
Abstract
We are evaluating the use of electroporation (EP) to deliver a novel DNA vaccine, p.DOM-PSMA27. This vaccine encodes a domain (DOM) of fragment C of tetanus toxin to induce CD4+ T cell help, fused to a tumor-derived epitope from prostate-specific membrane antigen (PSMA) for use in HLA-A2+ patients with recurrent prostate cancer. We report on safety and tolerability and on antibody response to DOM as a first indication of the effect of EP in patients. In this open label phase I/II, two-arm, dose escalation trial DNA was delivered either by intramuscular injection or by intramuscular injection followed by EP (DNA+EP), with five patients per dose level. Three vaccinations were given at 0, 4, and 8 weeks,with booster doses at 24 and 48 weeks; here we allowed crossover between study arms if supported by the safety and immunological data. In the 20 patients in the first two dose cohorts we observed that beyond brief and acceptable pain at the injection site, EP did not appear to add toxicity to the vaccination. We evaluated humoral responses to DOM. Low anti-DOM IgG antibody responses were observed after intramuscular injection of DNA without EP (at week 12: mean 1.7- vs. 24.5-fold increase over baseline with DNA+EP). These could be boosted by delivery of DNA+EP at later time points. Delivery of DNA+EP at all five vaccinations yielded the highest levels of anti-DOM antibody. Responses persisted to 18 months of follow-up. These data establish EP as a potent method for stimulating humoral responses induced by DNA vaccination in humans.
Introduction
DNA vaccination is a promising strategy for the induction of immunity, especially because the flexibility in design allows incorporation of antigen in an optimal form together with a range of molecules aimed to activate selected immune pathways (Rice et al., 2008). However, excitement waned when potent effects in preclinical models failed to translate into clinical efficacy. A contributing factor turned out to be the volume of fluid injected, as reduction of injection volume to less than 50 μl in mouse muscle leads to loss of response (Dupuis et al., 2000; Buchan et al., 2005). Simple scaling for human use, however, is not feasible as it would require that a > 100-ml volume be injected. It has been reported that the potency of DNA vaccines could be improved by delivery via electrical pulses: in vivo electroporation (EP) (Aihara and Miyazaki, 1998; Mathiesen and Lømo, 1998; Mir et al., 1998). The electrical pulses increase permeability of cell membranes for water-soluble substances (Swartz et al., 2001; Rols, 2006) and enhance DNA uptake both in muscle and nonmuscle cells. This results in local inflammation; increased recruitment of antigen-presenting cells (APCs) to the site of DNA inoculation; and increased duration, concentration, and level of antigen expression. All of these contribute to enhanced immunogenicity of plasmid-based vaccines (Aihara and Miyazaki, 1998; Mir et al., 1998; Mathiesen, 1999; Gronevik et al., 2005; Ahlen et al., 2007; Liu et al., 2008). Electroporation also appears to be particularly effective in recruiting innate “danger” pathways through TANK-binding kinase-1 (TBK1)-dependent and type-1 interferon receptor-mediated signaling (Ishii et al., 2008). We and others have shown that DNA delivery by EP into muscle enhances both humoral and cellular immunological responses (Mathiesen and Lømo, 1998; Widera et al., 2000; Zucchelli et al., 2000; Buchan et al., 2005) and restores the lack of immunogenicity of suboptimal volumes of DNA (Dupuis et al., 2000; Buchan et al., 2005). Our data also suggested that priming with DNA by intramuscular injection followed by boosting with EP is more potent than delivery with EP on two occasions (Buchan et al., 2005).
Data in rodents were soon confirmed in rabbits, other small mammals (Hirao et al., 2008; Mathiesen and Lømo, 1998), and nonhuman primates (Otten et al., 2004, 2006; Luckay et al., 2007; Hirao et al., 2008). Plasmid DNA delivered by EP also has been successful in stimulating immunity in farmed ruminants (Tollefsen et al., 2003; Scheerlinck et al., 2004), supporting the notion that with this approach body size is not a limiting factor. EP therefore also holds promise for human application and a number of groups are in early-phase clinical testing using DNA delivered by electroporation.
We have developed an electroporation device that is applicable for clinical use and accurately delivers DNA at the site of electroporation (Tjelle et al., 2006). The device was initially tested on healthy volunteers in a small feasibility study at Ullevaal University Hospital (Oslo, Norway) (Kjeken et al., 2004). The safety analysis generated from this pilot study allowed us to take the approach into the clinic to treat HLA-A2+ patients with prostate cancer, whose disease showed evidence of biochemical failure by rising PSA after radical treatment.
