
Abstract
We investigated a gene vaccine strategy against human papillomavirus (HPV)-induced cancer and premalignant diseases, using adeno-associated virus (AAV) vector encoding the viral E7 oncoproteins as the tumor antigens from HPV serotypes 16 (HPV16) and 18 (HPV18). Genetically inactivated E7 proteins were fused with a heat shock protein 70 (hsp70) to minimize the risk of cell transformation and enhance immune responses. The fusion protein gene was packaged in AAV serotype 1 or 2 (AAV1 or 2) for efficient in vivo gene expression. Our results showed that after a single intramuscular injection, the AAV1 vector elicited stronger HPV-specific cytotoxic T lymphocyte (CTL) responses and interferon-γ secretion when compared with the AAV2 vector. Prophylactic immunization with AAV1 protected 100% of the mice from tumor growth for more than 1 year, whereas all the control mice immunized with either a LacZ vector or saline grew large tumors and died within 6 weeks after inoculation of E7-positive tumor cell line TC-1. In addition, this single-dose AAV1 vaccination completely protected the mice against second and third challenges with higher numbers of TC-1 cells. Despite lower CTL responses against the E7 antigens, AAV2 vector prophylactic immunization was also sufficient to protect 100% of the mice against the initial and second tumor challenges and 70% of the mice against the third challenge. In addition, therapeutic immunization with AAV1 after palpable tumor formation inhibited tumor growth and caused tumor regression in some mice. Thus, our studies support the potential of AAV vectors as a genetic vaccine for the prevention and treatment of HPV-induced malignancies.
Introduction
Recombinant adeno-associated virus (AAV) is a 4.7-kb, single-stranded DNA viral vector. Unlike other viral vectors presently available, AAV is nonpathogenic and has minimal vector-related toxicity (Muzyczka, 1992). It also exhibits a broad host range of infectivity and can deliver the genes into a variety of target tissues for vaccination (Xin et al., 2003; Ponnazhagan, 2004; Johnson et al., 2005; Mehendale et al., 2008). Strong cellular immune responses against transgene products could be induced, depending on the nature of protein and the route of gene delivery and choice of vectors (Brockstedt et al., 1999; Liu et al., 2000; Xin et al., 2002; Wang et al., 2005, 2007).
Our previous study has suggested that immunization with an AAV serotype 2 (AAV2) vector encoding an HPV16 E7 (aa 49–57) peptide RAHYNIVTF linked to the Mycobacterium tuberculosis heat shock protein 70 (hsp70) was effective in preventing the growth of E7-expressing tumor cells, TC-1 cells, in mice (Liu et al., 2000). However, there are two major limitations that prevent this study from being translated into clinical application. First, in clinical setting, the use of a major histocompatibility complex (MHC)-restricted epitope as a vaccine is limited to a small population of the patients who have the appropriate human leukocyte antigen (HLA) haplotype. So far, only several HLA-A*0201–restricted epitopes of E7 have been identified and shown to initiate T-cell–mediated antitumor immune response in vitro and in vivo (Feltkamp et al., 1993; Ressing et al., 1995, 2000; Rudolf et al., 2001). Second, the specific HPV16 E7 peptide used in that particular study, RAHYNIVTF, is an H-2b–restricted CTL epitope, which is not applicable to human clinical trials. To circumvent these limitations, we chose the entire E7 proteins as the antigen, instead of a single H-2b–restricted E7 peptide which was used in previous studies (Liu et al., 2000). Further, we chose to simultaneously use both HPV16 and HPV18 E7 proteins fused with hsp70 as a single large fusion protein (1618E7hsp70), to potentially cover the majority (70–85%) of cervical cancer patients. Both E7 proteins have been genetically inactivated by abolishing the Rb-binding sites to eliminate the risk of oncogenesis (Boursnell et al., 1996). Finally, we investigated the robust and long-term protective effect of 1618E7hsp70 fusion protein delivered by AAV1 vector, which has been reported to be more robust to deliver the gene in muscle (Xiao et al., 1996, 1999).
Here we demonstrated that a single injection of AAV vectors encoding the 1618E7hsp70 gene could efficiently initiate a robust, specific, and persistent immune response against the E7-expressing tumor cells. We also showed that both the prophylactic and therapeutic efficacy of the AAV-1618E7hsp70 were highly significant in mice challenged with the TC-1 cells. Compared with AAV2 vector, AAV1 showed a superior capability in eliciting the immune response against the repetitive tumor challenges and inhibiting existed tumor growth. The robust transduction in the muscle tissue by the AAV1-1618E7hsp70 vector rendered more effective and long-term protective immune response against the HPV-related tumors.
Materials and Methods
Animals and cell lines
Female C57BL/6 mice (H-2b; 6–8 weeks old) were purchased from Jackson Lab (Bar Harbor, MA) and kept in the animal facility of the University of Pittsburgh. All animal procedures were performed according to approved protocols and in accordance with recommendations for the proper use and care of laboratory animals. The human embryonic kidney 293 cell line was obtained from the American Type Culture Collection (Rockville, MD). TC-1 cell line, prepared by transformation of C57BL/6 mice lung tumor cells with HPV-16 E6/E7 oncogene, was kindly provided by Dr. T.C. Wu (Johns Hopkins University, Baltimore, MD). EL4E7 cell line, an EL4 cell line transfected with HPV16 E7 oncogene, was kindly provided by Dr. Robert Tindle (Sir Albert Sakzewski Virus Research Centre, Australia). B16-1 cell line which was transformed by hepatitis B virus surface antigen (HBSAg) gene was maintained in our laboratory. TC-1 cells were grown in a medium developed by Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% fetal bovine serum (FBS), 50 U/ml penicillin, 50 μg/ml streptomycin, and 0.4 mg/ml G418. Other cells were maintained in Dulbecco's modified Eagle's medium (Gibco, Gaithersburg, MD) supplemented with 10% FBS (Gibco) and 1% penicillin–streptomycin.
