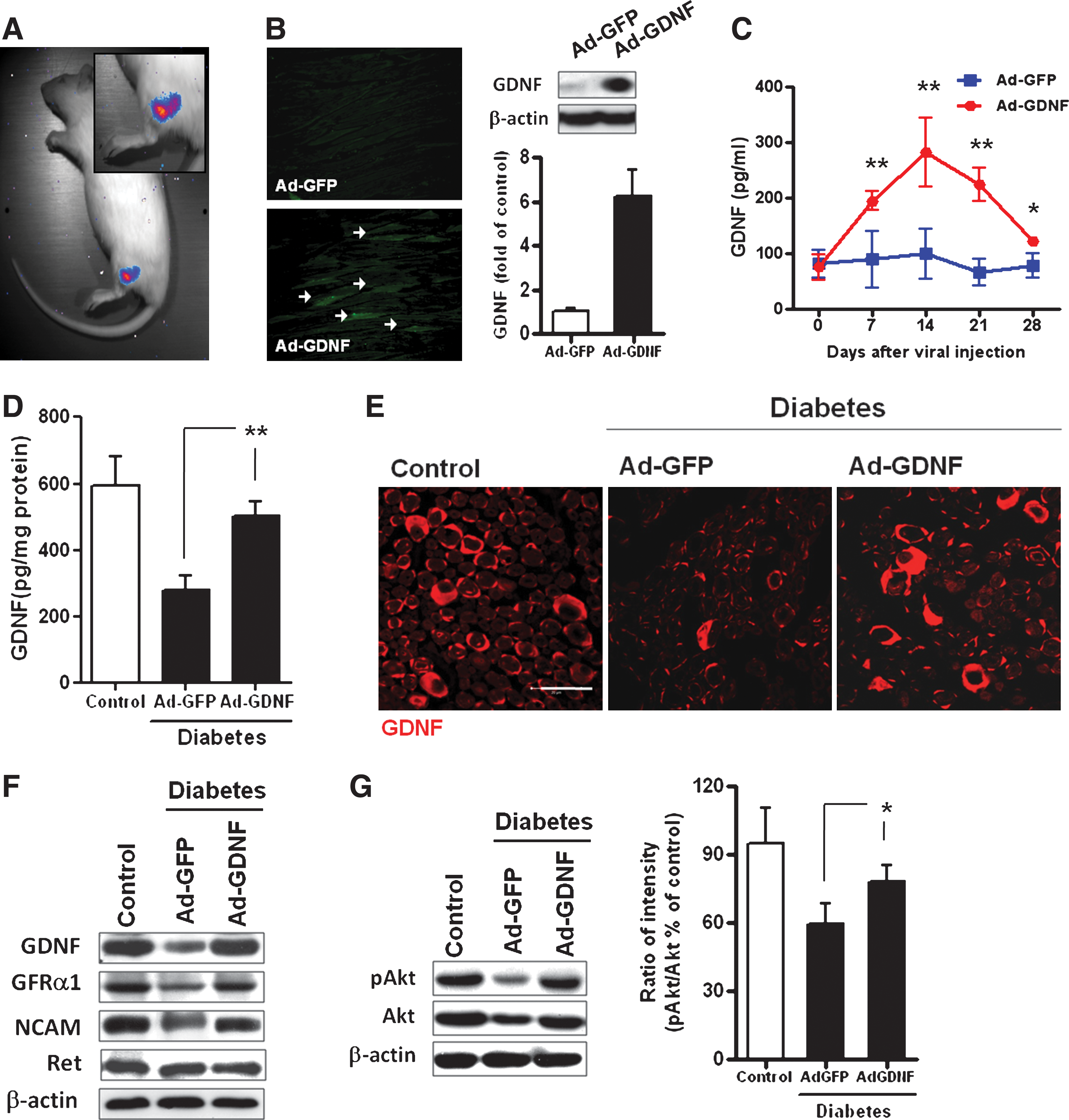
Effect of intramuscular GDNF gene transfer on GDNF expression and Akt phosphorylation in the sciatic nerve of diabetic rats. (
We subsequently investigated whether muscle-produced GDNF could be transported to the sciatic nerve of diabetic rats. Twenty-eigt days after gene delivery, ELISA analysis found that Ad-GDNF-treated rats exhibited a significant increment in GDNF content compared with saline- or Ad-GFP-treated rats (Fig. 4D). In addition, the restored GDNF level in Ad-GDNF-treated diabetic rats approximated the normal range of nondiabetic rats. Immunofluorescence analysis also revealed that GDNF gene transfer significantly increased the content of GDNF in the sciatic nerve of diabetic rats (Fig. 4E). This was accompanied by restored GFRα1 expression (Fig. 4F) and an elevated pAkt:Akt ratio (Fig. 4G). The enhanced Akt phosphorylation was further confirmed by increased pAkt-positive axons in the sciatic nerves (see Supplementary Fig. 3 at
Intramuscular GDNF gene delivery ameliorates neurological deficits in diabetic rats
To evaluate the efficacy of GDNF gene therapy for diabetic neuropathy, diabetic rats with confirmed neuropathy on day 14 were injected with adenoviral vectors via the muscular route on day 15, and then monitored for changes in electrophysiological and metabolic parameters for an additional 28 days. After gene delivery, Ad-GDNF-treated animals exhibited significant recovery in neurological parameters compared with saline- or Ad-GFP-treated diabetic rats (Fig. 5). The NCV of Ad-GDNF-treated rats (41.3 ± 2.1 and 44.9 ± 1.8 m/sec on day 28 and day 42, respectively) was significantly increased by a 20–30% increment over control on day 28 (38.1 ± 3.9 and 36.1 ± 3.1 m/sec for Ad-GFP and saline, respectively; p < 0.05) and day 42 (39.1.9 ± 2.2 and 39.5 ± 3.3 m/sec for Ad-GFP and saline, respectively; p < 0.05) (Fig. 5A). Furthermore, the H-reflex of Ad-GDNF-treated rats (0.422 ± 0.026 and 0.401 ± 0.034 on day 28 and day 42, respectively) was significantly reduced by 42% relative