For the DNA vaccine delivered here we have chosen to build on our preexisting preclinical and early clinical program of DNA fusion vaccines. These encode a strongly immunogenic helper domain (DOM), derived from fragment C of tetanus toxin (Rice et al., 2008), linked to an epitope sequence from the tumor antigen of choice. We have shown that this design overcomes tolerance, and induces high levels of tumor epitope-specific CD8+ T cells, able to suppress the growth of solid tumors in murine models (Rice et al., 2008). In our clinical study the vaccine target is prostate-specific membrane antigen (PSMA), and the vaccine encodes a 9-mer peptide (VLAGGFFLL) (Lu and Celis, 2002) from its transmembrane domain, PSMA27. We chose early recurrent prostate cancer as the clinical test bed, as patients have minimal disease load, good performance status in spite of incurable disease, and are likely to progress slowly over many months (Parker and Dearnaley, 1998; Soto et al., 2008). The aim of the study is to build a detailed picture of immunological and clinical effects over the 72 weeks each patient participates in the study. We report here the first data from this study. The use of electroporation in patients is safe, tolerable, and acceptable to vaccinees. The DOM–epitope fusion design allows assessment of induction of immunity against both the DOM protein and against the tumor-derived epitope. In this paper we provide details of the humoral responses against DOM, and the ability of EP to amplify this response in human subjects with cancer.
Materials and Methods
Trial design for vaccination study
The study is a phase I/II two-arm, open label, nonrandomized, two-center dose escalation study. Regulatory approval for use of the vaccine and device was obtained from the Medicines and Healthcare Products Regulatory Agency (London, UK), the national ethics committee responsible for the conduct of studies with genetic vaccines (Gene Therapy Advisory Committee [GTAC], London, UK), and the research ethics committee in both centers. The study was registered in the gene therapy trials database (UK-112;
Vaccine design and production
p.DOM-PSMA27 was constructed as previously published (Rice et al., 2001). Assembly primers (forward primer, AAGCTTGCCGCCACCATGGGTTGGAGCTGTATCATCTTC; reverse primer, AGCGGCCGCTTAGAGGAGAAAGAAGCCACCCGCCAGCACGTTACCCCAGAAGTCACGCAGGAA) were used to amplify DOM-PSMA27. The purified PCR product was cloned into pcDNA3, using HindIII and NotI. After sequencing the vector backbone and insert, the plasmid construct was produced in bulk, aliquoted, and filled in compliance with Good Manufacturing Practice at the Clinical Biotechnology Centre of the University of Bristol (Bristol, UK).
Electroporation equipment
Electroporation was carried out with an electroporation device consisting of a control unit and an injection/insertion unit (Tjelle et al., 2006). This uses two standard insulin syringes and 22-gauge needles 8 mm apart, with a maximal volume of delivery of 200 μl per syringe (i.e., 400 μl per injection). The device allows the injection to start at a defined depth of insertion (here 5 mm), and to further allow continuous injection of DNA for another 15 mm to ensure injection of the DNA solution into the muscle tissue. In the DNA vaccination study the needles were inserted to a depth of 2 cm in each patient. The needles also serve as electrodes and, after completion of the injection, a train of five pulses of 20 msec at 8.3 Hz, with a total duration of 0.5 sec, was delivered with a maximal current at 250 mA. The pulse generator was also set to limit the voltage to a maximum of 70 V. If the resistance in the muscle was above 280 Ω, less than 250 mA was delivered. Success of the delivery was confirmed clinically by observing three to five discrete muscle jerks during electroporation and by the automatic generation of an electronic file, which documents the flow of the current during vaccination.
