
Abstract
Interest has increased in the use of exogenous stem cells to optimize lung repair and serve as carriers of a therapeutic gene for genetic airway diseases such as cystic fibrosis. We investigated the survival and engraftment of exogenous stem cells after intratracheal injection, in a murine model of acute epithelial airway injury already used in gene therapy experiments on cystic fibrosis. Embryonic stem cells and mesenchymal stem cells were intratracheally injected 24 hr after 2% polidocanol administration, when epithelial airway injury was maximal. Stem cells were transfected with reporter genes immediately before administration. Reporter gene expression was analyzed in trachea–lungs and bronchoalveolar lavage, using nonfluorescence, quantitative, and sensitive methods. Enzyme-linked immunosorbent assay quantitative results showed that 0.4 to 5.5% of stem cells survived in the injured airway. Importantly, no stem cells survived in healthy airway or in the epithelial lining fluid. Using 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside staining, transduced mesenchymal stem cells were detected in injured trachea and bronchi lumen. When the epithelium was spontaneously regenerated, the in vivo amount of engrafted mesenchymal stem cells from cell lines decreased dramatically. No stem cells from primary culture were located within the lungs at 7 days. This study demonstrated the feasibility of intratracheal cell delivery for airway diseases with acute epithelial injury.
Introduction
Respiratory diseases remain one of the main causes of morbidity and mortality. Interest in optimizing repair of the lung by the use of stem cells (SCs) has increased. In particular, combining the ability of SCs to engraft into damaged lungs with their ability to serve as carriers of a therapeutic gene has great potential for the treatment of pulmonary fibrosis (Loebinger et al., 2008b) and genetic airway diseases such as cystic fibrosis. This developing therapeutic approach has been stimulated by early reports demonstrating that both embryonic SCs and SCs derived from adult bone marrow, including mesenchymal stem cells, can differentiate into respiratory cells in vitro, thus acquiring phenotypic and functional markers of airway and alveolar epithelial cells (Coraux et al., 2005; Wang et al., 2005; Rippon et al., 2006).
Systemic administration of adult SCs from the bone marrow of mice after total body irradiation (Pereira et al., 1995; Loebinger et al., 2008a; Sueblinvong et al., 2008) and/or pollutant reagent treatment has been reported (Beckett et al., 2005; MacPherson et al., 2006; Mei et al., 2007; Serikov et al., 2007; Xu et al., 2007; Loebinger et al., 2008 ) and showed that the administered SCs were engrafted mainly in alveolar spaces (Pereira et al., 1995; Beckett et al., 2005; Mei et al., 2007; Serikov et al., 2007; Xu et al., 2007; Loebinger et al., 2008a; Sueblinvong et al., 2008) and sometimes in conducting airway (Pereira et al., 1995; Krause et al., 2001; MacPherson et al., 2005, 2006; Loi et al., 2006; Serikov et al., 2007). The SC differentiation as pneumocytes (type I or II) or airway epithelial cells was evaluated mostly by various fluorescence techniques. Reported engraftment rates of adult bone marrow SC-derived cells ranged from 0% (Wagers et al., 2002) up to 20% (Krause et al., 2001) in the lungs, and from 0.025% (Loi et al., 2006) up to 4% (Krause et al., 2001) in conducting airway. These discrepancies have been partly attributed to the various fluorescence techniques used to detect the donor-derived epithelial cells, leading to significant artifacts (Loebinger et al., 2008a). Importantly, lung SC engraftment is currently estimated to be between 0.01 and 0.1% (Krause, 2005; Bruscia et al., 2006). Despite this low engraftment level, there is evidence that SCs transplanted postinjury have some therapeutic effects (Loebinger et al., 2008a).
In most of these studies, systemic administration of SCs required total body irradiation of the recipient to promote SC bone marrow engraftment, a difficult condition to apply in the clinical setting, especially in patients suffering from airway disease associated with chronic infections such as cystic fibrosis. Alternatively, considering the advantages of the intratracheal route to target the airway and the respiratory epithelium, more recent studies reported the intratracheal administration of adult SCs in reagent-injured lungs (Gupta et al., 2007; Wong et al., 2007). Using fluorescence techniques, SC engraftment was enhanced by the intratracheal route as compared with the intravenous route (Wong et al., 2007, 2009) but remained at 5–10% (Gupta et al., 2007; Wong et al., 2007, 2009). The intratracheal route was also used for the administration of differentiated cells and showed both positive (Serrano-Mollar et al., 2007) and negative results (Kuang et al., 2005; Gupta et al., 2007). SC or differentiated cell engraftment levels in these studies were assessed mostly by fluorescence techniques, which could explain these conflicting results.
The aims of our study were to evaluate the survival and engraftment of various types of exogenous SCs and differentiated cells after intratracheal injection. We used a murine model presenting acute epithelial airway injury without total body irradiation. This model was developed for gene therapy experiments on cystic fibrosis and consisted of intranasal injection of polidocanol detergent (Parsons et al., 1998). Quantification of cell survival rates and determination of cell location within lungs were performed by enzyme-linked immunosorbent assay (ELISA), biochemical assay, and polymerase chain reaction (PCR).
Materials and Methods
Animal model and intratracheal administration
Male, 8- to 10-week-old Swiss mice were obtained from Janvier Laboratories (Le Genest St Isle, France). Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Research, 1996). Mice were anesthetized with an intraperitoneal injection of xylazine (15 mg/kg) and ketamine (70 mg/kg), and placed supine. Briefly, intratracheal administration was performed with a 22-gauge catheter placed within an intubation cannula (Harvard Apparatus, Holliston, MA) that was removed immediately before injection. Airway injury was induced after anesthesia by intratracheal administration of 25 μl of 2% polidocanol [PDOC: polyoxyethylene (9) lauryl ether; Sigma-Aldrich, St. Louis, MO] (Borthwick et al., 2001; MacPherson et al., 2005) in phosphate-buffered saline (PBS) through the 22-gauge catheter. Cells (106 cells/25 μl) or PBS was intratracheally injected 24 hr after PDOC administration, using the same protocol. Mice were killed by an intraperitoneal lethal injection of thiopental (Nesdonal) 24 hr or 7 days after intratracheal administration.
Bronchoalveolar lavages
Bronchoalveolar lavages (BALs) were performed 24 hr after chloramphenicol acetyltransferase (CAT)-transfected cell administration in healthy or injured airway. Mice were anesthetized and the thoracic cavity was opened by careful dissection. The trachea was exposed, and a small transverse incision was made just below the level of the larynx. BAL was then performed with one dose of 1 ml of PBS, ensuring that both lungs inflated during the lavage process and that there was no leakage of lavage fluid from the trachea. BAL fluid was centrifuged at 400 × g for 5 min. The supernatant was removed and stored at −80°C until ELISA. Cell pellet was suspended in lysis buffer and stored at −80°C until ELISA.