to control on day 28 (0.466 ± 0.038 and 0.455 ± 0.047 for Ad-GFP and saline, respectively; p < 0.05) and by 62% relative to control on day 42 (0.464 ± 0.041 and 0.453 ± 0.031 for Ad-GFP and saline; p < 0.05) (Fig. 5B). However, there was no significant difference in plasma glucose level and body weight between Ad-GDNF-treated rats and rats in the control groups (Fig. 5C and D). Biochemical examination also revealed that peripheral GDNF gene delivery did not induce noticeable changes in the biochemical parameters of diabetic rats (see Supplementary Table 1 at

Effect of intramuscular GDNF gene delivery on STZ-induced neuropathy. After STZ injection (on day 0), the (
GDNF gene delivery rescues axonal demyelination and restores sensory neuropeptide level in sciatic nerve of diabetic rats
To elucidate whether GDNF therapy conferred protection against axonal demyelination during hyperglycemic insults, the extent of myelination of sciatic nerves of diabetic rats after GDNF gene delivery was examined by immunofluorescence analysis of the myelin marker P0, and the axonal fiber marker neurofilament 200. It was found that P0 as well as neurofilament 200 immunostaining was prominently decreased in diabetic rats compared with control rats (Fig. 6A). However, GDNF gene delivery for 28 days elicited an increase in both P0 and neurofilament 200 expression. Subsequently, morphometric analysis was performed to further confirm whether GDNF therapy conferred protection against diabetes-induced demyelination. By toluidine blue staining, it was revealed that diabetes increased the number of small-diameter axons but reduced the number of large-diameter axons in the sciatic nerve (Fig. 6B). Importantly, GDNF gene delivery significantly reverted diabetes-induced changes in the distribution of myelinated axons (p < 0.01), supporting the protection of myelinated axons by GDNF therapy. These results indicate that GDNF transported from transduced muscle cells exerts a neurotrophic effect to relieve myelination deficits in the peripheral nerves of diabetic rats.
