
Abstract
Type 1 diabetes (T1D) in both humans and nonobese diabetic (NOD) mice is a T cell-mediated autoimmune disease characterized by lymphocytic infiltration of pancreatic islets with subsequent destruction of the insulin-producing cells. The T regulatory (Treg) cell has been suggested to play an important role in controlling T cell-mediated inflammatory T1D. We previously demonstrated that induction of antigen-specific Treg cells in vivo by coimmunization with a DNA vaccine and its encoded protein can effectively inhibit T cell-mediated inflammatory diseases. To further demonstrate the potential of this strategy, we show here that coimmunization of NOD mice twice with DNA encoding proinsulin plus insulin protein prevents the onset of T1D and induces the impairment of antigen-specific T cell responses in a dose-dependent manner. We further show that the inhibitory function is due to the induction of TGF-β-producing CD4+CD25− islet-specific iTreg cells against the onset of T1D in NOD mice. Induced iTreg cells were observed only in the coimmunization group, but derived neither from the DNA vaccine nor the protein alone, suggesting that a biased helper T cell type 1 response plays no inhibitory role. A strategy based on coimmunization to induce a protective response against the onset of diabetes in NOD mice may lead to the development of an immunotherapeutic/preventive protocol against T1D in humans.
Introduction
Although we reported that induced Treg cells were able to inhibit antigen-specific T cell function in vivo following coimmunization with a DNA vaccine and its encoded protein antigen (Jin et al., 2005, 2008), autologous antigens for the coimmunization, particularly those that can cause autoimmune reactions, have not been tested. In this paper, we demonstrate that the induction of antigen-specific iTreg cells by coimmunization with recombinant insulin protein and a DNA vaccine encoding proinsulin leads to prevention of diabetes in NOD mice. We also show that such prevention is due, at least in part, to the suppressive effects of transforming growth factor (TGF)-β produced by iTreg cells.
Materials and Methods
Mice
Adult female BALB/c and NOD mice (6–8 weeks of age) were from the Animal Institute of the Chinese Medical Academy (Beijing, China). All mice were maintained under specific pathogen-free conditions with 12 hr:12 hr light:dark cycle.
Reagents
Human insulin protein and ovalbumin (OVA) were purchased from Sigma-Aldrich (St. Louis, MO). On the basis of the human proinsulin cDNA sequence (GenBank no. NM_000207), a pair of primers was designed and used to amplify its 750-bp full-length nucleic acid. The primers were as follows: primer 1, 5′-ATGGCCCTGTTGGTGCACTTCCTAC-3′; primer 2, 5′-TTAGTTGCAGTAGTTCTCCAGCTGG-3′. Total RNA was isolated from pancreatic tissues of a mature female mouse and purified with TRIzol reagent (Promega, Madison, WI). Amplification of the cDNA fragment was performed by reverse transcription-polymerase chain reaction (RT-PCR) according to the instructions of the manufacturer (TaKaRa RNA PCR kit; TaKaRa, Dalian, China). The purified PCR product was ligated into the pMD18-T vector (TaKaRa) after detection by electrophoresis in a 1.0% agarose gel (Promega). Recombinant plasmid was identified by restriction digestions and verified by sequencing analysis. The insert was subcloned into pVAX1 vector (Invitrogen, Carlsbad, CA) and designated pVAX-PI. Its expression was analyzed by RT-PCR with the previously described primers after 48 hr of transfection into HeLa cells.
Immunization
Female BALB/c mice (6–8 weeks old) and female NOD mice (6 weeks old) were immunized, according to various regimens, via the tibialis anterior muscle on day 0 and boosted on day 14. These regimens were as follows: insulin protein at 100 μg/mouse, pVAX-PI at 100 μg/mouse, coimmunization of 100 μg of insulin protein and 50 μg of pVAX-PI DNA vaccine, pcD3-VP1 at 100 μg/mouse, and pVAX at 100 μg/mouse. All substances were dissolved in saline at 1 mg/ml.
Antibody treatment
Monoclonal antibodies (mAbs) against mouse CD25 (TIB-222; American Type Culture Collection [ATCC], Manassas, VA) and foot-and-mouth disease virus (FMDV) VP1 were purified from hybridomas in our laboratory (Li, 2008). TGF-β antibody was obtained from Signalway Antibody (Pearland, TX).