Cell cultures
Undifferentiated mouse embryonic stem cells (ESCs, R1 cell line; Nagy et al., 1993) were cultured in dishes coated with 0.1% gelatin (Sigma-Aldrich). The culture medium was composed of Dulbecco's minimum essential medium with glucose at 4.5 g/liter (DMEM; Invitrogen, Carlsbad, CA) supplemented with 15% fetal calf serum (FCS; Invitrogen), 0.1% nonessential amino acids (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 2 mM l-glutamine (Invitrogen), 10−7 M 2-mercaptoethanol (Sigma-Aldrich), penicillin–streptomycin (100 U/ml and 100 μg/ml, respectively; Invitrogen), and leukemia inhibitory factor (LIF, 2000 U/ml; Sigma-Aldrich).
Undifferentiated murine mesenchymal stem cells (BMC9 cells; Dennis and Caplan, 1996) were cultured as previously described (Chateauvieux et al., 2007) in α-MEM with nucleosides (Invitrogen) supplemented with 10% FCS (Invitrogen), 2 mM l-glutamine (Invitrogen), and penicillin–streptomycin (100 U/ml and 100 μg/ml, respectively; Invitrogen).
A human hepatocellular carcinoma cell line (Hep 3B2.1-7, ATCC no. HB-8064; American Type Culture Collection [ATCC], Manassas, VA), transformed African green monkey kidney fibroblast cell line (COS-7, ATCC no. CRL-1651; ATCC), and murine osteosarcoma cell line (mOS-J; Joliat et al., 2002) were used for control experiments. Cells were cultured in DMEM with glucose at 4.5 g/liter and supplemented with 15% FCS, 2 mM l-glutamine, and penicillin–streptomycin (100 U/ml and 100 μg/ml, respectively).
Primary culture of adult mesenchymal stem cells
Total bone marrow was obtained from wild-type adult male Swiss mice and male Rosa26 lacZ mice (background C57BL/6J × 129S2; kindly provided by M.F. Gardahaut, Nantes, France) by flushing femurs and tibias with culture medium. Cells were plated at a density of 500,000 cells/cm2 in medium composed of α-MEM with nucleosides supplemented with 10% FCS, 2 mM l-glutamine, penicillin–streptomycin (100 U/ml and 100 μg/ml, respectively), and human fibroblast growth factor (FGF)-2 at 2 ng/ml (AbCys, Paris, France). The culture medium was changed on day 3 to remove nonadherent cells and was subsequently replaced weekly. The cells were grown for 2–3 weeks until confluent. Adherent cells were then detached with 0.5% trypsin–EDTA and plated at a density of 10,000 cells/cm2. Subsequent passages and seeding of the cells were performed at a density of 5000 cells/cm2. From passage 8, mesenchymal stem cell (MSC) cultures were characterized by fluorescence-activated cell-sorting (FACS) analysis (FACSCalibur instrument with CellQuestPro software; BD Biosciences, San Jose, CA) after incubation with anti-CD45–phycoerythrin (PE), anti-CD90–PE, anti-CD29–CyChrome, anti-Sca1–PE, anti-CD106–CyChrome (BD Biosciences) (Dominici et al., 2006).
In vitro cell transfection with chloramphenicol acetyltransferase or green fluorescent protein reporter gene and evaluation of gene transfer system efficacy
ESCs, BMC9 cells, and differentiated cells were transfected with pCIK-CAT (4.7 kb) and pEGFP-C1 (4.7 kb) plasmids encoding chloramphenicol acetyltransferase (CAT) protein and GFP, respectively, using synthetic vectors just before intratracheal administration (Pitard et al., 2001). DNA plasmids were complexed with ICAfectin 441 according to the manufacturer's instructions (In-Cell-Art, Nantes, France). After 2 hr, transfection complexes were removed by changing growth medium and cells were lysed for dosages or kept in culture for 24 hr or 7 days, or injected intratracheally as indicated previously.
MSCs from Swiss mice were transfected with pCIK-CAT and pEGFP-C1 plasmids, using nucleofection (Nucleofector solution; Amaxa Biosystems, Cologne, Germany), just before intratracheal administration (Aluigi et al., 2006). After 2 hr, growth medium was changed and cells were lysed for dosages or kept in culture for 24 hr or 7 days, or injected intratracheally as indicated previously. The percentage of GFP-expressing cells was evaluated 24 hr after GFP nucleofection, using FACS analysis.
The efficacy of gene transfer systems was evaluated by cytometry for GFP-expressing cells and by ELISA for CAT-expressing cells. The percentage of GFP-expressing cells was evaluated 24 hr after GFP transfection, using FACS analysis (FACSCalibur instrument with CellQuestPro software). CAT protein quantity per stem cell at 24 hr was then calculated on the basis of in vitro data: CAT protein quantity in cell lysis buffer was divided by the cell number and by the percentage of GFP-expressing cells (obtained by FACS analysis). Last, this value was used to determine the minimal number of CAT-expressing stem cells required to be detected by ELISA in vitro or in vivo, considering that the CAT ELISA detection threshold was 50 pg of CAT protein (see later).
In vitro stem cell transduction with nls-lacZ reporter gene
BMC9 cells were transduced with an amphotropic recombinant murine retroviral vector carrying the nls-lacZ reporter gene (lacZ gene encoding β-galactosidase [β-Gal]) coupled to a nuclear localization signal [nls]; kindly provided by N. Ferry, Nantes, France). This retroviral vector was obtained from the human producer cell line TELCeB6AF7 derived from the Te671 cell line as described by Aubert and colleagues (2002). Twenty-four-hour recombinant retroviral supernatant was harvested from the confluent producer cell line and filtered through a 0.45-μm (pore size) membrane. The titer of the β-Gal supernatant, determined by end-point dilution with Te671 target cells, was 2 × 108 transducing particles/ml. After BMC9 cell transduction, retroviral vector was removed by changing the growth medium and cells were lysed for dosages or kept in culture or injected intratracheally as indicated previously.
To evaluate the percentage of β-Gal-transduced BMC9 cells, cells were fixed in 4% paraformaldehyde, washed with PBS, and incubated for 2 hr at 37°C in 5 mM K4Fe(CN)6, 5 mM K3Fe3(CN)6, and 2 mM MgCl2 in PBS containing 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal [0.5 mg/ml] dissolved in N,N-dimethylformamide at 20 mg/ml before dilution into the reaction mixture; Sigma-Aldrich). Cells were identified as positive for β-Gal activity by blue nuclear staining after X-Gal reaction. The percentage of β-Gal-expressing cells in vitro was determined by counting the number of nuclear-blue cells in a total of 200 cells.