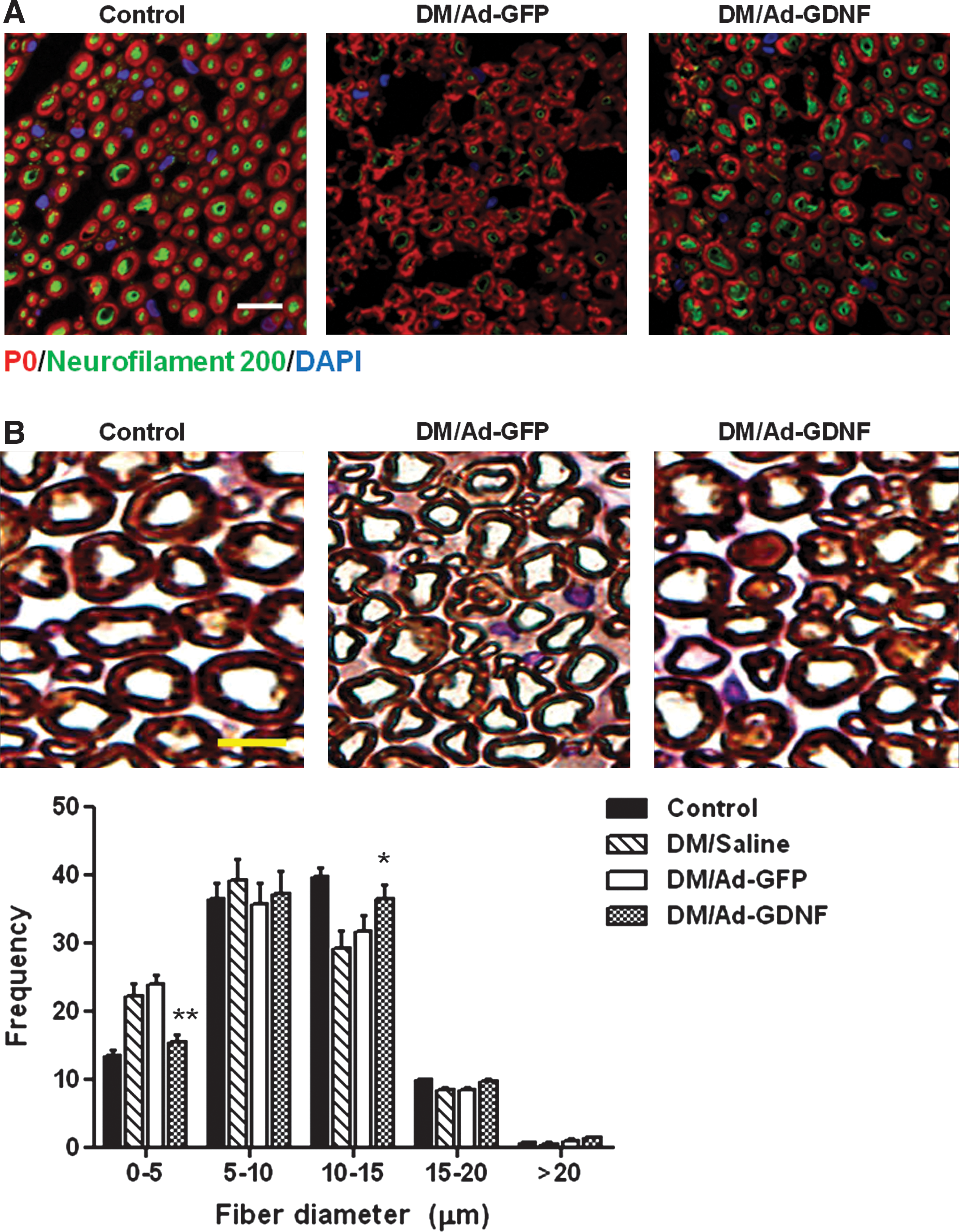
Effect of intramuscular GDNF gene delivery on axonal myelination in sciatic nerves of diabetic rats. (
We also examined whether GDNF gene delivery rescued the loss of sensory neuropeptides in the sciatic nerves of diabetes rats. After gene delivery for 28 days, ELISA revealed that the depleted CGRP level in the sciatic nerve of diabetic rats was restored to the normal threshold after intramuscular injection of Ad-GDNF, but not Ad-GFP or saline (Fig. 7A). Similarly, deprived SP expression in the sciatic nerve of diabetic rats was also increased by GDNF gene delivery (Fig. 7B). Therefore, GDNF gene delivery reversed the depletion of sensory neuropeptides in the sciatic nerve of diabetic rats.