Dose groups and vaccination and follow-up schedule
Vaccine was given either by intramuscular injection alone (arm I, DNA) or delivered intramuscularly by EP (arm II, DNA+EP) on three occasions at monthly intervals. Vaccines were given into alternate anterior thighs. Five patients were entered into the study per dose level and arm. The dose levels were 800, 1600, and 3200 μg/dose (arm I, DNA), or 400, 800, and 1600 μg by EP (arm II). Two further doses were given 24 and 48 weeks after the first dose. At week 24 continuation on the same arm or crossover to the other arm was allowed, depending on the immunological evidence gathered in the patients vaccinated to this point. Follow-up on study was at weeks 0, 2, and 4, monthly up to week 32, and every 2 months after that to week 72. After each vaccination visits were scheduled 2 and 4 weeks postinjection for clinical review and immunological sampling. For safety monitoring full blood counts, clotting, serum biochemistry, lactate dehydrogenase (LDH), creatine kinase (CK), and autoimmune profiles were monitored over time. After DNA+EP, LDH and CK were additionally measured on days +1 and +5 to assess biochemical evidence of muscle damage and recovery over time. An electrocardiogram (ECG) was produced before and after each vaccination with electroporation. Before dose escalation a safety review of study data was undertaken by the trial steering committee.
Subjective evaluation of patient reactions to vaccination
A visual analog scale (VAS) was used to capture the patients' experience of vaccination. The 10-cm scale was divided to allow assessment in 0.5-point increments (from 0 to 10, where 0 is “no pain” and 10 is “worst pain imaginable”). Using the VAS, patients were asked to grade their pain and discomfort at the time of vaccination, and 30 min and 48 hr postvaccination. Additional free text comments were invited. Pain was also assessed according to the National Cancer Institute (Bethesda, MD) Common Toxicity Criteria (CTC) scale version 3.0 (
Immunological monitoring
At each follow-up visit blood samples were taken and stored for immunological analysis. Antibodies against DOM were measured by enzyme-linked immunosorbent assay (ELISA), following formal validation of the assay (Mander et al., 2009). Baseline anti-DOM antibody values were additionally compared with values of antibodies against the parent protein fragment C (FrC) sequence, using a well-established assay that we had validated previously (Mander et al., 2009).
Generation of recombinant DOM protein
The DOM sequence, incorporating a 3′ histidine tag (6 × His), was cloned into the pcDNA3.1(+) vector (Invitrogen, Paisley, UK), using the HindIII and XbaI restriction enzyme sites. Plasmid DNA was then used to transfect suspension human embryonic kidney 293 cells (FreeStyle 293-F; Invitrogen) using 293fectin (Invitrogen) according to the manufacturer's instructions. Cultures were incubated at 37°C, 8% CO2, 125 rpm. After 72 hr the culture supernatants were harvested by centrifugation at 1700 rpm for 7 min and filtered over a 0.22-μm membrane under vacuum. The cells were placed back in culture for a further 72 hr and the supernatant was harvested again. Supernatants were concentrated 10 × by centrifugation at 2200 rpm at 4°C, using Vivaspin 20 (molecular weight cutoff, 10,000) concentrators (Sartorius, Epsom, UK). DOM protein was purified on a His · Bind column (Novagen, Madison, WI) in accordance with the manufacturer's instructions. Protein concentration was determined by bicinchoninic acid (BCA) protein assay (Perbio Science UK, Cramlington, UK) against known bovine serum albumin (BSA) standards. Purity of the recombinant protein was confirmed by gel electrophoresis and Western blot analysis (Amersham Biosciences UK, Little Chalfont, UK), with horseradish peroxidase (HRP)-conjugated anti-penta-His antibody (Qiagen, Crawley, UK) and using chemiluminescence plus reagents (Amersham Biosciences UK).