MSCs from Swiss mice were transduced with a lentiviral vector carrying the nls-lacZ reporter gene. The vector was derived from pHR′ LacZ (Zufferey et al., 1997) in which an nls from the simian virus 40 (SV40) T antigen was cloned in frame 5′ to the lacZ cDNA. The lentiviral vector was produced in human embryonic kidney 293T cells as previously described (Selig et al., 1999). After transduction with 50 multiplicities of infection (MOI), lentiviral vector was removed by changing the growth medium and cells were lysed for dosages or kept in culture or injected intratracheally as indicated previously. The percentage of β-Gal-transduced MSCs was evaluated by in vitro X-Gal staining and counting as described previously.
Reporter gene assay and estimation of survival rate
CAT-transfected cells, BAL cells and BAL fluid, or whole frozen trachea–lungs were homogenized in reporter lysis buffer (Roche Diagnostics, Indianapolis, IN) supplemented with protease inhibitors (Roche Diagnostics). After centrifugation at 10,000 rpm for 5 min, CAT quantity was measured in supernatant with a VICTOR2 multilabel counter (PerkinElmer Life Sciences, Waltham, MA), using a CAT ELISA kit according to the instructions of the supplier (Roche Diagnostics). Each sample was analyzed in duplicate. The CAT detection threshold was 50 pg. Protein content was measured with a bicinchoninic acid (BCA) protein assay kit (Pierce Biotechnology, Rockford, IL).
To estimate cell survival rate 24 hr after intratracheal administration, we first calculated CAT protein quantity per CAT-expressing cell, using in vitro data: CAT quantity in cell lysis buffer was divided by the cell number and by the percentage of GFP-expressing cells (obtained by FACS analysis). Last, the total in vivo CAT protein quantity per animal was divided by this in vitro value, in order to estimate the number of CAT-expressing cells per animal and subsequently the cell survival rate.
BMC9 cells and Rosa26 MSCs or whole frozen murine trachea–lungs were homogenized in 1 ml of reporter lysis buffer (Roche Diagnostics) supplemented with a protease inhibitor cocktail (Roche Diagnostics). After centrifugation at 10,000 rpm for 5 min, β-Gal activity was determined by enzymatic fluorimetric assay using 4-methylumbelliferyl-β-d-galactoside (4-MUG; Sigma-Aldrich) as a fluorescent substrate. Each cell and trachea–lung sample was analyzed in duplicate. The β-Gal activity detection threshold was 230 pg. Protein content was measured with a BCA protein assay kit. Additional experiments were done with lysates of β-Gal-transduced BMC9 cells or Rosa26 MSCs after three cycles of freezing and thawing. Viability was ascertained by trypan blue exclusion.
Detection of β-galactosidase activity
Tissues were fixed in 4% paraformaldehyde (at room temperature, 20 min) and rinsed three times with PBS. Immediately after fixation, trachea–lungs were stained by intratracheal infusion (though a 19-gauge needle; total volume, 5 ml) and immersion for 6 hr at 30°C in 5 mM K4Fe(CN)6, 5 mM K3Fe3(CN)6, and 2 mM MgCl2 in PBS containing X-Gal at 0.5 mg/ml (as indicated previously) to avoid the detection of endogenous β-Gal activity (Weiss et al., 1997). Tissues were again washed with PBS and immediately embedded in paraffin. Tissues were identified as positive for β-galactosidase activity by nuclear blue staining after X-Gal reaction. Sections (4 μm thick) were stained with nuclear fast red (Sigma-Aldrich).
Detection of nls-lacZ reporter gene
Total DNA from whole murine trachea–lungs was isolated for PCR analysis. Tissues were digested overnight with lysis buffer containing proteinase K (10 mg/ml; Sigma-Aldrich). Total DNA was extracted with phenol–chloroform–isoamyl alcohol (Sigma-Aldrich) and then precipitated in ethanol. Total DNA from 106 transduced MSCs was also extracted for PCR experiments. PCR were performed with primers that specifically bind the nls sequence and the lacZ gene encoded in the nls-lacZ lentiviral vector: 5′-GTA ACA ACT CCG CCC CAT-3′ and 5′-GAC AGT ATC GGC CTC AGG AA-3′.
Statistical analysis
Data are expressed as means ± SEM. Statistical significance was evaluated by analysis of variance (ANOVA) to compare control and treatment groups of three or more. Tukey's test was subsequently used for pair-wise comparisons. A nonparametric Mann–Whitney test was performed to compare two groups. Statistical significance was set at p < 0.05.
Results
Transient epithelial airway injury induced by intratracheal polidocanol administration
Intratracheal administration of 2% PDOC induced macroscopic lung injury at 24 hr with hemorrhage (Fig. 1b, k, and n). The major histological findings at 24 hr were acute injury of the murine airway epithelium with focal shedding areas observed at the epithelial surface, with only a remaining layer of basal cells or a total denudation of the basement membrane (Fig. 1e and h). However, in some regions, the lumenal layer of epithelial cells was just disrupted or sloughed whereas the basal cell layer and the basement membrane remained intact (data not shown), as already described after intratracheal or intranasal administration (Driscoll et al. [2000] and Southam et al. [2002], respectively). Inflammation was present in pulmonary parenchyma (Fig. 1k and n, arrowheads). Trachea–lung sections 7 days after PDOC administration demonstrated significant improvement of hemorrhage (Fig. 1c and l) and of airway injury, with epithelial surface again covered by cilia (Fig. 1f and i). The PDOC effect did not significantly alter animal survival (see Supplementary Table S1 at

Epithelial damage induced by intratracheal administration of polidocanol (PDOC). (
ESC and BMC9 cell survival in murine airway with acute epithelial airway injury
In a first set of experiments, embryonic stem cells (ESCs) and adult mesenchymal stem cells (BMC9 cells) from well-characterized cell lines were intratracheally injected. ESCs and BMC9 cells were injected 24 hr after PDOC administration, when epithelial airway injury induced by PDOC was acute (MacPherson et al., 2005). Stem cell survival was evaluated 24 hr after stem cell administration. To calculate the survival rate, we used a quantitative method based on in vivo expression of reporter genes by transplanted cells. Stem cells were transfected 2 hr before intratracheal injection in vitro with plasmids encoding the CAT or GFP reporter gene. CAT-transfected stem cells were then intratracheally injected, at a time when they did not yet express the foreign genes (see later). We hypothesized that only surviving stem cells would express in vivo CAT protein in trachea–lungs 24 hr after intratracheal cell injection.