Peripheral GDNF gene delivery restored the CGRP and SP levels in sciatic nerve of diabetic rats. Twenty-eight days after intramuscular injection of adenoviral vector, the (
Discussion
Summary and novel findings of this study
The present study simultaneously monitored structural and functional changes in the peripheral nerves of STZ-induced diabetic rats, thereby establishing that the critical timing for the onset of neuropathy is about 7–14 days after induction. After STZ-induced diabetes had been achieved, electrophysiological studies indicated that the primary neurological deficits in STZ-induced diabetic rats included a reduction in NCV and a delay in H-reflex. Electron microscopic analysis confirmed that the altered electrophysiological parameters were associated with degeneration of the sciatic nerves in rats, which closely resembles nerve dysfunction in diabetic patients. The reduction of GDNF and GFRα1 protein levels in the sciatic nerve is not observed until day 14 after STZ injection, which is well correlated with the loss of sensory neuropeptides and the development of neuropathic symptoms in diabetic rats. Importantly, intramuscular GDNF gene delivery restored GDNF content, GFRα1, Akt phosphorylation, and the levels of sensory neuropeptides in the sciatic nerve of diabetic rats. This was accompanied by a significant improvement in neurological parameters for at least 28 days. Collectively, diabetes-induced GDNF deprivation contributes to the axonal degeneration during the pathogenesis of diabetic neuropathy.
Role of GDNF signaling for development and treatment of diabetic neuropathy
Pathogenic mechanisms of diabetic neuropathy are multifactorial and related to chronic hyperglycemia (Vinik, 1999; Apfel et al., 2000). Loss of trophic support contributes to the clinical symptoms of small-fiber dysfunction and plays an important role in the pathogenesis of diabetic neuropathy in humans and experimental animals (Schmidt et al., 1986; Fernyhough et al., 1995, 1998). This study demonstrates that GDNF and its receptor complex are expressed in the peripheral nerves, particularly the Schwann cells. This is consistent with previous observations that Schwann cells express mRNA for GDNF and are presumed to be a source of GDNF for peripheral neurons (Springer et al., 1994; Choi-Lundberg and Bohn, 1995; Iwase et al., 2005). Moreover, we have shown that the GDNF signaling cascade including GDNF ligand, GFRα1 receptor, and downstream effector Akt are downregulated in the sciatic nerve, but not the spinal cord, when neuropathic symptoms are initially detected. This was not anticipated because GDNF and GFRα1 are upregulated in peripheral nerve transections (Trupp et al., 1995, 1997; Hammarberg et al., 1996; Hoke et al., 2000), in motor neuropathy in rats (Sagot et al., 1996), in various human neuropathies (Yamamoto et al., 1996), and in traumatized human nerves (Bar et al., 1998). Nevertheless, our data are consistent with studies that GDNF production is decreased in the peripheral tissues of diabetic mice and humans (Akkina et al., 2001; Christianson et al., 2003; Anand, 2004). Interestingly, unlike Ret and NCAM, the high-affinity GDNF receptor GFRα1 seems particularly vulnerable to diabetic challenge, probably because of its expression in Schwann cells, whereas Ret and NCAM are expressed mainly in the dorsal root ganglion. Finally, we propose the feasibility of using intramuscular GDNF gene transfer to restore the GDNF level in the sciatic nerve and to alleviate neuropathic deficits of diabetic rats. In fact, our finding on the protective role of the GDNF signaling complex for peripheral neurons is supported by a study using a transgenic model (Anitha et al., 2006), which reveals the pivotal role of GDNF/Akt signaling in the survival of enteric neurons during hyperglycemic insults. However, GDNF is the only trophic factor involved in diabetic neuropathy. Despite the fact that the GDNF level in the sciatic nerve was corrected to normal range, the neurological functions of diabetic rats were far from complete recovery after GDNF gene delivery, implicating the involvement of other trophic factors during hyperglycemia-induced neuropathy. Moreover, the mechanism underlying the reduction in GDNF availability, which may be a consequence of metabolic abnormalities or independent of hyperglycemia, remains to be elucidated.