Anti-DOM ELISA
A 96-well Maxisorp immunoplate (Nunc, Roskilde, Denmark) was coated with DOM–His protein (2 μg/ml in coating buffer [4.9 mM Na2CO3, 34.74 mM NaHCO3; pH 9.5]) (200 μl/well) and incubated at 4°C overnight. Plates were washed four times with PBS–0.001% Tween 20 (PBS-T) and blocked for 1 hr with shaking at 37°C with PBS–1% BSA. After two washes with PBS-T, patient serum samples were added to the plate in triplicate, diluted in PBS-T (concentration used determined by serum titration). A reference standard of tetanus antitoxin human immunoglobulin (National Institute of Biological Standards and Control, Potters Bar, UK) was double-diluted to provide a standard curve to measure the relative levels of anti-DOM antibodies. To validate the standard in each assay quality controls were run on every plate, made from pooled human sera and diluted at high, medium, and low ranges on the standard curve. Patient samples were diluted so that the complete series of time points were within the range of the standard. This varied from 1:50 to 1:800 depending on the preexisting level of anti-DOM antibodies and the magnitude of response. The plates were incubated for 1 hr with shaking at 37°C. After washing, the plate was incubated with HRP-conjugated polyclonal goat anti-human Fc IgG antibody (Sigma-Aldrich, Poole, UK) at a 1:2000 dilution in PBS-T (200 μl/well) for 1 hr with shaking at 37°C. After washing, the plate was incubated with a 200-μl/well concentration of substrate buffer: one o-phenylenediamine tablet (Sigma-Aldrich) in 25 ml of substrate buffer (24.3 mM citric acid, 51.4 mM NaHPO4; pH 5.5) plus 25 μl of H2O2. The reaction was stopped by adding 80 μl of 2.5 M H2SO4 to each well. The absorbance at 490 nm was measured with an Ultramark 680 ELISA microplate reader (Bio-Rad, Hercules, CA). Relative anti-DOM antibodies were then measured by comparison with the tetanus antitoxin human immunoglobulin standard curve, adjusted for the dilution factor in the assay and fold increase over the prevaccination baseline calculated. A positive response was defined as a :2-fold increase over the prevaccination baseline.
Statistical analysis
Mean values are presented throughout with standard error of the mean. Significance was determined by Student t test. A p value less than 0.05 was considered statistically significant.
Results
Patient data
Recruitment of prostate cancer patients to the vaccination study began in April 2005 and is complete; follow-up is ongoing at the highest dose level. All patients met the entry criteria for the study. Initially, 30 HLA-A2+ individuals were recruited to the study; however, 2 patients progressed after receiving only two vaccinations and were replaced as per study protocol. One patient received three vaccinations before progression and was followed up to week 16 and was evaluable for immune responses according to protocol. Twenty-nine patients completed the 72-week study follow-up period. Patients' ages ranged from 58 to 84 years, with a median age of 71 years. Three patients (patients 2, 17, and 19) were receiving stable hormonal treatment before and during study participation; the other patients had not received any systemic treatment for prostate cancer. Safety data are reported for the first 20 patients only (dose levels I and II) as the full, audited clinical data set for the third dose cohort is not yet available. No additional safety concerns have been identified by the investigators at the third dose level. Data on the humoral response to the DOM component of the vaccine is reported for 30 patients.
Safety and tolerance to DNA vaccine
Overall, 148 vaccine doses were administered, with 51 by DNA alone and 97 by DNA+EP. No crossover was allowed between the two study arms before the fourth dose at week 24; in this period, therefore, 15 patients received DNA by intramuscular injection only, 15 by DNA+EP. All patients (15) who had started with EP delivery of the vaccine continued on in this way. On the basis of the early immunological data that was gathered as the study proceeded, 11 of 15 patients who had started on the DNA-alone arm received the booster doses by DNA+EP. Two patients received the fifth booster dose at week 48 by DNA+EP, and two were not electroporated.
Safety data are reported for the first two dose cohorts (20 patients). All electroporation events for which electrical data were recorded (29 of 64 delivered) showed successful delivery of five pulses (data not shown). All patients received the five scheduled vaccinations; 94% of doses were given according to protocol schedule. Delays in five booster doses at 24 and 48 weeks, outside of the permitted time window of ±7 days, were caused by the need to accommodate patients' vacations (2 weeks late, two doses; 3 weeks late, two doses; 4 weeks late, one dose; 2 weeks early, one dose).
Vaccination of prostate cancer patients by both delivery strategies was well tolerated. Pain reported by the patient was graded according to the National Cancer Institute CTC scale version 3.0. Observation of any pain was graded as ≥WHO grade 1. Two patients reported WHO grade 2 pain. The first, caused by bleeding into the thigh muscle at the injection site after delivery by DNA+EP, settled over the next fortnight. The second event was an exacerbation of preexisting backache, triggered by the muscle contractions after DNA+EP. This settled within 48 hr. No pain >WHO grade 2 was observed. Pain was additionally assessed by a questionnaire after each vaccination (DNA alone and DNA+EP). The Visual Analog Scale (VAS) was used to assess pain immediately after vaccination, 30 min postvaccination, and 48 hr postvaccination (Table 1). The VAS was more sensitive at capturing the difference between delivery strategies compared with the WHO grading. EP was more painful than giving the vaccine intramuscularly only. EP-related pain varied widely (Table 1). Most patients who provided free text comments reported pain as mild; three patients reported that they had perceived the pain during EP as severe. In all patients, pain subsided rapidly within minutes. Assessment of pain 30 min postvaccination showed that pain had resolved or diminished to just 1 point on the VAS in 88% of patients (Table 1). At 48 hr after any vaccination 97% reported no more pain in the previous 48 hr. No patients declined EP after already having received an EP treatment.