Two hours after the beginning of cell incubation with both plasmids and synthetic vectors, transfection complexes were removed and reporter gene expression was evaluated. Neither CAT protein nor cell fluorescence was detected in stem cells by ELISA (Fig. 2b) and microscopy (data not shown), demonstrating that stem cells did not express CAT or GFP protein at the time of in vivo injection. In vitro reporter gene expression was also evaluated 24 hr and 7 days after incubation with plasmids and synthetic vectors. As evaluated by FACS analysis, 19.4 ± 0.2% of ESCs (n = 6) and 37.0 ± 4.4% of BMC9 cells (n = 6) expressed GFP in vitro at 24 hr (Fig. 2a). However, CAT and GFP were not detected 7 days after transfection (Fig. 2b), suggesting that transgene expression was transient. Using quantitative CAT ELISA results and cellular rates of GFP expression, we calculated that CAT-expressing ESCs and CAT-expressing BMC9 cells contained, at 24 hr, 0.03 ± 0.01 pg of CAT per cell (n = 10) and 0.08 ± 0.02 pg of CAT per cell (n = 6), respectively. Cell values were further used to evaluate the number of in vivo-surviving cells after intratracheal injection.
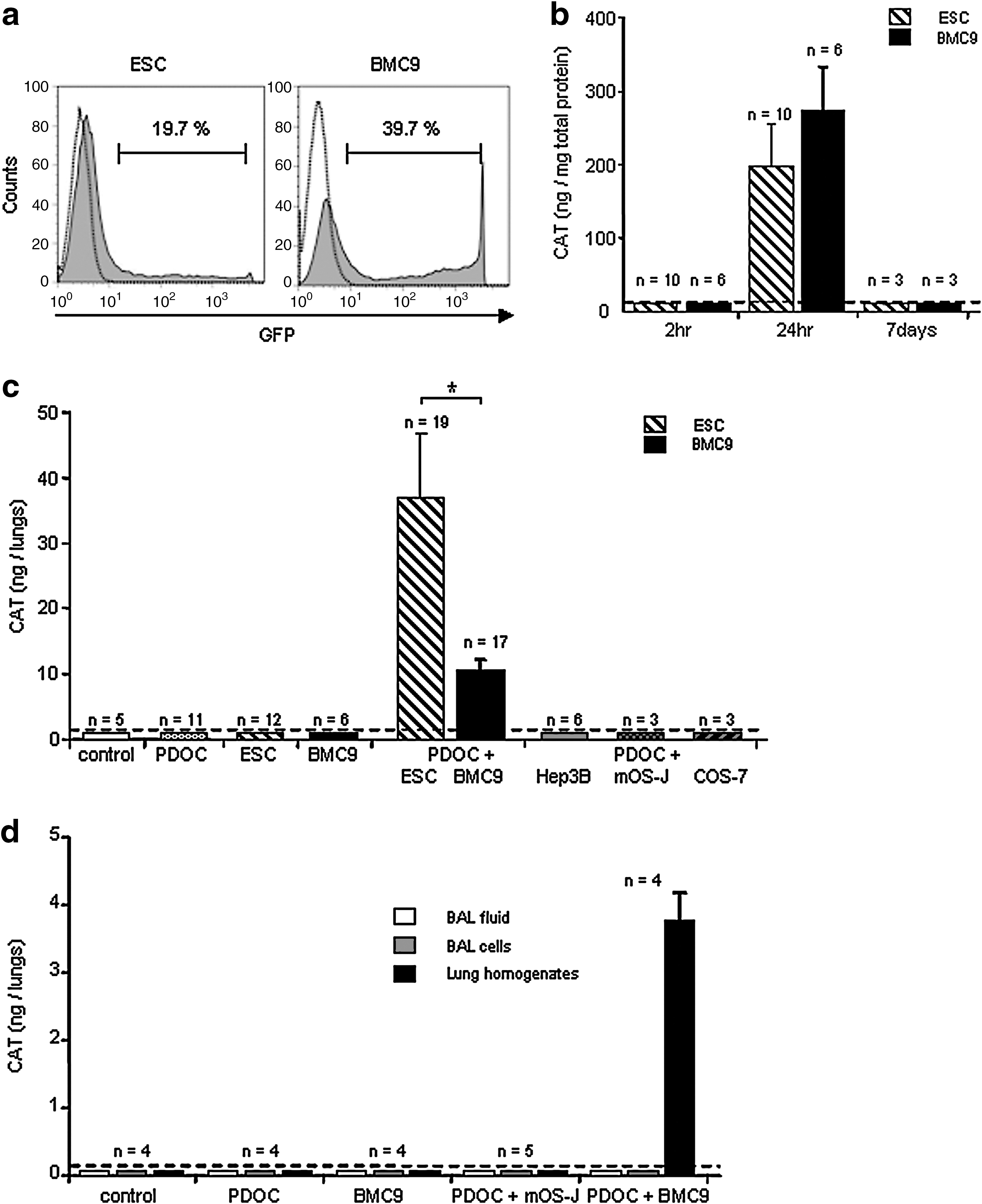
In vitro cell transfection with reporter genes and in vivo chloramphenicol acetyltransferase (CAT) gene expression in injured airway 24 hr after intratracheal stem cell administration. To monitor stem cell survival in vivo, murine embryonic stem cells (ESCs) and adult mesenchymal stem cells (BMC9 cells) were transfected in vitro with plasmids encoding the CAT or GFP reporter gene just before intratracheal administration. (
For intratracheal administration, CAT-transfected ESCs and CAT-transfected BMC9 cells were harvested 2 hr after the beginning of transfection and injected intratracheally. Mice were killed 24 hr after intratracheal administration; trachea–lungs were used in toto to quantify CAT protein by ELISA (Fig. 2c). No CAT protein was detected in control animals or animals injected with PDOC only. Importantly, CAT protein was detected in 19 of 31 animals (61%) intratracheally injected with PDOC and ESCs and in 17 of 21 animals (81%) intratracheally injected with PDOC and BMC9 cells, suggesting that ESCs and BMC9 cells survived within lungs and expressed reporter genes 24 hr after intratracheal injection. To estimate the number of surviving cells within the lungs at 24 hr, total CAT protein quantity per animal was divided by CAT quantity per cell in vitro. The survival rate of 106 injected stem cells was 3.69 ± 0.86% (n = 19), whereas this survival rate was significantly less with BMC9 cells (0.43 ± 0.12%, n = 17, Tukey's test; p = 0.02). CAT cell quantities were also used to calculate the in vivo cell detection threshold. A minimum of 10 (±1.2) × 103 ESCs and 4.3 (±1.1) × 103 BMC9 cells could be detected in one animal in vivo, corresponding to 0.4–1% of injected cells. These results suggest that even if the percentage of cells expressing reporter gene in vitro was low, this was still a sensitive method for in vivo detection of a small cell number.