T cell-proliferative response
Single-lymphocyte suspensions were obtained from spleens of immunized BALB/c mice on day 7 after the second immunization. Cells in RPMI 1640 medium (GIBCO, Eggenstein, Germany)–10% fetal bovine serum (FBS) were used to determine the T cell-proliferative response by carboxyfluorescein succinimidyl ester (CFSE) staining after insulin stimulation in vitro for 48 hr. CSFE-stained cells were analyzed with a FACSCalibur equipped with CellQuest Pro software (BD Biosciences, San Jose, CA) according to a previously described protocol (Jin et al., 2008).
Measurement of insulin protein-specific antibodies
The level of anti-insulin serum IgG antibodies was determined by enzyme-linked immunosorbent assay (ELISA) in 96-well plates coated with insulin protein at 5 μg/ml, followed by detection with a secondary goat anti-mouse IgG antibody conjugated with horseradish peroxidase (Bio-Rad, Hercules, CA). Absorbance at 450 nm was measured with an ELISA plate reader (Magellan; Tecan Austria, Grödig, Austria).
Isolation of CD4+CD25− T cells
Single-splenocyte suspensions were prepared from mouse spleen and T cell subtypes (CD4+CD25− or CD4+CD25+) were isolated and purified with a MagCellect mouse CD4+CD25+ T cell isolation kit according to the protocol of the manufacturer (R&D Systems, Minneapolis, MN). Briefly, CD4+ T cells were first isolated by negative selection and CD4+CD25+ and CD4+CD25− T cells were further separated by positive selection for CD25, using immunomagnetic beads and a magnetic separation rack (New England BioLabs, Ipswich, MA). The resulting CD4+, CD4+CD25+, and CD4+CD25− T cells were more than 90% pure as determined by flow cytometry (FACSCalibur; BD Biosciences).
RT-PCR
RNA of CD4+CD25− T cells was isolated from the spleens of NOD mice 7 days after the second immunization and from age-matched control NOD mice, using TRIzol reagent (Promega). RT-PCR was performed with hypoxanthine phosphoribosyltransferase (HPRT) and TGF-β primers according to the manufacturer's instructions (TaKaRa RNA PCR kit). PCR products on 2% agarose gels were visualized by ethidium bromide (EtBr) staining, and densities were analyzed by means of computer software (Alphaimager 2200; Alpha Innotech, San Leandro, CA). The mRNA level represented by each of the PCR samples was compared and plotted by comparing the intensity of the sample band to that of the HPRT band.
Inhibition of Treg cell function assay
Cells were cultured in complete RPMI 1640 medium (GIBCO) supplemented with 10% FCS. CFSE (5.0 μM; Invitrogen Molecular Probes, Eugene, OR)-labeled T lymphocytes at 5 × 104 cells per well, isolated from the spleens of 6-week-old NOD mice and used as the responder cells, were mixed with bone marrow-derived dendritic cells (DCs) of C57BL/6 mice as stimulators after treatment with mitomycin C (50 μg/ml; Sigma-Aldrich) at 10:1 in a flat-bottomed 96-well microtiter plate for 24 hr. The mixed lymphocyte reaction (MLR) was further added with CD4+CD25− T cells from spleens at 5 × 104 cells per well from the coimmunized NOD mice (1 × 105 cells per well; total volume of 200 μl/well) for coculture in a 5% CO2 incubator at 37°C for 48 hr. CFSE-labeled CD4+ T cell proliferation was analyzed by FACS. Results are expressed as the percent inhibition, calculated as follows: percent inhibition = [1 − number of (CD4+ T cells + CD4+CD25− T cells)/number of CD4+ T cells alone] × 100.
Flow cytometry
T cells were isolated and stained with the appropriate phycoerythrin (PE)- or fluorescein isothiocyanate (FITC)-conjugated mAbs in the presence of phosphate-buffered saline (PBS) for 30 min at 4°C. For intracellular staining, T cells were stimulated with insulin protein (10 μg/ml; Sigma-Aldrich) for 8 hr and subsequently treated with monensin (100 μg/ml; Sigma-Aldrich) for 2 hr in vitro. The cells were blocked with Fc Block (BD Biosciences) in PBS for 30 min at 4°C before being fixed with 4% paraformaldehyde and permeabilized with saponin (Sigma-Aldrich). The splenocytes were intracellularly stained with the appropriate concentrations of PE-labeled antibodies including anti-Foxp3 or CD25 antibody (eBioscience, San Diego, CA) and FITC-labeled anti-CD4 antibody (eBioscience) for 30 min at 4°C. The cells were washed and analyzed with a FACSCalibur equipped with CellQuest Pro software (BD Biosciences).