Subjective Evaluation of Pain after Vaccine Delivery
Abbreviation: EP, electroporation.
Biochemical measurements of CK, LDH, and creatinine are shown in Table 2 and Fig. 1. CK and LDH were evaluated as markers for muscle damage; serum creatinine values were monitored because renal tubules express low levels of PSMA (Silver et al., 1997). There was no significant difference between modalities of vaccine delivery (Fig. 1). The means of the baselines and of measurements taken throughout the vaccination study are comparable in both arms of the study and fall into the normal range. There are transient increases that are above the upper limit of norm (ULN) but <2.5 ULN (WHO grade 1); these return to normal levels by the next time point at day +2. A single observation at week 52 showed a CK level exceeding the WHO grade 1 cutoff of 2.5 × ULN = 642 IU/liter with a value of 685 IU/liter in a patient who had been vaccinated with DNA+EP at week 48. Creatine kinase levels were also evaluated on days 1 and 5 after vaccination with DNA+EP (Fig. 1). When an increase (compared with the day of vaccination) was measured the levels had returned back to normal levels by day 14 postvaccination. Measurements after DNA delivery by intramuscular injection without EP were not undertaken as no muscle toxicity was anticipated in this study, based on its absence in our previous clinical studies.
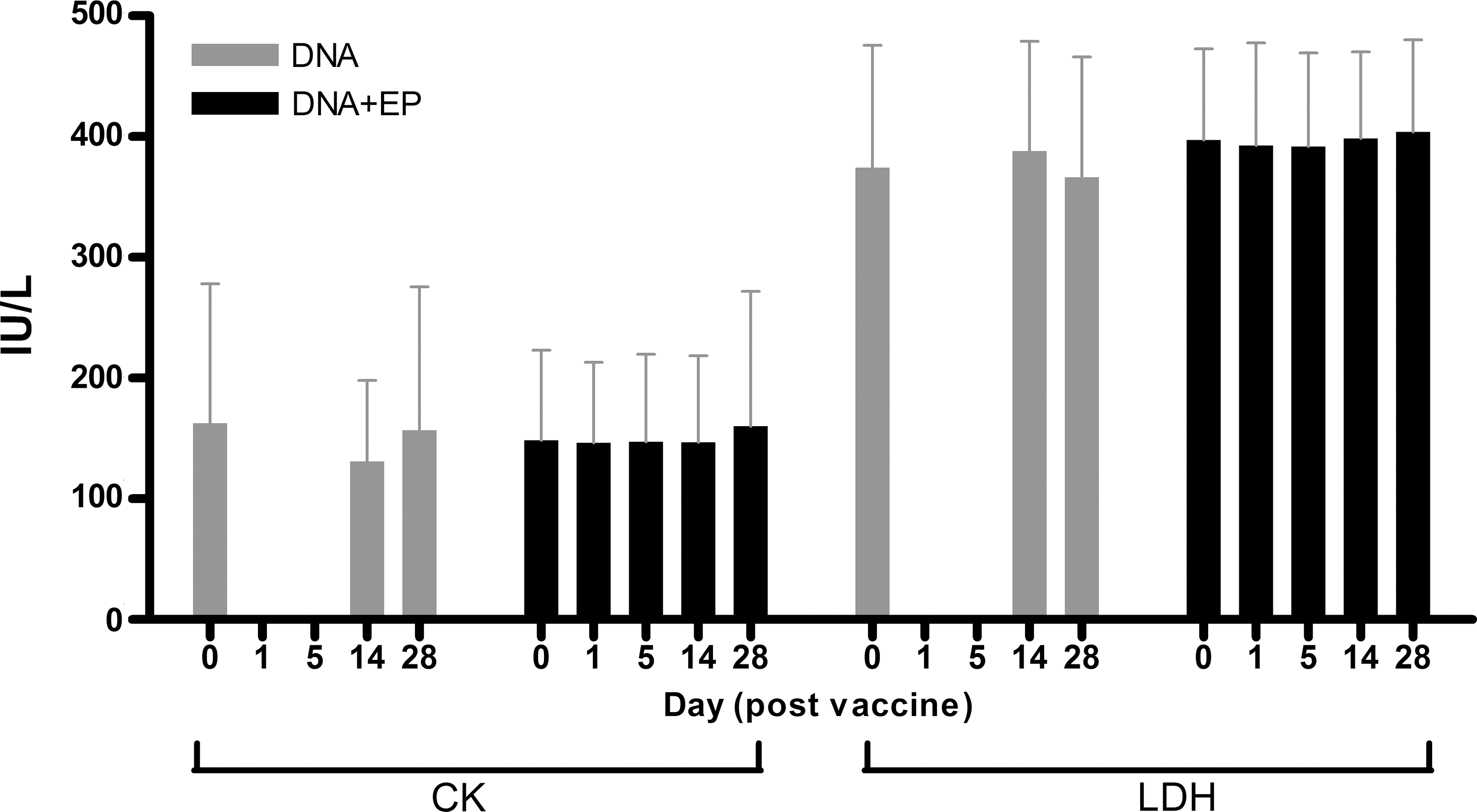
Comparison of CK and LDH levels between delivery modalities. Levels were measured on the day of vaccination and on days 1, 5, 14, and 28 postvaccination. Measurements on days 1 and 5 were undertaken only after delivery of DNA+EP. There was no significant difference between the levels measured after vaccination with DNA with or without EP. Values represent the mean value of CK or LDH (IU/liter) ± standard deviation, where n = 36 for DNA and n = 64 for DNA+EP.
Biochemical and Hematological Evaluation in Study Patients
Numbers of lymphocytes and neutrophils, and hemoglobin level, are shown in Table 2. The baselines and mean levels measured throughout the study in both arms of the study fell within the normal range. Individual measurements of lymphocytes below normal levels were from patients recruited to the study with a lower than normal lymphocyte count at baseline. Analyses for electrolytes (sodium, potassium, and calcium), liver function tests (albumin, alanine aminotransferase [ALT], alkaline phosphatase [ALP], bilirubin, γ-glutamyltranspeptidase [GGT]), red cell count, major histocompatibility complex (MHC), mean cell hemoglobin concentration (MCHC), autoimmune profile, rheumatoid factor, and anti-muscle and anti-DNA antibodies showed no significant changes over time in either the DNA-alone or DNA+EP arm of the study (data not shown). No changes were observed in the ECGs taken before and after each electroporation.
IgG antibody responses against DOM sequence
Thirty patients have been assessed for anti-DOM antibodies. The levels of anti-DOM at baseline were comparable and low for 28 patients, with a mean of 0.22 relative antibody units (RAU) (range, 0.01–1.38; Fig. 2). Two patients had a comparatively high level of anti-DOM antibody with 9.08 and 13.7 RAU, respectively, at baseline. Although we have found in the past that anti-FrC antibodies correlate closely with anti-tetanus toxoid antibodies in humans (Mander et al., 2009), there was no significant correlation between preexisting anti-FrC antibody levels and levels of anti-DOM antibodies in the patients analyzed here (data not shown). It appears therefore that DOM delivered by DNA is a new antigen for B cell recognition in the majority of patients, and the DOM component from tetanus toxoid appears to be poorly visible to B cells in patients under normal circumstances.

Baseline levels of anti-DOM and anti-FrC antibodies. Anti-DOM and anti-FrC levels of antibody were measured by ELISA in each patient at the baseline week of study before vaccination.
Twenty-one of 30 (70%) developed a :2-fold increase in IgG antibodies to DOM (summarized in Table 3). There was considerable variability in the antibody responses between patients (Fig. 3A and B). Of the 21 responders, 12 were vaccinated by DNA+EP every time, and 9 patients were vaccinated with DNA three times followed by two DNA+EP boosts. In the period from week 0 to week 24, when patients each had 3 monthly injections by DNA+EP (15 patients) or DNA alone (15 patients), the maximal mean increase in anti-DOM antibody responses at week 12 was 25-fold in patients treated with DNA+EP, compared with 1.5-fold in patients receiving DNA alone (Fig. 3A, DNA+EP vs. DNA, significant at p = 0.012). This increased to 45- and 1.9-fold for DNA+EP and DNA alone, respectively, when only responders were analyzed.