Influence of differentiation state and airway environment on ESC and BMC9 cell survival
To determine whether cell survival ability in the injured murine airway was specific to stem cells, differentiated cells of various origins (Hep 3B, mOS-J, and COS-7 cells) were transfected in vitro with CAT plasmids under conditions similar to that of ESC and BMC9 cell transfection (data not shown). CAT-transfected differentiated cells were then intratracheally injected 2 hr after the beginning of incubation with CAT plasmid. No CAT protein was detected in any animal injected with PDOC and differentiated cells at 24 hr (Fig. 2c), suggesting that only stem cells survived after intratracheal injection into injured airway. CAT-transfected ESCs and BMC9 cells were injected into healthy control animals. No CAT protein was detected in any animal with a healthy airway 24 hr after stem cell injection (Fig. 2c), suggesting that stem cell survival was favored by airway injury.
Cell survival in epithelial lining fluid
To evaluate whether cells expressing CAT reporter protein were present within the epithelial lining fluid or after phagocytosis by alveolar macrophages, bronchoalveolar lavages (BALs) were performed at 24 hr under in vivo conditions, when phagocytosis was expected to be maximal (e.g., when CAT ELISA results were negative), using mesenchymal stem cells (BMC9 cells) in healthy airway and differentiated murine cells (mOS-J) in injured airway. As further controls, BALs were also performed in control and PDOC-treated animals and in animals intratracheally injected with PDOC and BMC9 cells. No CAT protein was detected in any BAL fluid or BAL cell sample from any animal (Fig. 2d), suggesting that if stem cells or differentiated cells were phagocytosed by alveolar macrophages, such phagocytosis did not alter CAT protein measurement or cell survival detection. Importantly, in animals intratracheally injected with PDOC and BMC9 cells, high levels of CAT protein were measured in trachea and lung homogenates whereas no CAT protein was detected in BAL fluid or BAL cell samples, suggesting that CAT-expressing cells were present mostly within the lungs and did not survive in the epithelial lining fluid (Fig. 2d).
BMC9 cell location at 24 hr and 7 days
We next investigated BMC9 cell location at 7 days, when the airway epithelium was known to be spontaneously regenerating (MacPherson et al., 2005). As CAT expression using synthetic vectors was transient, we used integrative viral vectors allowing long-term foreign gene expression. BMC9 cells were transduced with a retroviral vector encoding the nls-lacZ gene. As evaluated by X-Gal staining, 57.2 ± 0.1% of BMC9 cells expressed nuclear β-galactosidase (β-Gal) when they were injected (Fig. 3a; n = 6). β-Gal activity in transduced BMC9 cells was significantly increased just before injection as compared with endogenous β-Gal activity in BMC9 cells (Fig. 3b; Mann–Whitney test, p = 0.02) and remained stable at 7 days (data not shown). Animals were assessed for β-Gal activity 24 hr and 7 days after intratracheal administration, using the 4-MUG method (Fig. 3c). In contrast to previous experiments, β-Gal-transduced BMC9 cells expressed the nls-lacZ gene at the time of intratracheal injection. Therefore, as a further control, we injected lysates of β-Gal-transduced BMC9 cells, and no significant increase in β-Gal activity was observed as compared with endogenous β-Gal activity (Tukey's test, p = 0.98). In animals intratracheally injected with PDOC and β-Gal-transduced BMC9 cells, 24 hr after injection β-Gal activity significantly increased as compared with that of control animals (Tukey's test, p = 0.003) and animals injected with PDOC and lysate of β-Gal-transduced BMC9 cells (Tukey's test, p = 0.004), confirming results with CAT gene transfer. However, 7 days after stem cell injection β-Gal activity was similar to that of control animals (Tukey's test, p = 0.98) and animals injected with PDOC and lysate of β-Gal-transduced BMC9 cells (Tukey's test, p = 0.94), suggesting that the number of surviving BMC9 cells in the airway epithelium decreased significantly between 24 hr and 7 days.
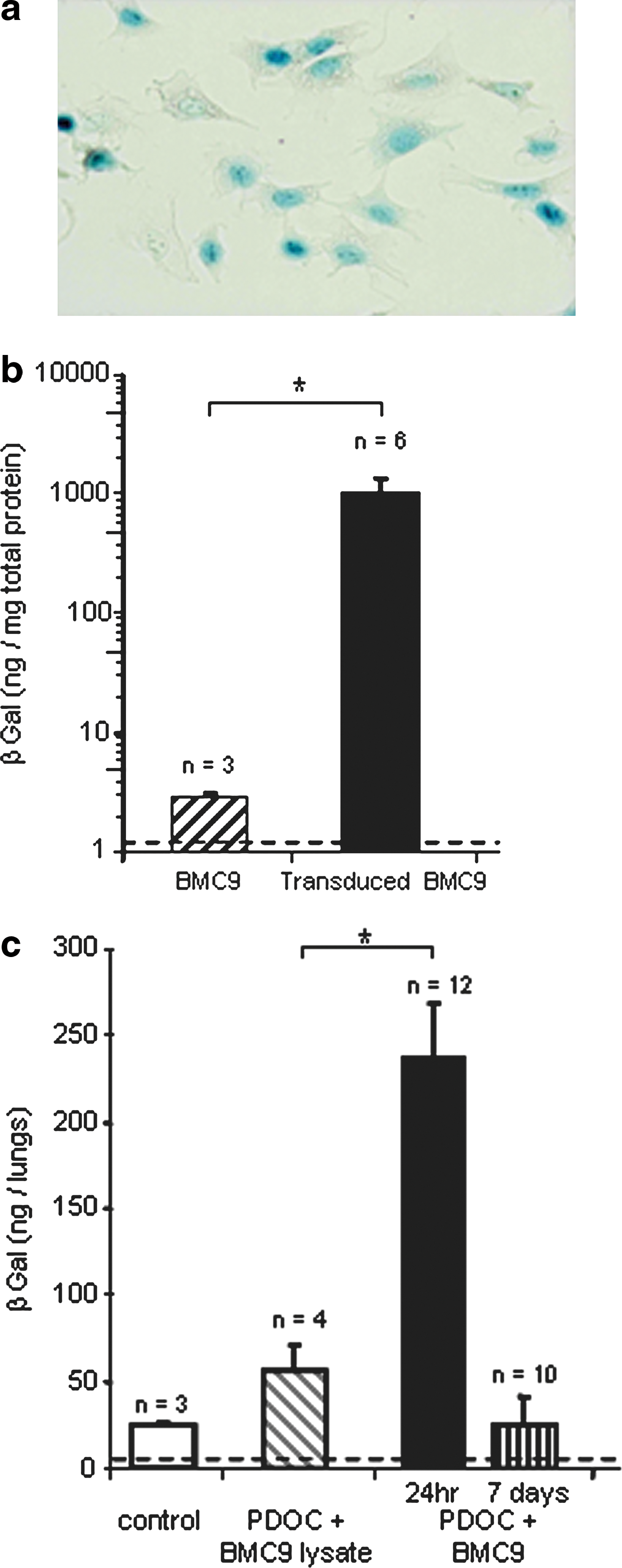
In vitro and in vivo detection of β-Gal activity 24 hr or 7 days after β-Gal-transduced BMC9 intratracheal administration. BMC9 cells were transduced in vitro with a retroviral vector encoding the nls-lacZ gene before in vivo administration. (