Histopathology
After immunization, the pancreata of 25-week-old NOD mice were fixed in formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) or with anti-insulin and anti-glucagon antibodies (Boster Biotech, Wuhan, China). Sections were analyzed under a light microscope to determine histological changes.
Diabetes monitoring
NOD mice were monitored once per week to evaluate glycemia, using a blood glucose meter (Yicheng Biostrips; Yicheng, Beijing, China). Animals were considered diabetic when the blood glucose (BG) level exceeded 300 mg/dl. All mice presenting three consecutive positive hyperglycemia tests were culled.
Statistics
The Student t test was used for data analysis. Differences were considered to be statistically significant at p < 0.05.
Results
T cell responses are impaired by coimmunization with DNA and protein vaccines
We previously demonstrated that coimmunization with DNA vaccine and its cognate protein antigen can induce antigen-specific iTreg cells. To test whether this holds true for coimmunization with a DNA vaccine encoding proinsulin and insulin protein (designated pVAX-PI+Ins), we examined the proliferative responses of T cells from BALB/c mice 7 days after coimmunization twice at a biweekly interval. The T cell-proliferative response was profoundly inhibited in the pVAX-PI+Ins-immunized group compared with other groups (Fig. 1A), suggesting that coimmunization of an autoantigen, insulin in this case, could impair the antigen-specific T cell-proliferative response.

Effects of coimmunization on T cell proliferation and serum IgG. BALB/c mice (three per group) were immunized intramuscularly with 100 μg of insulin protein and/or 100 μg of plasmid pVAX-PI and boosted 14 days later. (
To determine whether the use of a DNA construct encoding proinsulin must be matched with the insulin protein used in the coimmunizing regimens, several mismatched coimmunizations were used as follows. Mice were coimmunized either with VP1 protein derived from foot-and-mouth disease virus plus pVAX-PI (pVAX-PI+VP1), or with pVAX empty vector plus insulin protein (V+Ins). None of the combinations was able to inhibit insulin-specific T cell proliferation (Fig. 1B). This indicates that antigens used in the coimmunizing regimens must contain an overlapping sequence.
To optimize the dosing of each regimen, pVAX-PI at various doses (25, 50, 100, 200, and 400 μg) was coimmunized with 100 μg of insulin, respectively. A maximal inhibition of T cell proliferation was observed with pVAX-PI at 50 μg and insulin at 100 μg (Fig. 1C). This dose was then used throughout the rest of the study.
However, the level of anti-insulin IgG antibodies in these immunized animals was not directly affected by the coimmunization (Fig. 1D). Results indicated that inhibition directly affects T cell proliferation but not antibody production.
Coimmunization induces a protective response against T1D in NOD mice
Having demonstrated that coimmunization of autoantigen can inhibit T cell proliferation, we next tested whether the protocol could be used to prevent the development of T1D in NOD mice. Female NOD mice were twice coimmunized intramuscularly with various regimens when they were 6 and 8 weeks old. Blood glucose levels of the mice were monitored every week and marked as the onset of diabetes once the level reached 300 mg/dl for two consecutive weeks. As expected, the NOD control mice started to show elevated levels of blood glucose at about 10 weeks of age, and the incidence of diabetes increased to more than 65% by 24 weeks of age; the same was true of the mice vaccinated with pVAX-PI or insulin. Interestingly, mice in the coimmunized group (pVAX-PI+Ins) did not show diabetes until they were 15 weeks old, at which time only 1 of 16 NOD mice raised its BG level and became T1D (Fig. 2A). This result not only demonstrates that coimmunization with a proinsulin DNA and insulin protein vaccine induces a protective response against T1D in NOD mice, but also suggests that the protection is not likely due to the induction of a biased Th1 response relative to the Th2 response, because pVAX-PI alone did not induce the protection.