Mutagenesis of E7 gene and plasmid construction
The HPV16 and HPV18 E7 genes were derived from plasmids pCP4/HPV16E7 and pZIP18E7 by polymerase chain reaction (PCR) using the following primers: 5′-GCCGCCATGCATGGAGATACACCT-3′ and 5′-AGATGGTTTCTGAGAACAGATGGG-3′ for HPV16 E7; 5′-ATGCATGGACCTAAGGCAACA-3′ and 5′-AGACTGCTGGGATGCACACCAC-3′ for HPV18 E7. These primers were designed to change the E7 stop codon TAA into TCT. The modified HPV16 and HPV18 E7 genes were subcloned into pBluescript KS(+) (Stratagene, La Jolla, CA). The Rb-binding sites in E7 were further altered by PCR with the following primers: 5′-TACGGTTATGGGCAATTAAATGAC-3′ and 5′-GAGATCAGTTGTCTCTGG-3’ for HPV16 E7; 5′-ACGGGCAATTAAGCGACTCAGA-3′ and 5′-GACCTAGAAGGTCAACCGGAA-3′ for HPV18 E7. The PCR products were self-ligated to produce plasmids pBSKS(+)16E7(m) and pBSKS(+)18E7(m). To ligate the modified HPV16 E7 and HPV18 E7 gene in tandem, PCR reactions were first performed to amplify a part of the ampicillin gene in the plasmid backbone and the modified E7 gene. The fragment containing HPV16 E7 was amplified with 5′-GGTTGTCAGAAGTAAGTTGGCCGCA-3′ and 5′-AGATGGTTTCTGAGAACAGATGGG-3′ from pBSKS(+)16E7(m). The HPV18 E7 was amplified with 5′-ATGCATGGACCTAAGGCAACA-3′ and 5′-ATCGGAGGACCGAAGGAGCTAACCG-3′ from pBSKS(+)18E7(m). The two PCR fragments were ligated to each other to form plasmid pBSKS(+)16E718E7(m), in which the ampicillin gene was recovered and the HPV16 E7 and HPV18 E7 were joined together. The hsp70 fragment was obtained by PCR with the primers 5′-CCGCTCGAGATGGCTCGTGCGGTCGGGATC-3′ and 5′-CCGCTCGAGTCAATCAGCCGAGCCGGG-3′, from PXX/E7112-hsp70. Both primers contain a XhoI site. The PCR products were digested with XhoI and ligated into the XhoI site of pBSKS(+)16E718E7(m) to create pBSKS(+)-1618E7hsp70. This vector was then digested with BamHI/ApaI, and the sticky ends were filled in and ligated into the filled NotI site in PC1 plasmid to produce pC1-1618E7hsp70. The HPV16 and 18 E7 and hsp70 fusion gene were excised from pC1-1618E7hsp70 by EcoRI/HpaI digestion. After filling in, this fragment was inserted into the filled NotI/SalI site of the AAV vector pXX-UF1 by blunt-end ligation to form the AAV vector plasmid UF1-1618E7hsp70 (Fig. 1B).

Construction and expression of AAV-1618E7hsp70 vector containing HPV16 E7 and HPV18 E7 Rb-binding site mutations. (
Generation of AAV stocks
AAV-1618E7hsp70 and AAV-LacZ vectors were generated by calcium phosphate triple-plasmid cotransfection methods as described previously (Xiao et al., 1998). The viruses were purified through double cesium chloride gradient centrifugations. AAV particle titers were determined by dot blot. The vector titers were in the range of 1 × 1012 to 1 × 1013 viral particles per milliliter.
Detection of E7 gene expression by immunofluorescent staining
Two hundred ninety-three cells in six-well plates were transfected with pC1-1618E7hsp70 plasmid (2 μg DNA per well) or infected with AAV1-1618E7hsp70 vector (1 × 105 AAV particles per cell) in the presence of wild-type adenovirus (200 particles per cell). Forty-eight hours after transfection or 24 hr after infection, cells were fixed with 4% paraformaldehyde for 15 min and permeabilized in PBS–0.2% Triton X-100 for 5 min at room temperature. E7 antigen was stained with goat anti-HPV-16 E7 or anti-HPV-18 E7 polyclonal antibodies (1:100; Santa Cruz Biotechnology, Santa Cruz, CA). The secondary antibody was mouse anti-goat monoclonal antibodies conjugated with Cy3 (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA).
In vivo tumor preventive and therapeutic experiments
For the tumor prevention study, C57BL/6 mice (n = 9–10 per group) were intramuscularly injected with 2 × 1011 particles of AAV1-1618E7hsp70 or AAV2-1618E7hsp70 into the gastrominius muscle of hind limbs. Two age-matched control groups were injected with either 2 × 1011 particles of AAV1-LacZ or 200 μl PBS. Five weeks after immunization, each mouse was inoculated subcutaneously with 1 × 105 TC-1 tumor cells in the dorsal region of the neck. The tumor-free mice were inoculated with 2 × 105 TC-1 cells for the second and third challenge at 12 and 24 weeks after first inoculation, respectively. For PBS and AAV1-LacZ treatment groups, the age-matched mice were added for negative control.
To test the specific prevention effect, 6–8-week-old C57BL/6 mice were intramuscularly injected with 2 × 1011 particles of AAV1-1618E7hsp70 or 200 μl PBS (n = 10 per group). Five weeks after injection, five mice in each group were challenged with 1 × 105 TC-1 cells, and the others were challenged with 2 × 104 B16-1 melanoma cells engineered to express Hepatitis B virus (HBV) surface antigen.
For the tumor treatment study, mice were first inoculated subcutaneously in the neck with 1 × 105 TC-1 cells. When the tumors were palpable (8–10 days after tumor inoculation), mice were intramuscularly injected with 2 × 1011 particles of AAV1-1618E7hsp70, AAV2-1618E7hsp70, AAV1-LacZ particles, or 200 μl PBS into the hind leg gastrominius muscles (n = 10 per group).
Tumor formation was observed by palpation every 3 days. Tumor volume was calculated by measuring tumor diameters with a caliper, using the following formula: V = (A × B 2)/2, where V is the volume, A is the larger diameter, and B is the smaller diameter. All the mice were humanely sacrificed when the tumor size approached ∼6000 mm3.
CTL assays
CTL activity was measured using Cytotox96-Nonradioactive Cytotoxicity Assay kit (Promega, Madison, WI) according to the manufacturer's protocol. Five or 52 weeks after immunization, mice splenocytes, as effector cells, were collected and cultured in RPMI supplemented with 10% FBS, 50 U/ml penicillin/streptomycin, 2 mM
Interferon-γ assays
The supernatants of cultured splenocytes were harvested and assayed for the presence of interferon-γ (IFN-γ) using ELISA kits (R&D system, Minneapolis, MN) according to the manufacturer's protocol. The plate was read with a standard ELISA reader (Molecular Devices, Sunnyvale, CA) at 450 nm.