To locate BMC9 cells after intratracheal injection into injured airway, we performed X-Gal staining of in toto trachea–lungs at 24 hr and 7 days (Fig. 4). No blue staining was observed in trachea–lungs (Fig. 4a) and histological sections (Fig. 4b–d) from any control animal (Fig. 4a; n = 3), animal injected with PDOC (Fig. 4b; n = 3), or animal intratracheally injected with β-Gal-transduced BMC9 cells only (Fig. 4c; n = 5). No cell with nuclear blue staining was observed in any animal injected with PDOC and lysate of β-Gal-transduced BMC9 cells (Fig. 4d; n = 5), suggesting that cells with nuclear blue staining expressed the nls-lacZ gene de novo. In contrast, macroscopic and microscopic strong nuclear blue staining was observed in trachea and pulmonary lobes at 24 hr (Fig. 4e–m; n = 16) and 7 days (Fig. 4n–p; n = 7) in each animal intratracheally injected with PDOC and β-Gal-transduced BMC9 cells. At 24 hr, macroscopic analyses showed blue spots (≤5 spots) in trachea from 8 of 16 animals (Fig. 4e and f) and blue spots in one pulmonary lobe (15 of 16; Fig. 4g and h) or two pulmonary lobes (1 of 16; Fig. 4i and j). Blue spots were also observed in large bronchi from 5 of 16 animals (Fig. 4i and j). Histological analyses confirmed the macroscopic result for each sample, showing β-Gal-positive cells on histological sections where blue spots had been observed. Clusters of cells with blue nuclei were observed in the lumen of injured trachea (Fig. 4k) and injured bronchi or bronchioles (Fig. 4l and m), but not in lung parenchyma (Fig. 4l). At 7 days, blue spots were observed in seven of seven animals, in the lumen of bronchioles (Fig. 4n–p). No blue cell was observed in trachea or in pulmonary parenchyma (data not shown). Clusters of blue cells were sometimes found in polyp-like structures, located in bronchioles and large bronchi. Polyp-like structures were not observed in any animal receiving PDOC only. Further quantitative and statistical analyses demonstrated that the development of polyp-like structures at 7 days was not due to stem cell administration but rather to a second intratracheal administration (PBS or BMC9 cells) 24 hr after PDOC injection (see Supplementary Table S2 at

BMC9 cell location 24 hr and 7 days after intratracheal injection, using X-Gal staining. (
MSC survival in murine airway with acute epithelial airway injury
In a second set of experiments, to avoid in vivo cell survival linked to cell line transformation, we repeated previous experiments with the same experimental design, using murine MSCs from primary cultures. MSC cultures were characterized according to Dominici and colleagues (2006) and Chateauvieux and colleagues (2007). FACS analysis demonstrated that MSCs were CD45– (Fig. 5a; 0.4 ± 0.1%, n = 4), CD90– (0.43 ± 0.2%, n = 6), CD29+Sca-1+ (Fig. 5a; 96.9 ± 1.3 and 98.1 ± 0.7%, respectively, n = 4), and CD106+ (96 ± 0.8%, n = 4).

In vitro cell transfection with reporter genes and in vivo CAT gene expression 24 hr after intratracheal administration into injured airway of murine adult mesenchymal stem cells (MSCs) from primary cultures. (
MSCs were transiently transfected in vitro with plasmids encoding the CAT or GFP reporter gene. In contrast to ESCs and BMC9 cells, MSC transfection with synthetic vectors was ineffective (data not shown), and therefore nucleofection was used to obtain a significant gene transfer rate. As evaluated by FACS analysis, 56.8 ± 2.9% of MSCs expressed GFP in vitro at 24 hr (Fig. 5c; n = 3), but CAT and GFP were not detected 7 days after nucleofection (Fig. 5b), suggesting that foreign gene expression was again transient and that nucleofection could not be used for 7-day in vivo experiments. CAT-transfected MSCs contained 0.35 ± 0.02 pg of CAT per cell at 24 hr (n = 6). CAT-transfected MSCs were then intratracheally injected 2 hr after the beginning of incubation with CAT plasmid. In control mice and mice injected with PDOC only, no CAT protein was detected at 24 hr (Fig. 5d). Importantly, CAT protein was detected in each animal injected with PDOC and MSCs (Fig. 5d; n = 8; Tukey's test, p < 0.001), suggesting that MSCs from primary cultures survived in injured airway and expressed CAT protein after intratracheal administration. The MSC survival rate was 5.52 ± 1.9% (n = 8), and a minimum of 710 ± 45 CAT-expressing MSCs could be detected in one animal in vivo, corresponding to 0.07% of injected cells, confirming CAT ELISA sensitivity for in vivo experiments.
MSC location at 24 hr and 7 days
To investigate whether MSCs could be located into the airway epithelium 7 days after cell injection, we first used MSCs in primary cultures from β-Gal+ transgenic (Rosa26) mice, thus continuously expressing cytoplasmic β-Gal (Fig. 6a). Using the 4-MUG method (Fig. 6c), in vitro β-Gal activity was significantly increased in Rosa26 MSCs as compared with MSCs from Swiss mice (Fig. 6c; Mann–Whitney test, p = 0.03). Mice were assessed for β-Gal activity 24 hr and 7 days after intratracheal administration of Rosa26 MSCs or of a lysate of Rosa26 MSCs (Fig. 6d). As observed in previous experiments with β-Gal-transduced BMC9 cells, β-Gal activity was similar in murine airway from control animals and animals intratracheally injected with lysates of Rosa26 MSCs (Tukey's test, p = 0.68). In animals intratracheally injected with PDOC and Rosa26 MSCs, β-Gal activity was significantly increased as compared with that of both control groups (Tukey's test, p = 0.01). However, in contrast to previous experiments with β-Gal-transduced BMC9 cells, no blue staining was observed at 24 hr in trachea–lungs from animals injected intratracheally with PDOC and Rosa26 MSCs, and Rosa26 MSCs could not be located on histological slides (data not shown). Finally, β-Gal activity in murine injured airway returned to baseline 7 days after Rosa26 MSC intratracheal injection (Tukey's test, p = 0.85).