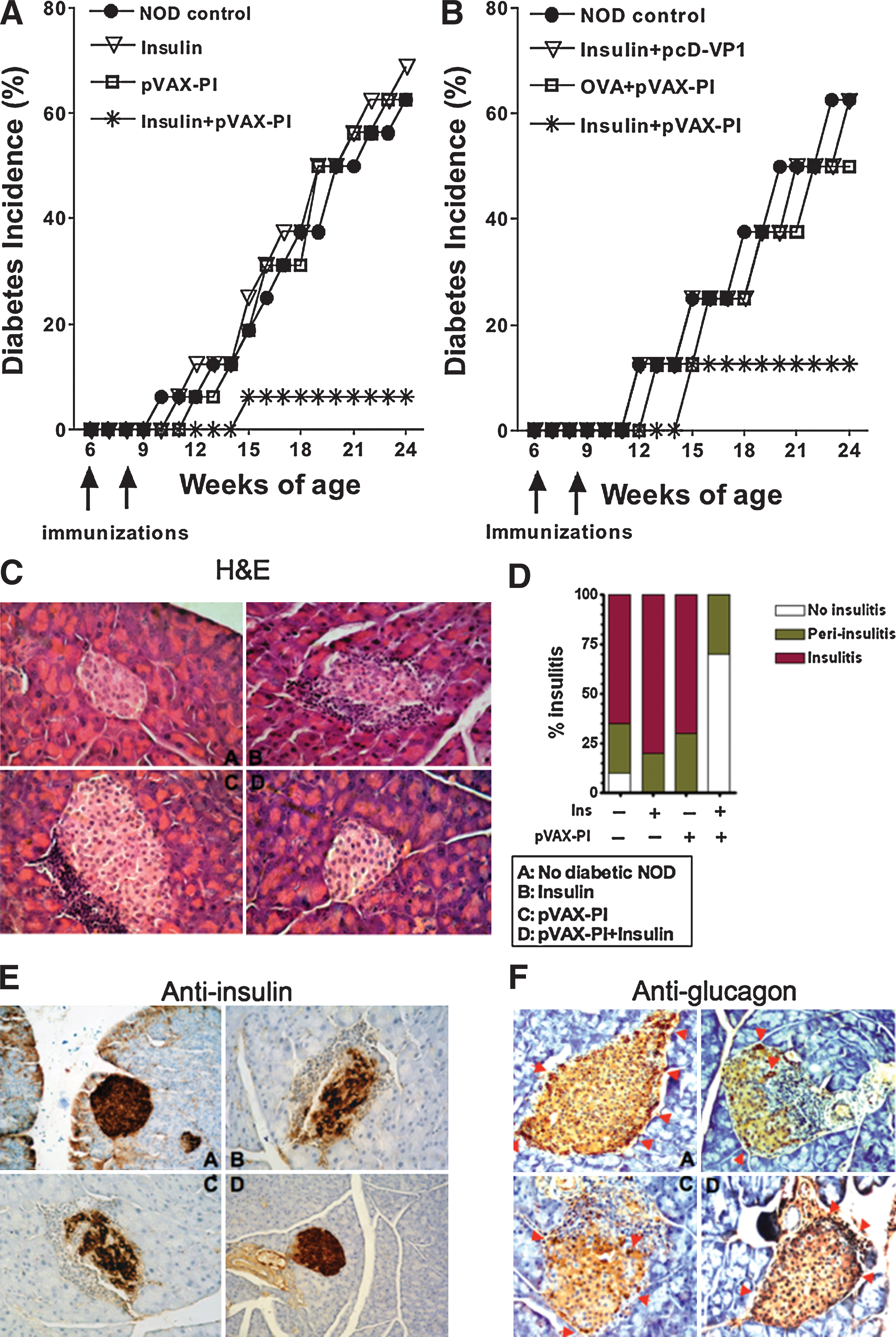
Diabetes is prevented by coimmunization. (
To confirm that the coimmunization regimen must be matched in sequence as observed for the T cell impairment described previously, pVAX-PI mixed with OVA (mismatched protein antigens), or insulin protein mixed with a DNA construct encoding VP1 (mismatched gene), was used to immunize NOD mice according to the same vaccination protocol. No similar protections were induced (Fig. 2B). This result indicates that the matched antigen sequences must play an important role to induce the protective response, and further supports the notion that the Th1 versus Th2 response did not play a protective role in our study.