Statistics
Statistical analysis was performed using the Fisher t test; a difference was considered significant at p < 0.05.
Results
Efficient in vitro expression of the HPV1618 E7 fusion protein
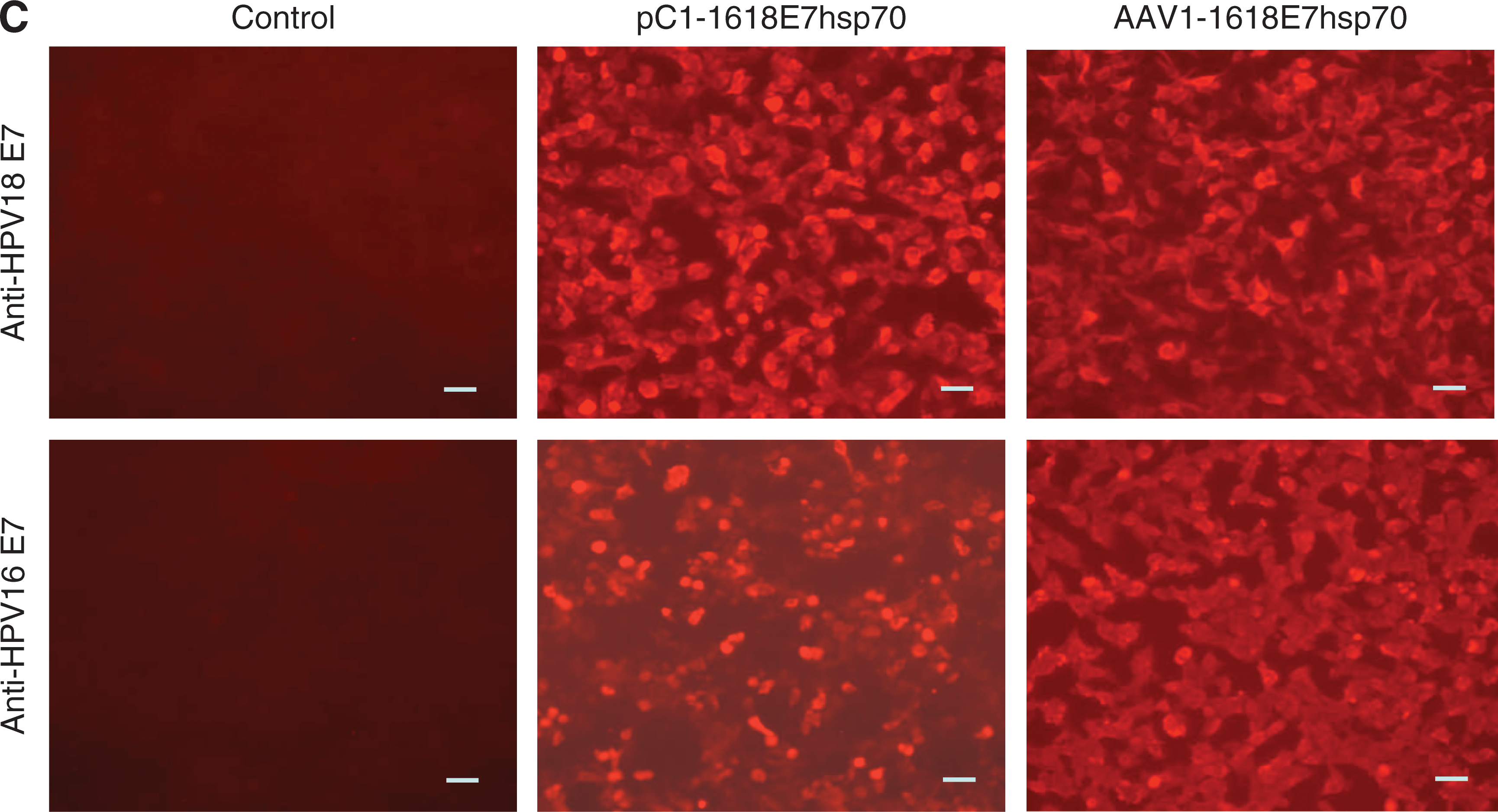
After making the 1618E7hsp70 fusion protein construct, we first tested its expression in vitro. Human 293 cells were either transfected with the pC1-1618E7hsp70 plasmid or infected with the AAV-1618E7hsp70 viral particles (see Materials and Methods section for details). Immunofluorescent staining against the E7 protein was performed to evaluate the expression of the transfected plasmid or infected viral vector. Strong fluorescence was observed in either the AAV-1618E7hsp70-infected (Fig. 1C, upper panels), or the pC1-1618E7hsp70-transfected (Fig. 1C, lower panels) 293 cells. Both HPV16 E7 (Fig. 1C, middle panels) and HPV18 E7 (Fig. 1C, right panels) antigens were detectable in the cytoplasm of the cells. No fluorescence was detected in the control 293 cells (Fig. 1C, left panels). The results indicated that HPV16 and HPV18 E7 antigens could be efficiently expressed by the 1618E7hsp70 fusion construct.
Prophylactic protection against E7-expressing tumor cells elicited by AAV-1618E7hsp70 immunization
To determine if the AAV-1618E7hsp70 immunization could generate protective immunity against tumor challenges, we injected either AAV1 or AAV2 vectors into the C57BL6 mice. Five weeks later, the mice were inoculated with 1 × 105 TC-1 cells and then challenged at indicated time points. As shown in Fig. 2 and Table 1, PBS- or AAV1-LacZ-treated mice developed palpable tumors within 2–4 weeks after TC-1 cell inoculation. In contrast, all the mice vaccinated with 2 × 1011 vector genome particles of either AAV1- or AAV2-1618E7hsp70 showed no evidence of tumor development after tumor cell inoculation for at least 12 weeks, at which time the second tumor cell challenge was administered (see below). These data strongly suggest that both AAV1 and AAV2 vectors were able to provide efficient protection against the TC-1 cell inoculation.