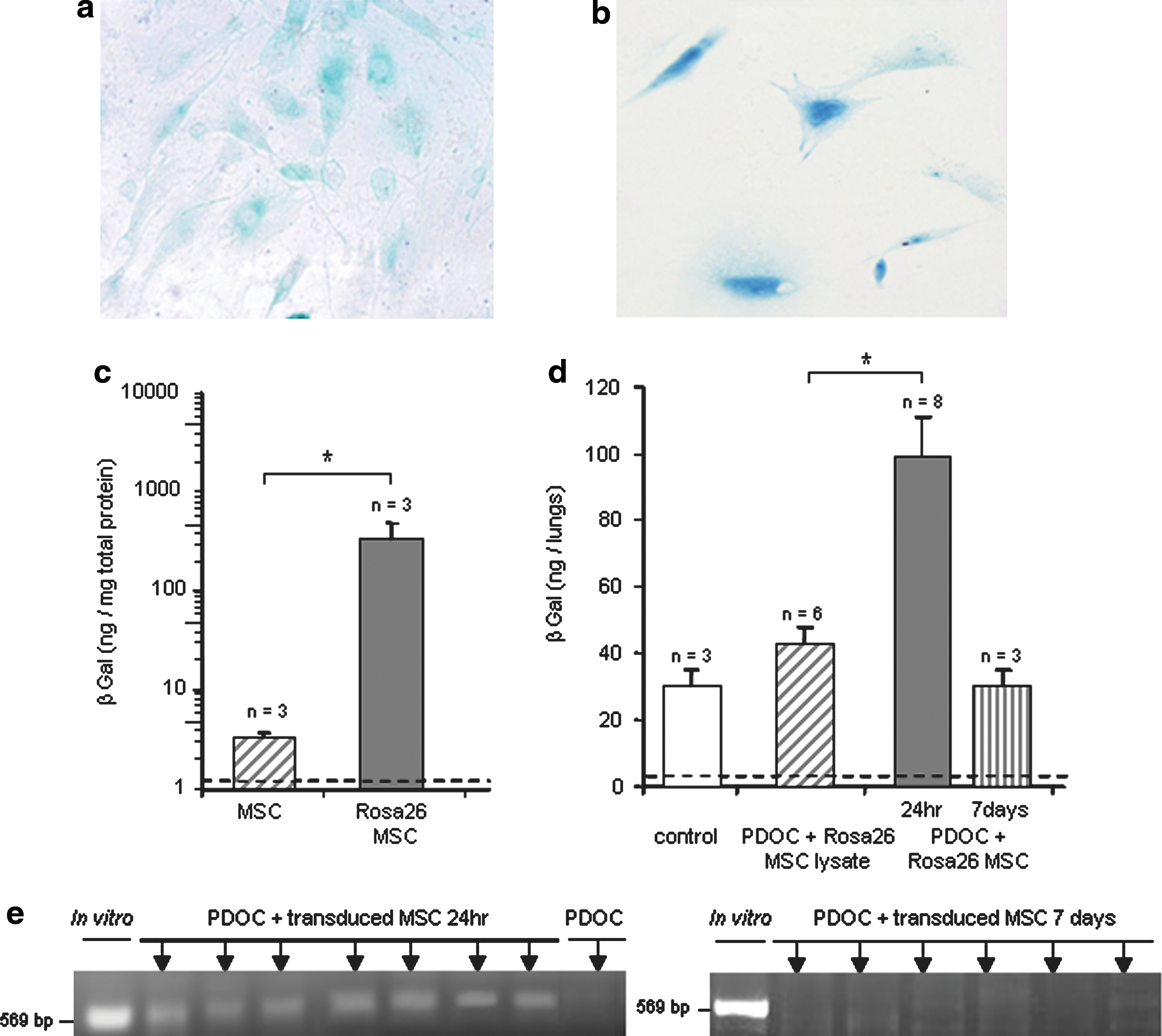
In vitro and in vivo detection of Rosa26 MSCs and β-Gal-transduced MSCs 24 hr and 7 days after administration. MSCs from wild-type mice were transduced with a lentiviral vector encoding the nls-lacZ gene. (
In a last set of experiments, as previously performed with BMC9 cells, we overexpressed the nls-lacZ gene in MSCs from wild-type Swiss mice, to locate them in vivo at 24 hr and 7 days. MSC transduction with the β-Gal retroviral vector remained ineffective. Therefore, MSCs from Swiss mice were transduced before intratracheal injection with a lentiviral vector encoding the nls-lacZ gene (Conrad et al., 2007). After lentiviral transduction, 20.1 ± 3.0% of MSCs expressed β-Gal (Fig. 6b; n = 4). Nevertheless, no blue staining was observed in trachea–lungs from animals injected intratracheally with PDOC and β-Gal-transduced MSCs at 7 days (data not shown). Because rapid downregulation of foreign gene expression under the control of the cytomegalovirus promoter has been described in lungs (Pringle et al., 2005), we used PCR concurrently to detect the presence of the nls-lacZ gene even without gene expression. PCR experiments using primers specific to the nls-lacZ gene were performed 24 hr and 7 days after β-Gal-transduced MSC injection (Fig. 6e). At 24 hr, PCR was positive in trachea–lungs from each animal injected with PDOC and β-Gal-transduced MSCs (Fig. 6e; n = 7), confirming that β-Gal-transduced MSCs were present 24 hr after injection into injured airway. However, no nls-lacZ gene was detected in animals 7 days after β-Gal-transduced MSC injection, suggesting that no β-Gal-transduced MSCs from primary culture survived at this time once the epithelium is regenerated (Fig. 6e; n = 6).
Discussion
Using independent methods based on reporter gene transfer, we demonstrated the short-term survival of various types of exogenous stem cells, including embryonic and adult mesenchymal stem cells, after intratracheal injection into murine airway presenting acute epithelial airway injury and without total body irradiation. In contrast to differentiated cells, 0.43 to 5.5% of stem cells were capable of surviving within the injured lungs at 24 hr. Importantly, no SCs survived in healthy airway or in the epithelial lining fluid. Biochemical staining showed that transduced MSCs were located in the lumen of conducting airway at 24 hr and at 7 days. However, the in vivo amount of engrafted MSCs decreased dramatically with time. No primary MSCs were located within lungs at 7 days.