Insulitis is a key indicator of beta cell destruction resulting from inflammatory T cell infiltration. We evaluated the severity of insulitis in NOD mice by categorizing the residual islets as insulitis, periinsulitis, and no insulitis on the basis of lymphocyte infiltration. After coimmunizations pancreatic tissues were removed from 25-week-old NOD mice and microsectioned. NOD mice, less than 8 weeks old and free of insulitis, served as negative controls. As shown in Fig. 2C and D, severity scores ranging from mild periinsulitis to no insulitis were observed in the group coimmunized with pVAX-PI+Ins, whereas NOD mice immunized with pVAX-PI alone or with insulin alone exhibited severe insulitis and periinsulitis to a similar degree as observed in NOD mice of the same age.
Furthermore, we examined the expression of insulin and glucagon in islets by sectioning the pancreatic tissue of NOD mice and staining the sections with anti-insulin and anti-glucagon antibodies. Normal expression of insulin and glucagon was observed in the pVAX-PI+Ins group, but the number of insulin- or glucagon-expressing cells in pancreatic tissue was dramatically reduced in the pVAX-PI-alone and insulin-alone groups as compared with the control NOD mice (Fig. 2E and F). These data suggest that coimmunization with pVAX-PI+Ins vaccines reduces the filtration of autoreactive T cells into the islets of NOD mice.
Taken together, coimmunization with pVAX-PI+Ins vaccines induces protective responses against T1D in NOD mice.
Protection against T1D in NOD mice is dependent on induction of CD4+CD25− Treg cells
Naturally occurring CD4+ Treg (nTreg) cells that express the interleukin-2 receptor α (IL-2Rα) chain (CD4+CD25+) are well characterized and play a central role in dampening peripheral self-reactive T cell responses and maintain tolerance in several models of autoimmunity (Shevach, 2000; Singh et al., 2001). It would be interesting to explore the possibility that CD4+CD25+ nTreg cells are induced by coimmunization to play their observed protective role. We first