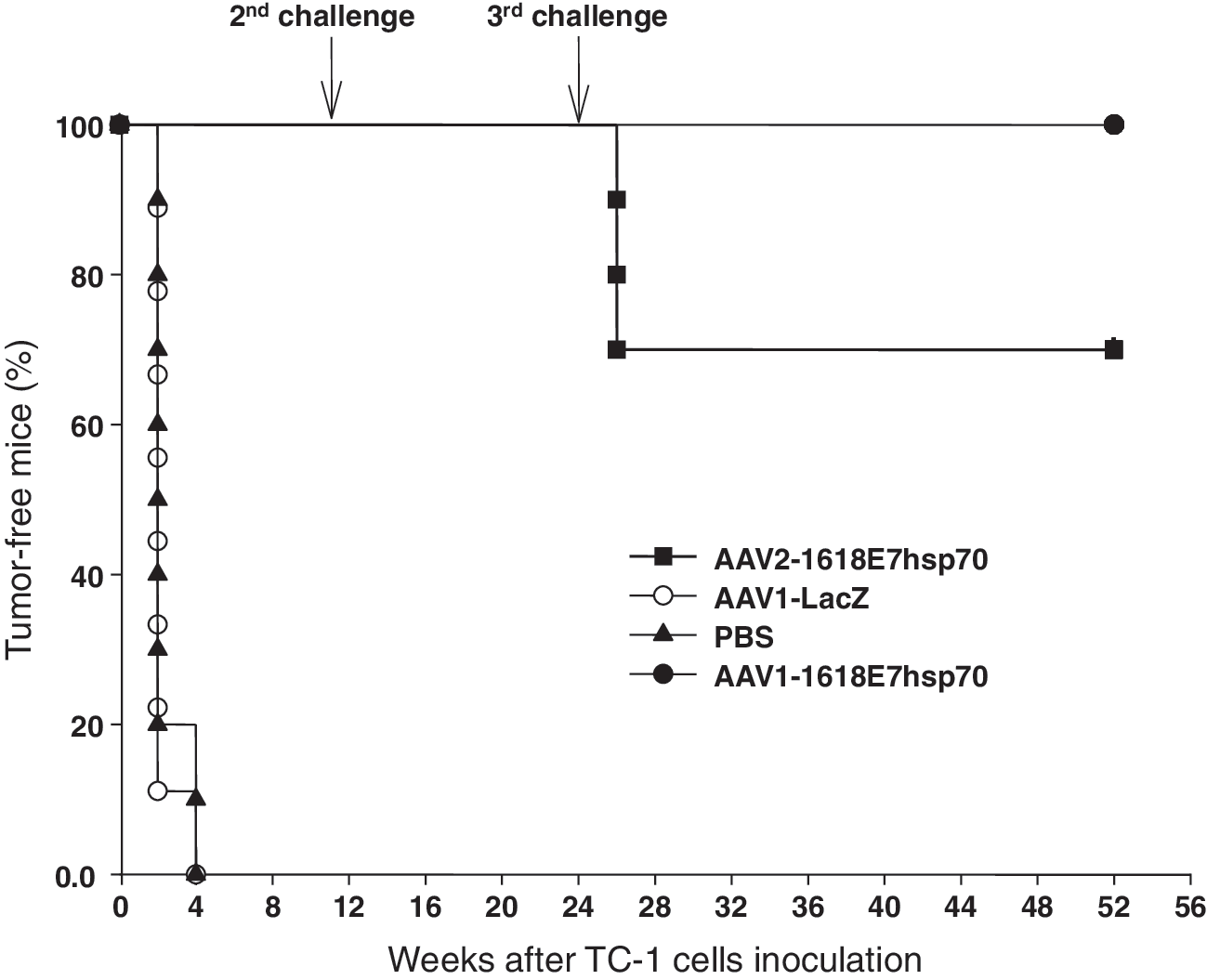
Effective and long-term prevention of TC-1 tumor growth in mice vaccinated with AAV-1618E7hsp70 vectors. Female C57BL/6 mice were injected with 2 × 1011 particles of AAV1-LacZ (n = 9; open circles), AAV1-1618E7hsp70 (n = 10; closed circles), or AAV2-1618E7hsp70 (n = 10; closed squares). Control mice were injected with 200 μl PBS (n = 10; closed triangles). Five weeks after immunization, 1 × 105 TC-1 cells were inoculated subcutaneously in the neck area of each mouse. Tumor growth was monitored every 3 days. At 12 and 24 weeks after first TC-1 cell inoculation, mice vaccinated with AAV1- or AAV2-1618E7hsp70 were given second and third challenges with 2 × 105 TC-1 cells. Mice were sacrificed when tumor size approached 6000 mm3. The percentages of tumor-free mice are shown.
Numbers of tumor-free mice after multiple TC-1 cell inoculation are shown.
Abbreviations: AAV, adeno-associated virus; hsp70, heat shock protein 70.
Mice were intramuscularly vaccinated with 2 × 1011 particles of AAV1-LacZ, AAV1-1618E7hsp70, AAV2-1618E7hsp70, or 200 μl PBS, respectively. At 5 weeks postimmunization, 1 × 105 TC-1 tumor cells were inoculated subcutaneously in the neck. At 12 weeks and 52 weeks after the first challenge, mice were given second and third challenges with 2 × 105 TC-1 cells.
For the second and third challenge experiments, all the mice in the PBS and AAV1-LacZ control groups were only given a single (first) challenge at the time when the AAV1-1618E7hsp70 and AAV2-1618E7hsp70 groups received their second and third challenges. However, the mice in all groups were “immunized” with the vectors or PBS at the same age and were age matched.
To further determine if persistent protection was induced, the AAV1- and AAV2-1618E7hsp70-treated mice were subjected to a second challenge with 2 × 105 TC-1 cells at 12 weeks after the first challenge. Five age-matched mice from both the PBS and AAV1-LacZ control groups were included. After the second challenge, all control mice developed tumors, whereas all the AAV1- or AAV2-1618E7hsp70-vaccinated mice did not (Fig. 2 and Table 1). Interestingly, when a third challenge was administered at 52 weeks after the first challenge, all AAV1-1618E7hsp70-treated mice remained tumor-free. However, 3 of 10 AAV2-1618E7hsp70-treated mice developed tumors. These results showed that, compared with AAV2, AAV1-1618E7hsp70 induced a more potent and persistent immune protection effect against the E7-expressing tumor cells.
