
Abstract
Fanconi anemia (FA) is an inherited genetic disease characterized mainly by bone marrow failure and cancer predisposition. Although gene therapy may constitute a good therapeutic option for many patients with FA, none of the clinical trials so far developed has improved the clinical status of these patients. We have proposed strategies for the genetic correction of bone marrow grafts from patients with FA, using lentiviral vectors (LVs). Here we investigate the relevance of the expression of FANCA to confer a therapeutic effect in cells from patients with FA-A, the most frequent complementation group in FA. Our data show that relatively weak promoters such as the vav or phosphoglycerate kinase (PGK) promoter confer, per copy of FANCA, physiological levels of FANCA mRNA in lymphoblastoid cell lines, whereas the cytomegalovirus and, more significantly, spleen focus-forming virus (SFFV) promoters mediated the expression of supraphysiological levels of FANCA mRNA. Insertion of the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) or a mutated WPRE into the 3′ region of PGK-FANCA LVs significantly increased FANCA mRNA levels. At the protein level, however, all tested vectors conferred, per copy of FANCA, similar and physiological levels of the protein, except SFFV LVs, which again conferred supraphysiological levels of FANCA. In spite of their different activity, all tested vectors mediated a similar phenotypic correction in FA-A lymphoblastoid cell lines and also in hematopoietic progenitors from patients with FA-A. On the basis of the efficacy and safety properties of PGK LVs, a PGK LV carrying FANCA and a mutant WPRE is proposed as an optimized vector for the gene therapy of patients with FA-A.
Introduction
Although gene therapy was considered a good alternative for patients with FA who lack an HLA-identical related donor, none of the retrovirus-mediated gene therapy protocols so far conducted in FA has resulted in clinical benefits (Liu et al., 1999; Walsh et al., 2001; Kelly et al., 2007). This fact, together with safety concerns related to the use of gammaretroviral vectors (Hacein-Bey-Abina et al., 2003a,b; Ott et al., 2006; Grez et al., 2007), indicate the need to improve the design of these protocols, both in terms of the manipulation of the hematopoietic grafts and in terms of vector design.
In previous studies we proposed that, in contrast to other monogenic diseases, hematopoietic grafts from patients with FA should be subjected to minimal ex vivo manipulation during the transduction process (Jacome et al., 2006). In addition, to improve the efficacy of transduction of FA hematopoietic stem cells (HSCs) and also to limit risks of insertional oncogenesis, lentiviral vectors (LVs) were used to transduce unselected, not in vitro-prestimulated, bone marrow (BM) samples from patients with FA (Jacome et al., 2009).
In the present study we investigate the functional properties of LVs expressing various levels of FANCA, the gene that is more frequently mutated in patients with FA (Levitus et al., 2004; Taniguchi and D'Andrea, 2006; Casado et al., 2007; Rosenberg et al., 2008). In particular, we show the influence of the promoter and of the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) in both the ectopic expression of FANCA and in the therapeutic efficacy of the vector.
Materials and Methods
Lentiviral vector design
Lentiviral vectors carrying the cDNA encoding FANCA under the control of the vav1 promoter (vav) (Almarza et al., 2007), the human phosphoglycerate kinase promoter (hPGK), the cytomegalovirus promoter (CMV), or the spleen focus-forming virus promoter (SFFV) were constructed. As controls, LVs carrying the enhanced green fluorescent protein gene (EGFP) under the control of the SFFV promoter or PGK promoter (kindly provided by L. Naldini, San Raffaele-Telethon Institute for Gene Therapy, Milan, Italy) were used. In the case of PGK-FANCA LVs, constructs harboring the wild-type WPRE or a mutated version of this element (WPRE*) (Schambach et al., 2006) were generated.
Supernatants: production and titration
293T cells (CRL-11268; American Type Culture Collection [ATCC], Manassas, VA) and HT-1080 cells (CRL-12103; ATCC), used to produce and titrate lentiviral vectors, were grown in Dulbecco's modified Eagle's medium (DMEM; GIBCO Laboratories/Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO) and with 2 mM glutamine and 0.5% penicillin–streptomycin (GIBCO Laboratories/Invitrogen). Vector stocks of third-generation self-inactivating LVs pseudotyped with gibbon ape leukemia virus transmembrane protein (GALV-TR) envelope were prepared by four-plasmid calcium phosphate-mediated transfection in 293T cells, essentially as described (Dull et al., 1998).
Functional titers by flow cytometry or quantitative polymerase chain reaction (qPCR) of infective LVs were determined in HT-1080 cells, plated at 3.5 × 104 cells per plate in 24-well plates and infected with various dilutions of either LV overnight (Meza et al., 2006).
Transduction of lymphoblastoid cell lines from patients with FA-A
Epstein–Barr virus (EBV)-transformed lymphoblastoid cell lines (LCLs) were generated and maintained in RPMI 1640 supplemented with 15% FBS, 2 mM glutamine, and 0.5% penicillin–streptomycin. The transduction of LCLs with LVs pseudotyped with GALV-TR was conducted in plates precoated with RetroNectin (Takara Bio, Shiga, Otsu, Japan) at 2 μg/cm2 (Casado et al., 2007). To determine the viability of FA LCLs, cells were exposed to increasing concentrations of mitomycin C (MMC, 0 to 1000 nM; Sigma-Aldrich) in fresh medium. Five days afterward, cell viability was determined by flow cytometry, using the propidium iodide test (Casado et al., 2007).