Rapid onset of diabetic neuropathy in STZ-injected rats
To our knowledge, the present study provides the first evidence that hyperglycemia for merely 14 days is sufficient to induce functional and structural changes in the peripheral nervous system of rats. Although contrary to the clinical perception, our results are in agreement with previous studies that mechanical hyperalgesia (Fox et al., 1999) and reduction in endoneurial blood flow (Cameron et al., 1991; Coppey et al., 2000) can be detected in rats as early as 1 week after STZ injection. In addition, the correlation between downregulation of sensory neuropeptides and the initiation of neuropathic symptoms also supports the notion that early change in gene expression profiles contributes to neuropathic deficits after hyperglycemia. Therefore, diabetic neuropathy can be reproducibly induced in experimental animals within weeks instead of years. The plausible explanation for the rapid onset of neuropathic deficits could be the lack of insulin treatment given these rapidly growing, juvenile rats, again strengthening the importance of glucose control for diabetic patients.
Advantages and limitation of peripheral GDNF gene therapy
The present study strongly advocates the beneficial effects of peripheral GDNF gene transfer to improve neurological functions in diabetic rats. Continuous GDNF production by gene transfer may constitute a promising alternative treatment for diabetic neuropathy because of its high potency and safety, except for lowered appetite when administration is done via the central route (Gill et al., 2003). In addition, GDNF overexpression is not known to contribute to cancer progression whereas continuous expression of vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), or insulin-like growth factor (IGF)-I may potentially lead to carcinogenesis. Our results supported the potential of peripheral GDNF gene delivery to intervene in the progression of diabetic neuropathy and possibly to alleviate the established, chronic neuropathy. The minimal effective dose and the duration of GDNF gene therapy remain to be characterized. Because gene delivery of neurotrophic factors such as nerve growth factor (NGF), IGF-I, neurotrophin (NT)-3, HGF, and VEGF also alleviates diabetic neuropathy, combinatory gene delivery of GDNF with other neurotrophic factors may result in additional benefits for amelioration of diabetic neuropathy (Pradat et al., 2001; Goss et al., 2002; Chattopadhyay et al., 2005; Kato et al., 2005). In transgenic mice overexpressing GDNF in beta cells, Mwangi and colleagues unveiled that GDNF increased beta cell mass by promoting proliferation and insulin content by conferring protection to beta cells against thapsigargin challenge (Mwangi et al., 2008). Thus, GDNF constitutes a novel therapeutic target to increase insulin and preserve beta cells for diabetes. In this study, however, GDNF gene delivery did not reverse the plasma glucose level or increase the circulating insulin level in STZ-injected rats, suggesting that exogenous GDNF supply failed to rescue or restore STZ-depleted beta cells. Nevertheless, it should be worthwhile to investigate whether GDNF exerts benefits to control glucose levels in animals with beta cell dysfunction leading to obesity or type 2 diabetes.
Conclusion
In conclusion, our study clearly indicates that a defective GDNF signaling pathway and downregulation of sensory neuropeptides take place in the peripheral nerves and constitute early events in the pathogenic mechanism underlying neuropathic deficits in an animal model of experimentally induced type 1 diabetes. Intramuscular GDNF gene transfer restores the levels of GDNF and sensory neuropeptides in the sciatic nerves, attenuates axonal degeneration, and ultimately improves the neurological functions of diabetic rats. Therefore, GDNF and molecules in the GDNF signaling pathway are potential therapeutic targets for diabetic neuropathy.
Footnotes
Acknowledgments
This work was supported in part by grants from the National Science Council, Taiwan (NSC 94-2752-B-075B-001-PAE and NSC90-2320-B-075B-005 to M.H.T., NSC 91-3112-B-037003 to S.L.H., and NSC 92-2314-B-037-058 to S.J.S.), Kaohsiung Veterans General Hospital, Taiwan (VGHNSU98-002), National Cheng Kung University, Taiwan (OUA 96-3-7-247), and National Sun Yat-Sen University-Kaohsiung Medical University Joint Research Center and Asia-Pacific Ocean Research Center.
Author Disclosure Statement
No competing financial interests exist.