Therapeutic efficacy against existing tumors by AAV-1618E7hsp70 immunization
We next evaluated the therapeutic effects of AAV1- and AAV2-1618E7hsp70 immunization in mice with existing TC-1 tumors. As showed in Fig. 3, treatment with AAV1- or AAV2-1618E7hsp70 significantly inhibited tumor growth, whereas treatment with AAV1-LacZ or PBS controls rendered no inhibition effects. It was also important to note that AAV1-1618E7hsp70 was more effective in inhibiting tumor growth as the tumor volumes in the AAV1-1618E7hsp70 treatment group were markedly smaller than those in the AAV2-1618E7hsp70 treatment group at each time point (p < 0.05). The experiment was terminated at 39 days after tumor cell inoculation because of excessive tumor growth in the AAV-LacZ and PBS control mice, which had to be humanely sacrificed. However, more than 50% of the AAV1-1618E7hsp70-treated mice survived beyond 66 days and most were essentially tumor-free (data not shown). These results again showed that AAV1 was more efficient than AAV2 in inducing the antitumor immune responses, consistent with those obtained in the protective group at the third round of TC-1 cell inoculation (Fig. 2).
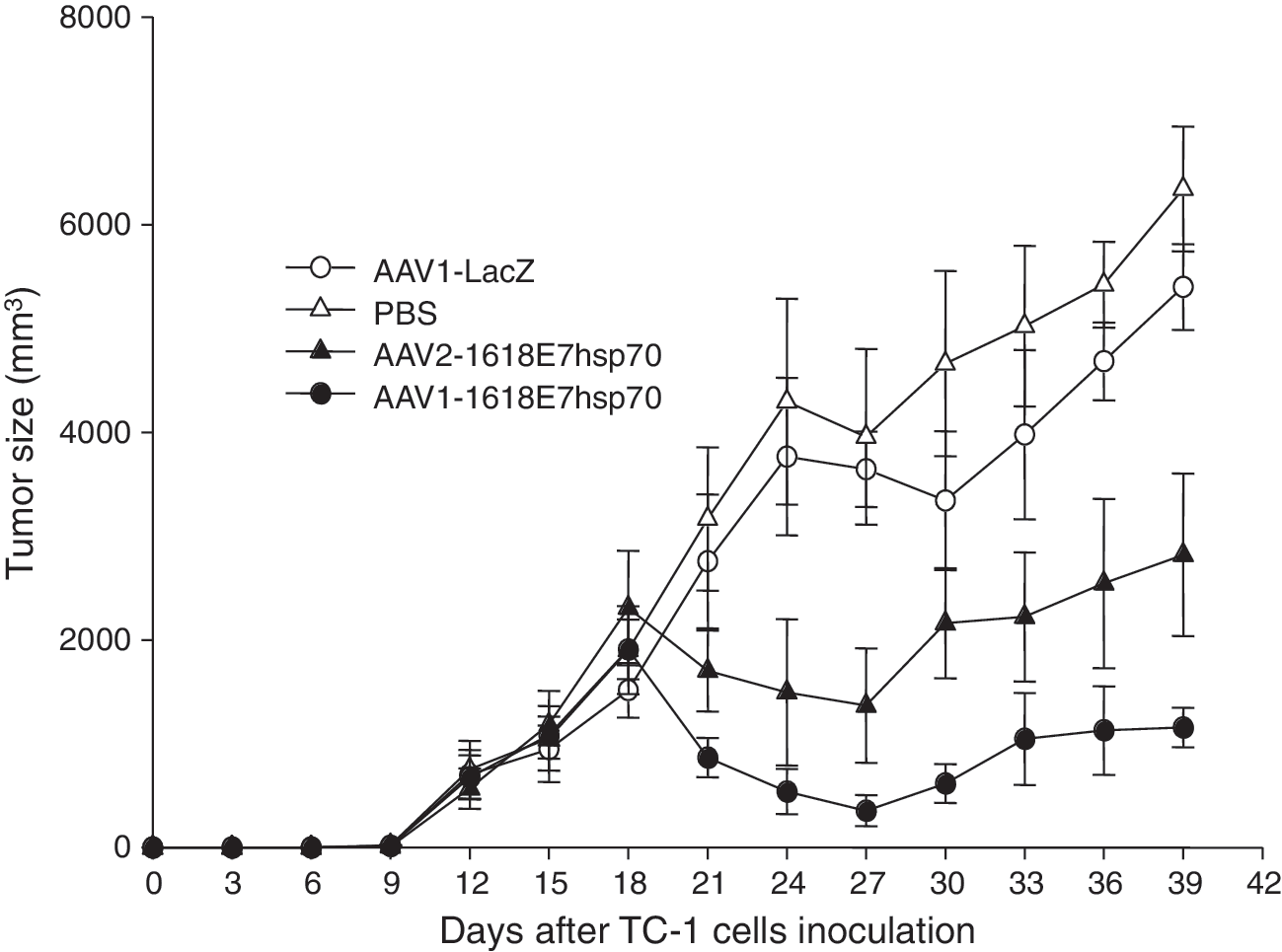
Inhibition of tumor growth by AAV-1618E7hsp70 immunization after tumor cell inoculation. TC-1 cells (1 × 105) were inoculated subcutaneously in the neck area of female C57BL/6 mice. Eight to 10 days after inoculation, when the tumor became palpable, mice were injected with 200 μl PBS, 2 × 1011 particles of AAV1-LacZ, AAV1-1618E7hsp70, or AAV2-1618E7hsp70 (n = 10 per group). Tumor volume was measured every 3 days and the mice were sacrificed when the tumor size approached 6000 mm3. Data were plotted as the mean ± SEM. SEM, standard error of the mean.
Antigen-specific tumor prevention after AAV-1618E7hsp70 immunization
To test the specificity of the protective immune responses triggered by the AAV-1618E7hsp70 against E7-expressing tumors, we challenged the preimmunized C57B/L6 mice with either 2 × 104 B16-1 cells, a mouse melanoma cell line that was engineered to express HBSAg, or 1 × 105 TC-1 cells at 5 weeks after AAV1-1618E7hsp70 or PBS injection. As expected, all mice challenged with the control B16-1 cells rapidly developed large tumors, regardless of whether they were treated with PBS or AAV1-1618E7hsp70 (Fig. 4). All the PBS-treated mice that were challenged with the TC-1 cells also developed large tumors, whereas no tumor was developed in mice previously treated with the AAV1-1618E7hsp70 (Fig. 4). These results demonstrate that strong protection against the TC-1 challenge by AAV1-1618E7hsp70 immunization was specific against the E7-expressing tumors.