analyzed the frequency of CD4+CD25+ T cells by FACS before and after coimmunization. We observed that the frequency of CD4+CD25+ T cells did not change significantly after coimmunization (Fig. 3A), although the expression of Foxp3 was higher in the coimmunized group (Fig. 3B). We further confirmed this by neutralizing the function of CD4+CD25+ T cells via intravenous injection of anti-CD25 mAb into these coimmunized mice, and used anti-VP1 mAb as a control. The anti-CD25 mAb blockade resulted in the progression of one additional mouse (of eight in the coimmunization group) into diabetic status (Fig. 3C), indicating that CD4+CD25+ nTreg cells play a minor role in the observed inhibition of diabetic progression. Together, these results suggest that iTreg cells playing the inhibitory role belong to the CD4+CD25− subtype.
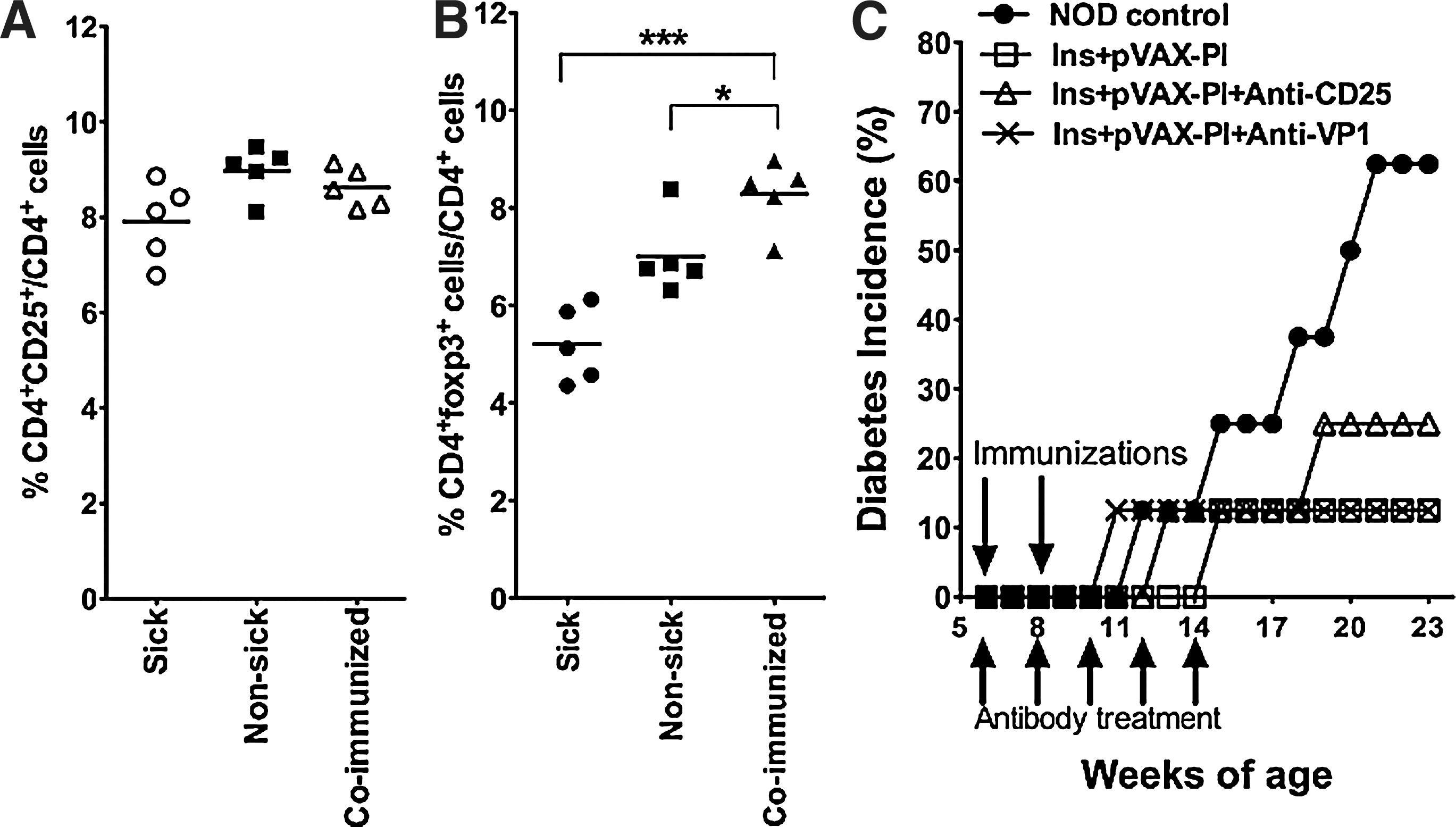
Induction of CD4+CD25− iTreg cells is responsible for prevention. CD4+ T cells were double-stained with anti-CD4 and anti-CD25 (
TGF-β plays a suppressive role against T1D
As TGF-β is an antiinflammatory factor and implicated as one of the mediators of iTreg cell suppression described previously (Fu et al., 2004), we examined the correlation of the regulatory function of CD4+CD25− iTreg cells and expression of the TGF-β gene by RT-PCR analysis in CD4+CD25− iTreg cells derived from coimmunized and control NOD mice. The results show that higher level expression of TGF-β was observed in CD4+CD25− iTreg cells from coimmunized mice compared with nonsick NOD mice and sick NOD mice (Fig. 4A and B). To further demonstrate the importance of TGF-β, CD4+CD25− iTreg cells were purified after coimmunization and used to inhibit T cell proliferation in a MLR. The inhibition of MLR was exhibited and partially blocked by the addition of an anti-TGF-β antibody at a low dose (10 μg/ml) and more effectively at a higher dose (50 μg/ml; Fig. 4C). This result suggests that TGF-β produced by CD4+CD25− iTreg cells plays a suppressive function, at least in part, against the onset of T1D in NOD mice.