Comparison of anti-DOM antibodies in all patients grouped by modality of vaccine delivery. Values represent the mean fold increase in anti-DOM antibodies compared with baseline ±SEM. Arrows on the x axis show the timing of vaccine delivery. (
Overall Humoral Responses to DOM
Abbreviations: D, DNA; E, DNA+EP.
When plotting the antibody data across the whole time course of 72 weeks, DNA+EP again generated a higher level of response (Fig. 3B). Delivery of five DNA+EP vaccinations increased the level of antibody response compared with vaccination with DNA followed by two DNA+EP boosts (p = <0.0001) (Fig. 3B). The mean maximal fold increase in all patients who received five vaccines with DNA+EP was 55 (n = 15; range, <2- to 393-fold increase) and 20 in patients who received DNA alone on three occasions followed by two DNA+EP boosts (Fig. 3B) (n = 11; range, 3- to 74-fold increase). This increased to 70-fold (DNA+EP, n = 12) and 23-fold (DNA + two DNA+EP boosts, n = 9) when only the responders were analyzed. There was no definite effect of dose on anti-DOM responses (Table 2). The highest number of responders was observed in the first dose level; however, the numbers within each group are small and the allocation to the groups was not randomized.
Individual responses were also analyzed. In 10 of 21 responders anti-DOM responses exceeded the baseline by 2- to 20-fold (Fig. 4A and B). In 11 further patients the increase exceeded 20-fold; this was seen only after delivery of DNA+EP (5 doses DNA+EP, 6 patients; 3 doses DNA and 2 booster doses DNA+EP, 5 patients) (Fig. 4C and D). Electroporation appeared to be able to induce visible increases in anti-DOM antibody levels after each vaccination (Fig. 4B and D). This effect was particularly dramatic in patients PSMA04 and PSMA32 (Fig. 4D), who received DNA+EP on five occasions; however, it was also visible in patient PSMA01 after three doses of DNA alone (Fig. 4C). In this individual boosting with DNA+EP further increased the response at 24 and 48 weeks.

Individual patient data of anti-DOM IgG levels. A positive response is defined as a 2-fold increase over the baseline level. Arrows below the x axis show the timing and modality of vaccinations. (
The kinetics of the antibody responses at 24 and 48 weeks induced by DNA+EP, after three doses of DNA only, closely mirrored those observed in patients treated with DNA+EP on five occasions, but at mean values, which were more than 50% lower (Fig. 3B). At the end of the study, the level of antibodies measured at week 72 remained highest in those individuals receiving all five vaccinations with EP (Fig. 3B).
We identified nine nonresponders: three patients had received DNA+EP on five occasions, and two patients had three doses of DNA followed by two DNA+EP (Fig. 4E). Four nonresponders received DNA alone on four or five occasions (two patients, DDD/DD; two patients, DDD/DE).
Discussion
There has been a clear problem in translating the impressive performance of DNA vaccines in mice to larger animals and human subjects. For intramuscular injection, the volume of injected fluid appears to be a strong determinant of the response, likely because of changes in cellular uptake in muscle mediated by hydrostatic pressure (Dupuis et al., 2000). Electroporation can compensate for a low nonoptimal volume, apparently by increasing both the transfection level and the degree of inflammation (Ahlen et al., 2007), and by exploiting innate immune effector mechanisms (Ishii et al., 2008). Improved delivery is particularly important for the induction of antibody, where DNA vaccines have been found to be inadequate (Ulmer et al., 2006), making the approach less attractive for prophylactic vaccination, where antibody is the usual goal. Strategies to improve the performance of DNA vaccines have included intradermal injection via various devices (O'Hagan et al., 2006; Wang et al., 2008), with apparent induction of protective levels of antibody against influenza in human subjects being reported (Drape et al., 2006).