In our study, several gene transfer systems were used, according to our aims and to cell types. Using careful timing, nonviral vectors were useful to allow expression of the foreign gene only after intratracheal cell injection. Gene delivery with viral vectors was used for in vivo studies at 7 days, but we had to use different viral vectors to transduce MSCs from cell lines and primary MSCs. Depending on the gene transfer system and the cell type, gene transfer efficacy was variable in vitro. Studies with retroviral vectors showed high efficacy and stable MSC transduction (up to 97%; Sales et al., 2007) whereas studies with lentiviral vectors showed variable efficacy between murine MSCs (up to 50%) (Ricks et al., 2008; Santoni de Sio et al., 2008; Xu et al., 2008) and human MSCs (up to 93%; Chan et al., 2005). These data confirmed that primary murine MSCs are more difficult to transduce than human MSCs.
Engraftment of exogenous stem cells into the lungs has been reported in several in vivo studies, but the cell engraftment rate remains controversial (Loebinger et al., 2008a). Our results highlight the main advantage of using ELISA and biochemical methods to detect and quantify a small number of administered cells in the whole organ. Although the lacZ gene is widely used to locate stem cells, some reports have shown that standard protocols for X-Gal staining can lead, especially in the lungs, both to false positive results as well as to failure to adequately detect β-Gal-expressing cells (Weiss et al., 1997). In our study, although β-Gal activity significantly increased after intratracheal injection of MSCs from Rosa26 mice, surviving Rosa26 MSCs could not be located in vivo on histological slides, as already described in another report (MacPherson et al., 2005). The lacZ gene coupled with a nuclear localization signal had already been used in several studies from our group (Lemarchand et al., 1994; Chapelier et al., 1996) and others (Mastrangeli et al., 1993) and was shown to allow easy distinction of endogenous from exogenous β-Gal activity. Using the nls-lacZ gene, we were able to detect and locate nuclear β-Gal-expressing cells, even when β-Gal activity in the whole lungs was not significantly different from baseline.
We investigated whether stem cells would be capable of locating specifically on the epithelial side of the airway after intratracheal administration. BAL experiments showed that stem cells expressing reporter protein did not survive in the epithelial lining fluid. X-Gal staining showed that stem cells were localized into the conducting airway in vivo. By injecting stem cells into healthy airway, we also confirmed the major role of the injured environment in the engraftment (MacPherson et al., 2005). Stem cells may not engraft into healthy airway because of mucociliary clearance mechanisms that could impede access to airway epithelium, as described for in vivo gene transfer to the lungs (Weiss, 2002). We speculated that mucociliary clearance would be disrupted in polidocanol (PDOC)-treated airway and that the freshly denuded basement membrane may also specifically promote stem cell adherence and engraftment in the airway epithelium (Engelhardt et al., 1992; Nikolova et al., 2006). In the first set of experiments, murine ESCs were intratracheally injected and a significant survival cell rate was observed, although the in vitro ESC transfection rate was low. This confirms the great potential of ESC pluripotency for regenerative medicine and in particular airway injury. Nevertheless, ESCs for cell-based therapy have met with ethical, moral, and political challenges, and with inherent risks associated with immune rejection (Chidgey et al., 2008). In further experiments, we focused our attention on MSCs because they are already used in the clinical setting (Uccelli et al., 2008). Our ELISA data indicated that the MSC survival rate varied according to whether cultures were immortalized or primary, probably because of the different gene transfer systems we used. Survival rates (from 0.4 to 5.5%) 24 hr after intratracheal administration were higher than those described after intravenous administration (from 0.01 to 0.1%) and were in agreement with those usually described in stem cell therapy protocols, including cardiology (Robey et al., 2008) and diabetology (Bottino et al., 2003) protocols, and lung injury models (Gupta et al., 2007; Wong et al., 2007, 2009).
We hypothesized that MSC incorporation in the lungs at 7 days could be masked by downregulation of transgene expression in the lungs because of the common cytomegalovirus enhancer/promoter element in the viral vector constructs (Alton et al., 1999; Pringle et al., 2005). The negative results of PCR experiments on the nls-lacZ gene at 7 days demonstrated that this was not the case. Allogeneic rejection seems unlikely to be responsible for the lack of survival of differentiated cells at 24 hr because ESCs, although known to be targeted for rejection by the immune system (Chidgey et al., 2008), survived at 24 hr. Nevertheless, allogeneic MSC rejection cannot be ruled out at 7 days.
Another hypothesis to explain negative MSC results at 7 days concerns the spontaneous regeneration of airway epithelium, which may have hampered MSC incorporation into the airway epithelium. At 24 hr, that is, when there is desquamation of the epithelial surface and a denuded basement membrane, the PDOC murine model is a good model to evaluate stem cell survival and presence in injured airway; but stem cell differentiation into airway epithelial cells is unlikely at this time. Seven days after PDOC administration, the epithelium was spontaneously regenerated, demonstrating that PDOC did not alter the lung's endogenous ability to regenerate. Therefore, the PDOC model may not be an adequate model to evaluate stem cell capability in regenerating lung epithelium, because the endogenous repair mechanisms may impede the MSC contribution to achieve structural lung regeneration. This limitation was described in the study by Wong and colleagues (2007), in which naphthalene-injured airway epithelium was still undergoing rapid cell turnover and regeneration and the number of administered stem cells also decreased with time. A more appropriate animal model to study stem cell contribution to structural lung regeneration would be a model with chronic epithelial airway injury and a recurrent loss of the epithelial surface, due to a lack, or exhaustion, of endogenous progenitor cells. This hypothesis was evaluated in animal models with permanent retinal epithelial degeneration such as the RCS rat model (D'Cruz et al., 2000) and the rhodopsin knockout mouse (Humphries et al., 1997). MSC engraftment and epithelial regeneration involving MSCs were observed in these animal models (Arnhold et al., 2007; Inoue et al., 2007). For lung tissue, a murine model overexpressing the β subunit of the epithelial Na+ channel (ENaC +/+) and showing epithelial degeneration in newborn animals (Mall et al., 2008) may be more appropriate to evaluate the MSC contribution to lung regeneration.
Finally, an important limitation of our study was also the lack of phenotypic characterization of surviving cells. This characterization of stem cell-derived epithelial cells has been hampered by methodological problems, in the lungs as in many other organs such as the heart (Murry et al., 2004) or the brain (Castro et al., 2002), and remains difficult and controversial. In this regard, additional studies using complex transgenic models with reporter genes under the control of lung-specific promoters in vivo will be essential.
Footnotes
Acknowledgments
The authors thank Nicolas Ferry (Nantes, France) for providing the nls-lacZ retroviral vector and Marie-France Gardahaut (Nantes, France) for providing the Rosa26 mice. The authors are grateful to Beatrice Delasalle for statistical analyses. This work was supported in part by a grant from Vaincre la Mucoviscidose (Paris, France).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.