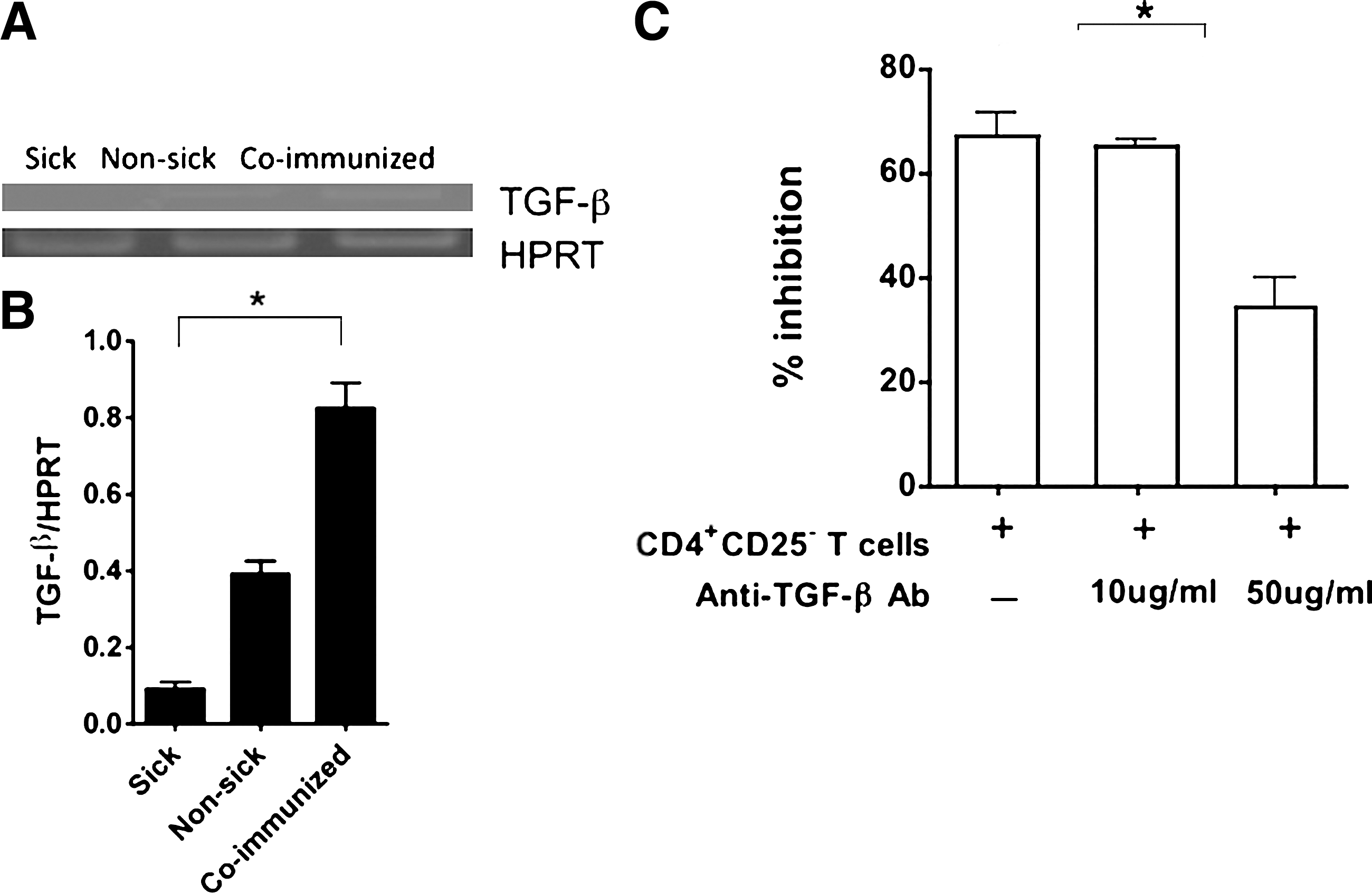
The suppression of CD4+CD25− Treg cells is transforming growth factor (TGF)-β dependent. (
Discussion
In type 1 diabetes (T1D), insulin is one of the key factors that provoke pathogenic T cells. On being provoked, the pathogenic T cells attack and destroy insulin-secreting beta cells in the pancreatic islets of Langerhans, leading to elevated glucose levels and the onset of diabetes (Vialettes et al., 1982). Daily insulin injection, although troublesome, is currently used to control the disease. Development of an alternative therapeutic strategy would be advantageous and beneficial to patients. Antigen-specific Treg cells are considered to be an attractive and potent means to control this type of disease (Green et al., 2003; Bluestone et al., 2008). Practical induction of antigen-specific Treg cells in the periphery has limited this strategy to clinical trials (Jaeckel et al., 2008). Although we discovered the induction of antigen-specific iTreg cells by means of our coimmunization protocol (Jin et al., 2005, 2008), autologous antigens for the coimmunization, particularly those that can cause autoimmune reaction such as the insulin involved in the diabetes, have not been tested. Thus, we look a step further to explore whether coimmunization with insulin protein and a proinsulin DNA vaccine can induce antigen-specific iTreg cells, and more importantly, whether these iTreg cells are able to suppress inflammation caused by pathogenic T cells in an antigen-specific fashion, which could ultimately prevent diabetes in the NOD mouse model.