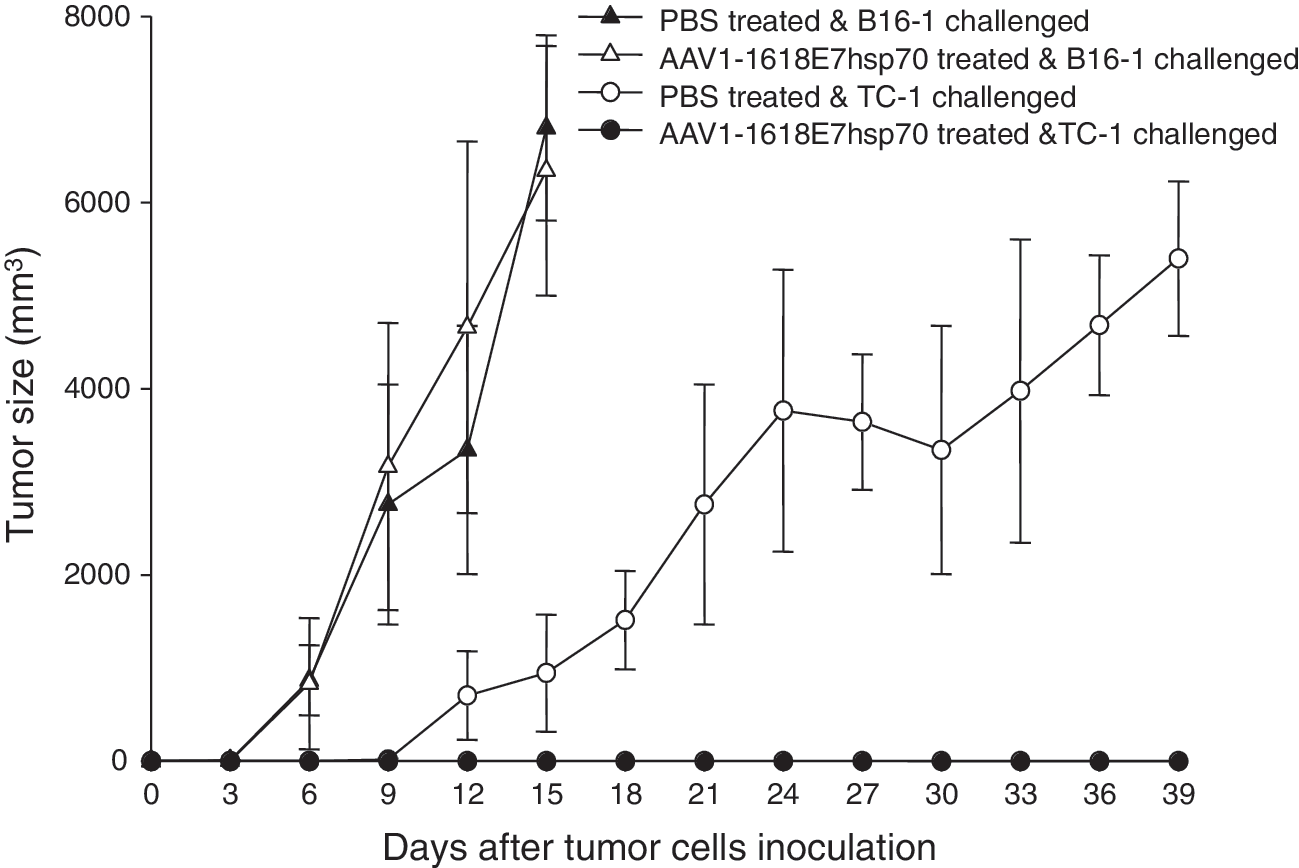
Specific antitumor effect elicited by AAV1-1618E7hsp70 immunization against TC-1 cells but not B16 melanoma cells. Female C57BL/6 mice were vaccinated intramuscularly with 2 × 1011 particles of AAV1-1618E7hsp70 or 200 μl PBS (n = 10 per group). Five weeks after immunization, five mice from each group were inoculated subcutaneously with 2 × 104 B16-1 cells or 1 × 105 TC-1 tumor cells. Tumor volume was measured every 3 days and the mice were sacrificed when the tumor size approached 6000 mm3. Data were plotted as the mean ± SEM.
Generation of enhanced E7-specific CTL reactions by AAV-1618E7hsp70 immunization
To verify the hypothesis that AAV-1618E7hsp70 induced cell-mediated immune response against E7-expression cells, we measured cytotoxic T-cell activity against an E7-expressing cell line EL4E7 (Fernando et al., 1998), which was found to be a more reliable target cell line for in vitro E7-CTL assays. Our results showed that at 5 weeks following immunization, mice vaccinated with either AAV1- or AAV2-1618E7hsp70 vector displayed markedly higher levels of cytolysis against EL4E7 tumor cells than the PBS or AAV1-LacZ control groups (p < 0.05; Fig. 5A). In addition, mice in the AAV1-1618E7hsp70-vaccinated group demonstrated significantly higher levels of CTL activity than those in the AAV2-1618E7hsp70-treated group (p < 0.05; Fig. 5A), again supporting that stronger immune responses were generated by the AAV1 vector.