For the intramuscular route, EP has clearly improved antibody levels induced in large animals and nonhuman primates (Laddy et al., 2009). In human subjects EP-mediated delivery of plasmid DNA encoding interleukin (IL)-12 into melanoma lesions (Daud et al., 2008) has been reported, but the question of induction of antibody was not addressed. The next step to intramuscular delivery required a clinically acceptable device, which is now available (Tjelle et al., 2006). A pilot study in six volunteers supported that although transiently painful, human use of EP was feasible (Kjeken et al., 2004) and suggested that electroporation would be acceptable to patients also. These data prompted us to undertake a clinical phase I/II trial in patients with prostate cancer. We built on our preclinical and clinical experience with DNA fusion vaccines, encoding a sequence from tetanus toxin (DOM) and linked to a tumor antigen-derived epitope. For this study we used a vaccine directed against an epitope identified from PSMA, PSMA27 (Lu and Celis, 2002). Our goal is to induce CD8+ T cell responses capable of killing tumor cells. However, codelivery of the encoded DOM protein allowed investigation of the induction of antibody in the patients. Key end points were the safety and immunogenicity of the vaccine, delivered either by simple intramuscular injection or by injection followed by electroporation. We used a two-arm, nonrandomized approach with escalating DNA doses and follow-up at the highest dose level is ongoing in both arms of the study. We identified no safety concerns with either delivery strategy. This is not surprising for the DNA vaccine per se, as there is now a substantial body of evidence supporting the safety of DNA given by intramuscular injection (Rice et al., 2008; Wloch et al., 2008). We find that electroporation also is a feasible strategy in the clinic. Although clearly painful, the duration of pain postelectroporation is short and lasts only minutes. It also did not affect the willingness of patients to continue participation in the study and we were able to deliver DNA in both arms of the study on time and according to protocol without analgesia. No significant differences in biomarkers for muscle damage, kidney function, other biochemical tests, autoimmune profiles, or hematological results were seen with DNA or DNA+EP.
Evaluation of the immunological responses to DOM is presented here. We show significant evidence of improved humoral immune responses to DNA vaccination, when delivered by EP. Although DNA given intramuscularly but without EP induces some antibody responses, DNA+EP is considerably more effective. It also appears that, by the current schedule, multiple dosing with DNA+EP induces higher levels of antibody than the prime–boost approach with initial DNA delivery followed by DNA+EP, in contrast to what we had observed in mice.
The “gold standard” for antibody induction has to date has been protein-based vaccination or injection of attenuated or inactivated pathogen. We find that DNA+EP brings the levels of humoral responses into a range comparable to protein vaccination. Although further comparative study is required, fold increase after DNA+EP against the pathogen-derived DOM sequence reaches levels similar to responses with tetanus toxoid in healthy volunteers (Di Genova et al., 2006). The responses to DOM observed here take longer to develop (in the best responder 30-fold at week 4, 204-fold at week 8; Fig. 4D); this could be explained in part by low or absent preexisting levels of anti-DOM antibodies, suggesting that DOM may prime a new B cell response not previously triggered by tetanus toxoid vaccination.
The data support optimism for use of DNA+EP as a strategy for vaccination against infection and cancer. The current trial is aimed at inducing tumor-specific CD8+ T cell responses (against PSMA27) and evaluation of these responses is ongoing. Meanwhile, measurement of antibody responses against the DOM component provides data on the performance of the DNA fusion vaccine in raising antibody, and demonstrates the amplifying effect of EP. Physical strategies to improve delivery of DNA are attractive and avoid the involvement of viral vectors, which face the problem of preexisting antivector immunity. Electroporation is acceptable by patients with cancer and, as devices improve further, may be considered for prophylactic vaccination where antibody is the goal, but where coinduction of T cell responses could be a useful addition.
Footnotes
Acknowledgments
The authors thank Dr. Catherine Heath for allowing the recruitment of patients from her clinics, and Dr. Paul Kerr and the research nurses Mr. James Dobbyn and Mrs. Bernadette Johnson for helping with the running of the study. Dr. Rune Kjeken from Inovio provided support in the regulatory submission for the device and provided training to the study centers in its use. Mrs. Julie Gwilt provided critical support with the administration and regulatory aspects of the trial. Dr. Paul Lloyd-Evans and Professor David Anstee produced the clinical-grade vaccine at the Clinical Biotechnology Centre (Bristol, UK). The project was supported by funding from Cancer Research UK, Inovio, the Allan Willett Foundation, and the Southern Prostate Cancer Collaborative.
Author Disclosure Statement
L. Low, A. Mander, K. McCann, and D. Dearnaley have no conflict of interest to declare. F.K. Stevenson and C.H. Ottensmeier are shareholders in Genvax, which have licensed the IP to the DOM epitope vaccine design. C. Ottensmeier has received an unrestricted research grant from Inovio. T. Tjelle and I. Mathiesen are employees of Inovio, and hold the rights to the electroporation equipment used for this study.