In this study, we have demonstrated that simultaneous coimmunization of NOD mice with DNA encoding proinsulin plus insulin protein induces a preventive response against the onset of T1D (Fig. 2A). Disease onset is characterized by the profound elevation of the level of blood glucose (>300 mg/dl) for more than two consecutive weeks in correlation with massive lymphocyte infiltration, and destruction of the expression of insulin in beta cells (Fig. 2E) and glucagon in alpha cells (Fig. 2F). The vaccinated mice remained disease-free over 30 weeks until the end of this study, after being coimmunized twice (during weeks 6 and 8), suggesting a significant and sustainable protection was induced by the coimmunization. This prevention may also be due to the successful induction of insulin-specific CD4+CD25− Treg cells, because the mismatched regimens (i.e., insulin+pcD-VP1, and OVA+pVAX-PI) failed to suppress the progression of T1D in NOD mice (Fig. 2B). Although CD4+CD25+ nTreg cells can suppress T1D progression, its suppression has been demonstrated to be a global effect rather than antigen specific (Pop et al., 2007). The blockade of anti-CD25 antibodies in vivo allowed the progression of one more animal (for a total of two of eight coimmunized NOD mice) to diabetic status (Fig. 3C), suggesting a minor protective role. Furthermore, CD4+CD25+ nTreg cells decrease over time, which is concurrent with the evolution of insulitis from periinsulitis, to the clinical onset of diabetes (Besin et al., 2008). The loss of CD4+CD25+ nTreg cell activity is part of the reason for disease progression in autoimmune disease-prone individuals.
TGF-β has been shown to play a pivotal suppressive role in Treg cell function (Prud'homme et al., 2007). Our results show that TGF-β plays a suppressive function in vivo because anti-TGF-β antibodies effectively abrogate its suppressive function on induced CD4+CD25− iTreg cells (Fig. 4C). This is supported by several previous reports that only CD25− T cells from NOD mice, but not those from normal mice, are TGF-β dependent (Fu et al., 2004). Therefore, TGF-β-dependent CD4+CD25− Treg cells may be preferentially developed in autoimmune disease-resistant animals.
Although the use of DNA vaccine encoding proinsulin alone has been shown to prevent diabetes in NOD mice in some previous publications, this type of approach has created contradictory results (Tisch et al., 2001; Every et al., 2006). Its protection against diabetes was accompanied by a Th2-biased response, and more often activation of Th2 cells contributes to pathogenic progression of autoimmune diseases and allergies (Prud'homme et al., 2007). A strategy to use mucosal delivery of proinsulin DNA has been shown to induce Treg cells that prevent NOD mice from developing diabetes (Every et al., 2006). The nasal route for DNA delivery has been shown to achieve effects only with difficulty (McCluskie and Davis, 1998). One of the other approaches, similar to ours, showed that proinsulin DNA vaccine administered with another DNA vaccine encoding the cytotoxic T lymphocyte antigen (CTLA)-4 ligand can trigger the negative effects of T cell activation and protect NOD mice from the onset of diabetes in (Glinka et al., 2006). However, the induction of Treg cells in that context was not determined.
We previously demonstrated that induction of CD4+CD25− Treg cells is due to induction of tolerogenic dendritic cells with low expression of CD40 and high expression of IL-10 (Jin et al., 2008). The mechanism of the protective effect of our coimmunization strategy on type 1 diabetes is still unknown. Future studies should determine whether the Treg cells suppress islet-specific CD8+ T cells directly through TGF-β–TGF-β receptor interaction.
In conclusion, we have demonstrated in this study that coimmunization with insulin protein and proinsulin DNA vaccine can induce antigen-specific iTreg cells, and that the induction of iTreg cells effectively prevents the onset of type 1 diabetes in NOD mice. This approach may lead to the development of an immunotherapeutic/preventive protocol in humans against T1D, and more importantly, may provide a novel approach to induce peripheral tolerance in an antigen-specific manner against other autoimmune diseases.
Footnotes
Acknowledgments
This work was supported in part by SKLAB funding (2008SKLAB05-02) and by the Chinese National Natural Science Foundation (30930068) (B.W.). The authors are grateful to Dr. Shijun Zheng for valuable suggestions and a critical review of this manuscript; the authors also thank Dr. Jane Q.L. Yu and Mr. Zhonghuai He for assistance in this work.
Author Disclosure Statement
No conflicts of interest are claimed.