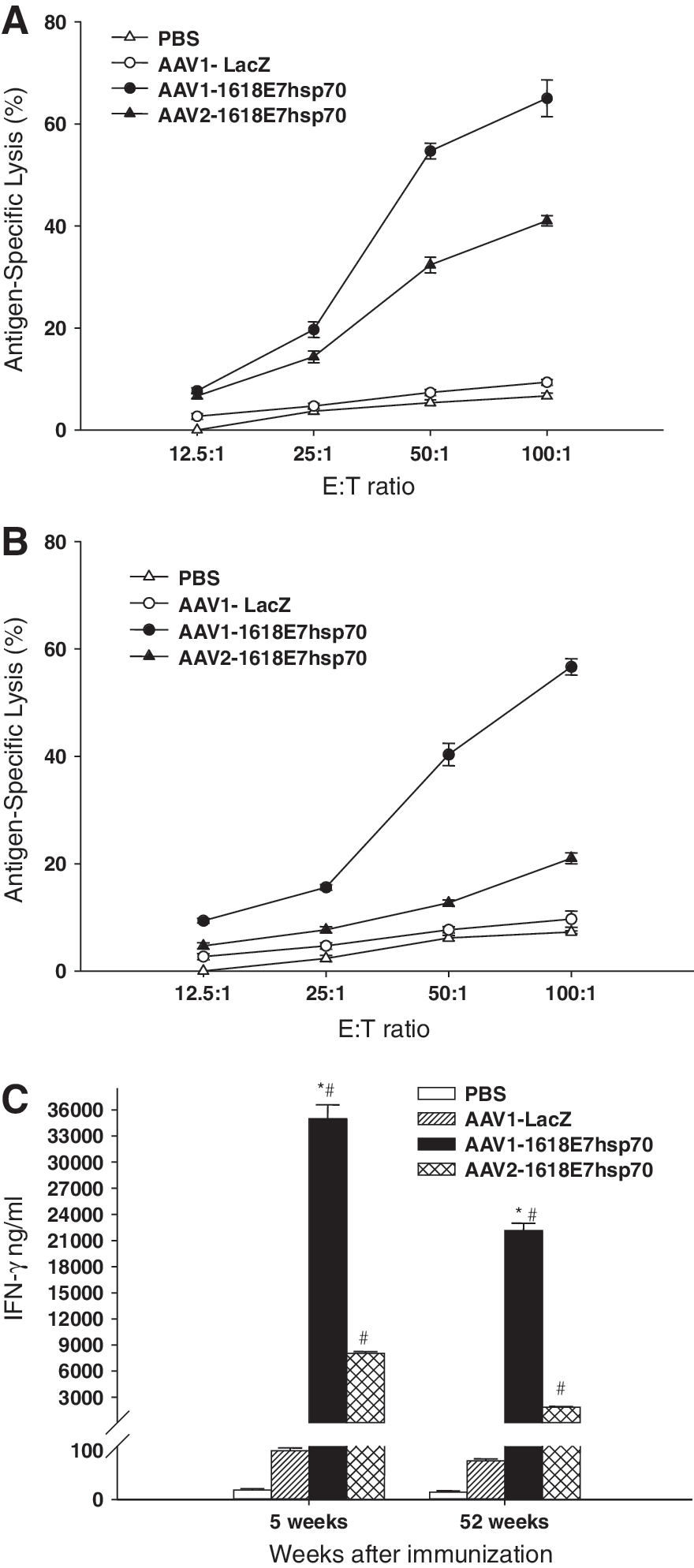
In vitro cytotoxic T lymphocyte and interferon secretion assays showed enhanced activities in AAV-1618E7hsp70 vector-immunized mice. At 5 weeks (
We next investigated the CTL activities at 52 weeks following immunization. Significantly higher levels of cytolysis against target cells were observed in both AAV1- and AAV2-1618E7hsp70 vector-vaccinated groups, compared with control groups (p < 0.05; Fig. 5B), although noticeably lower levels were observed when compared with the levels at 5 weeks following immunization, especially for AAV2-vaccinated group. This result indicates that AAV vectors, especially AAV1, could prime persistent cellular immune responses against tumor antigen.
We also measured the levels of IFN-γ secreted by the splenocytes isolated from the AAV-1618E7hsp70 vector-treated group and the control mice. The splenocytes were stimulated and coincubated with the target EL4E7 cells for IFN-γ production. Mice in the AAV-1618E7hsp70 group showed significantly higher IFN-γ secretion than those in the control groups (p < 0.05; Fig. 5C). Again, the AAV1-1618E7hsp70-treated group showed much higher amounts of IFN-γ secretion (35,010 ± 904 and 22,150 ± 480 pg/ml at 5 and 52 weeks, respectively) than those in the AAV2-1618E7hsp70-treated group (8045 ± 105 and 1868 ± 40 pg/ml at 5 and 52 weeks, respectively) (p < 0.05).
In summary, both enhanced CTL activity and high secretion of IFN-γ support the hypothesis that AAV-1618E7hsp70 elicited a strong and long-lasting cytotoxic T-cell immune response. Comparatively, AAV1-1618E7hsp70 is more potent in inducing and maintaining such an immune response against the E7-expressing tumor.
Discussion
This study has two major objectives: The first objective was to test whether strong antitumor effects can be achieved with the E7-hsp70 fusion protein, which has broader antigen targets for both HPV16 and HPV18 and molecular adjuvant effects by hsp70. The second objective was to test whether the AAV1 vector could achieve higher transgene expression and elicit stronger antitumor immune responses than AAV2, by means of intramuscular vaccination. We first tested the efficacy of this novel AAV vaccine in immunocompetent mice challenged with TC-1 tumor cells. Tumor development was observed in 100% of the control mice. In contrast, both the AAV1- and AAV2-1618E7hsp70-vaccinated mice were protected from tumor growth. The antitumor CTL activities in both AAV1- and AAV2-treated groups lasted for up to 1 year (duration of the experiments) with increased secretion of IFN-γ, but the activities were much higher in the AAV1-treated group (Fig. 5). Further, we confirmed that the AAV-1618E7hsp70-induced antitumor effect is highly antigen specific, as tumor protection effect was only observed against the TC-1 cell challenge. Finally, we found that administration of the AAV-1618E7hsp70 vaccine following establishment of TC-1 tumors could significantly reduce tumor growth. Taken together, these results demonstrate that AAV-1618E7hsp70 is a potent preventive and therapeutic vaccine against E7-expressing tumors in mice by a simple single-dose intramuscular immunization.
The initiation and maintenance of protective T-cell immune response against tumor cells are largely dependent on the quantity and duration of the presence of tumor-specific antigens. Sufficient and relatively durable antigen expression is an important requirement in the selection of a gene delivery system for DNA vaccines. It has been reported that the level of E7 vaccine protein expression correlates with both the CTL activity in vitro and the anti E7-expressing tumor efficacy in vivo, after immunization with plasmid DNA that expresses HPV16 E7 (Steinberg et al., 2005). Our previous study showed that Lac-Z gene, delivered by AAV2 vectors, can be expressed at high levels in skeletal muscles for long periods (Xiao et al., 1996). We have also demonstrated that a single intramuscular injection of AAV2 encoding an HPV16 E7 peptide fused with hsp70 effectively inhibited the growth of TC-1 tumor cells in mice (Liu et al., 2000). Recently, a large number of new serotypes of AAV have been isolated from human and nonhuman tissues (Xiao et al., 1999; Gao et al., 2002). Although they share a great deal of homology in the amino-acid sequences of the capsids, different serotype vectors have demonstrated diverse cellular tropisms (Gao et al., 2002). Specifically, the transduction efficiency of AAV1 in muscle has been found to be tens of folds higher than that of AAV2 vectors (Chao et al., 2000; Arruda et al., 2004), which is part of the reason we chose to investigate AAV1 in this report. It also corroborates in part the superior antitumor vaccination effects induced using AAV1 vector in this study. Also, antigen-specific T-cell immune responses are also dependent on the efficiency of antigen presentation by antigen-presenting cells (APCs), such as the dendritic cells (DCs), through MHC class I and class II pathways. It has been reported that human monocyte-derived DCs could be efficiently transduced by AAV vector (Ponnazhagan et al., 2001), and DCs infected with AAV2-encoded HPV-16 E7 gene elicit a strong CTL activity after only 7 days of priming (Chiriva-Internati et al., 2002). Antigen gene expression in the DCs would trigger MHC class II-mediated antigen presentation, whereas antigen gene expression in the skeletal muscles most plausibly would trigger antigen cross presentation via the MHC-I–restricted pathway. It is also possible that direct transduction of APCs has taken place; however, this needs further investigation. The shortcoming of the ex vivo approach in clinical practice is the tedious patient-specific cell isolation and manipulation under stringent Good Manufacturing Practice (GMP) for each patient. On the other hand, a generic cervical cancer gene vaccine for direct intramuscular injection should be more convenient and cost effective, provided that it is also efficacious. In addition, unlike the conventional vaccines that need repeated boosts for achieving long-term antigen-specific T-cell immune responses, a single injection of AAV1-1618E7hsp70 in this study was sufficient to generate a potent and long-term protective effect.
To target more oncogenic HPV genotypes and more antigenic epitopes, we selected full-length E7 of both HPV 16 and 18 fusion genes as the vaccine antigen gene, as these two viruses are the most common HPV genotypes in cervical cancers, accounting for 70–85% of the patients (van den Brule et al., 1989). There are two key issues during the construction of the E7-based anti-HPV vaccine. Safety is the first concern because E7 is an oncogenic protein that can bind and facilitate Rb-protein degradation and induce cellular transformation. Therefore, wild-type E7 cannot be directly used as a vaccine gene. To minimize risk, we introduced point mutations in the Rb-binding site of the HPV 16 and 18 E7 genes. This strategy was based on a method previously described (Boursnell et al., 1996), which demonstrated that the mutated E7 proteins lost the ability to transform but retained their immunogenicity. In addition, this in vitro experiment has demonstrated that the point mutations did not abolish the E7 expression, although its fusion with hsp70 has changed E7's primary cellular localization from nucleus to cytoplasmic (Fig. 1C), which should further abolish the oncogenic capacity. The safety of mutated E7 proteins has been shown by other studies and is being tested in human clinical trials (Borysiewicz et al., 1996; Adams et al., 2001). The second key issue is to increase the antigenicity of the vaccine gene product. E7 encodes a nuclear protein and can be rapidly degraded, resulting in poor processing and presentation by MHC molecules on the cell membrane and accounting for the mild immunogenicity and immune system evasion of HPV (Frazer, 2004). Our results showed that covalent linkage of the E7 to the adjuvant hsp70 protein has kept it outside the nucleus and possibly more accessible for antigen processing and presentation. Antigen fusion with hsp70 has proven powerful as a strategy to increase the immunogenicity of E7 and to induce robust cellular immune responses (Ji et al., 1999; Cheng et al., 2001a). The hsp70 protein is an intracellular chaperone molecule responsible for folding and translocation of newly synthesized polypeptides throughout various cellular compartments (Craig et al., 1994; Bukau et al., 2000). The mechanism through which hsp70 enhances tumor cell immunogenicity is related to MHC processing and presentation of endogenous tumor antigens directly to tumor-specific T cells (Todryk et al., 2003; Chaput et al., 2004). Wu and colleagues have elegantly demonstrated that hsp70 is a potent molecular adjuvant for anti-HPV DNA vaccines (Chen et al., 2000a,b; Cheng et al., 2001b; Hsu et al., 2001). Our previous studies have also shown that hsp70 can increase the immunogenicity of transgene production and induce an effective CD8+ CTL response (Liu et al., 2000). We need to point out that a limitation of this study is the lack of an established tumor model that expresses the HPV18 E7 antigen. As a result, we could not directly examine T-cell responses and therapeutic effects against the HPV18 E7 antigen, because the syngeneic tumor cell line expressing that protein is not currently available.
It also became apparent during this study that, although both the AAV1 and AAV2 vaccines initiated persistent and robust protection against the development of E7-induced tumor, the protective effects of AAV1 vector were superior over those of AAV2 vector. Although all the mice vaccinated with AAV1-1618E7hsp70 resisted three challenges of TC-1 tumor cells, 3 of 10 AAV2-1618E7hsp70-treated mice finally gave in and developed tumors after the third round TC-1 cell challenge (Table 1). Further, tumor-specific lysis by the T cells from AAV1-treated mice was more effective than that induced by AAV2 at each postvaccination time point (Fig. 5A and B), suggesting that AAV1-mediated strong expression in the muscle boosted antigen cross-presentation and maintained the specific CD8+ T-cell memory for a longer period of time. Consistently, splenocytes from AAV1-vaccinated mice produced much higher IFN-γ than those vaccinated with AAV2 during in vitro culture (Fig. 5C). Previous studies showed the importance of IFN-γ in mediating a potent antitumor effect (Dighe et al., 1994; Kaplan et al., 1998). The potency of the HPV16E7hsp70 DNA vaccine found in normal mice was lost in the IFN-γ knockout C57BL/6 mice (Cheng et al., 2003). Finally, vaccination with the AAV1 vector rendered a stronger protective effect to the tumor-bearing mice than the AAV2 vector (Figs. 2 and 3). A recent report also showed that AAV1 elicited stronger CTL responses than AAV2 in mice (Lin et al., 2007). We attribute the higher efficacy of the AAV1-1618E7hsp70 vector over its AAV2 counterpart to the much higher gene transfer efficiency in skeletal muscle by AAV1, which resulted in higher antigen gene expression and more effective antigen processing facilitated by the hsp70 fusion protein via the MHC-I–restricted pathway in the muscle tissue.
In conclusion, a single injection of AAV1-1618E7hsp70 has produced a robust, specific, and persistent preventive and therapeutic effect against E7-expressing tumors in mice, indicating that AAV1-1618E7hsp70 is a promising vaccine for immunotherapy of HPV-associated cancers, and potentially of precancerous lesions, because of persistent E7 gene expression in HPV-infected patients. Thus, this study supports continued development of AAV viral vectors for the prevention and clinical treatment of HPV-associated neoplastic diseases.
Footnotes
Acknowledgment
This work was in part supported by an award to X. Xiao from the PNC Foundation, Pittsburgh, PA.
Author Disclosure Statement
No competing financial interests exist.