Transduction of BM cells from patients with FA-A
Bone marrow samples were depleted of erythrocytes with hydroxyethyl starch (HES; Grifols Laboratories, Barcelona, Spain) as previously described (Jacome et al., 2009). For the transduction of erythrocyte-depleted BM cells, samples were resuspended in X-VIVO 10 supplemented with thrombopoietin (TPO, 5 ng/ml; R&D Systems, Minneapolis, MN), stem cell factor (SCF, 15 ng/ml; Peprotech, London, UK), Flt3 ligand (Flt3-L, 5 ng/ml; Invitrogen, Carlsbad, CA), and etanercept (anti-tumor necrosis factor [TNF]-α fusion protein, 10 μg/ml; Immunex, Seattle, WA) and transduced for 16 hr in plates preloaded with LV supernatants (Jacome et al., 2009). Fourteen days later, colonies grown in the absence and the presence of 10 nM MMC were scored, to determine the proportion of progenitors that became resistant to the drug.
Analysis of lentiviral vector copy number
DNA corresponding to 1 million LCLs transduced with the various LV vectors was extracted, using a DNeasy blood and tissue kit (Qiagen, Valencia, CA). The number of LV proviruses in these cells was investigated with primers specific for hFANCA cDNA (FANCA-F, 5′-GCTCAAGGGTCAGGGCAAC-3′; and FANCA-R, 5′-TGTGAGAAGCTCTTTTTCGGG-3′) and detected with the TaqMan probe FANCA-P (5′-FAM-CGTCTTTTTCTGCTGCAGTTAATACCTCGGT-BHQ1-3′) in a Rotor-Gene 3000 (Corbett Research Products, Foxboro, MA) (Navarro et al., 2006). Hematopoietic progenitors from patients with FA-A were picked up from methylcellulose plates and DNA was extracted with TRIzol reagent (Invitrogen) in accordance with the manufacturer's instructions. Lentiviral vector copy number was calculated by qPCR detection of hFANCA normalized to the human albumin gene (hALB), using primers hALB-F (5′-GCTGTCATCTCTTGTGGGCTGT-3′) and hALB-R (5′-ACTCATGGGAGCTGCTGGTTC-3′) and detected with the TaqMan probe hALB-P (5′-TR-CCTGTCATGCCCACACAAATCTCTCC-BHQ2-3′). Standard curves were generated with pRRLcpptPGKGFP-WPRE-Alb (Charrier et al., 2007) and PGK-FANCA plasmids.
Analysis of FANCA mRNA
The expression of human FANCA mRNA was analyzed by real-time quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) of cDNA obtained from total RNA, either from LCLs or colony-forming cells (CFCs) from patients with FA-A. Untransduced samples or samples transduced with EGFP LVs were used as negative controls. The relative transgene expression of FANCA was determined by the 2–ΔΔC t method as previously described (Livak and Schmittgen, 2001), using FANCA primers and probes described in the previous section. For housekeeping control expression human β-actin was used: β-actin-F, 5′-ATTGGCAATGAGCGGTTCC-3′; and β-actin-R, 5′-CACAGGACTCCATGCCCA-3′; and β-actin-P, 5′-TR-CCCTGAGGCACTCTTCCAGCCTTCC-BHQ2-3′ probe.
Western blot analysis and immunofluorescence studies
Western blot analyses were performed with nuclear extracts from LCLs (5 × 106 per sample) collected by centrifugation and washed twice in phosphate-buffered saline (PBS) as previously described (Almarza et al., 2007). Relative quantification of the protein level was done with Quantity One (Bio-Rad, Hercules, CA). For immunofluorescence studies, cells were fixed with 3.7% paraformaldehyde in PBS, permeabilized, and stained with mouse monoclonal anti-FANCD2 (Santa Cruz Biotechnology, Santa Cruz, CA) as primary antibody. Anti-mouse Alexa 488nm (Molecular Probes, Leiden, The Netherlands) was used as secondary antibody (Almarza et al., 2007).
Results
In this study we aimed to investigate the level of expression of FANCA that was necessary to correct the phenotype of FA-A cells. To this end, LVs expressing FANCA under the control of various promoters (vav, PGK, CMV, and SFFV) were constructed (Fig. 1A). In addition, we also investigated the influence of the WPRE, both on the expression level of FANCA and on the therapeutic efficacy of the LVs. In all instances LVs were packaged with the chimeric GALV-TR envelope. Titers of 1–2 × 106 transducing units (TU)/ml were routinely obtained, and transductions were conducted at estimated multiplicities of infection (MOIs) of 1–2 TU/cell. Under these conditions transduction efficacies ranged between 38.6 and 77.9%, as deduced from flow cytometric analyses of LCLs transduced with EGFP-expressing LVs.
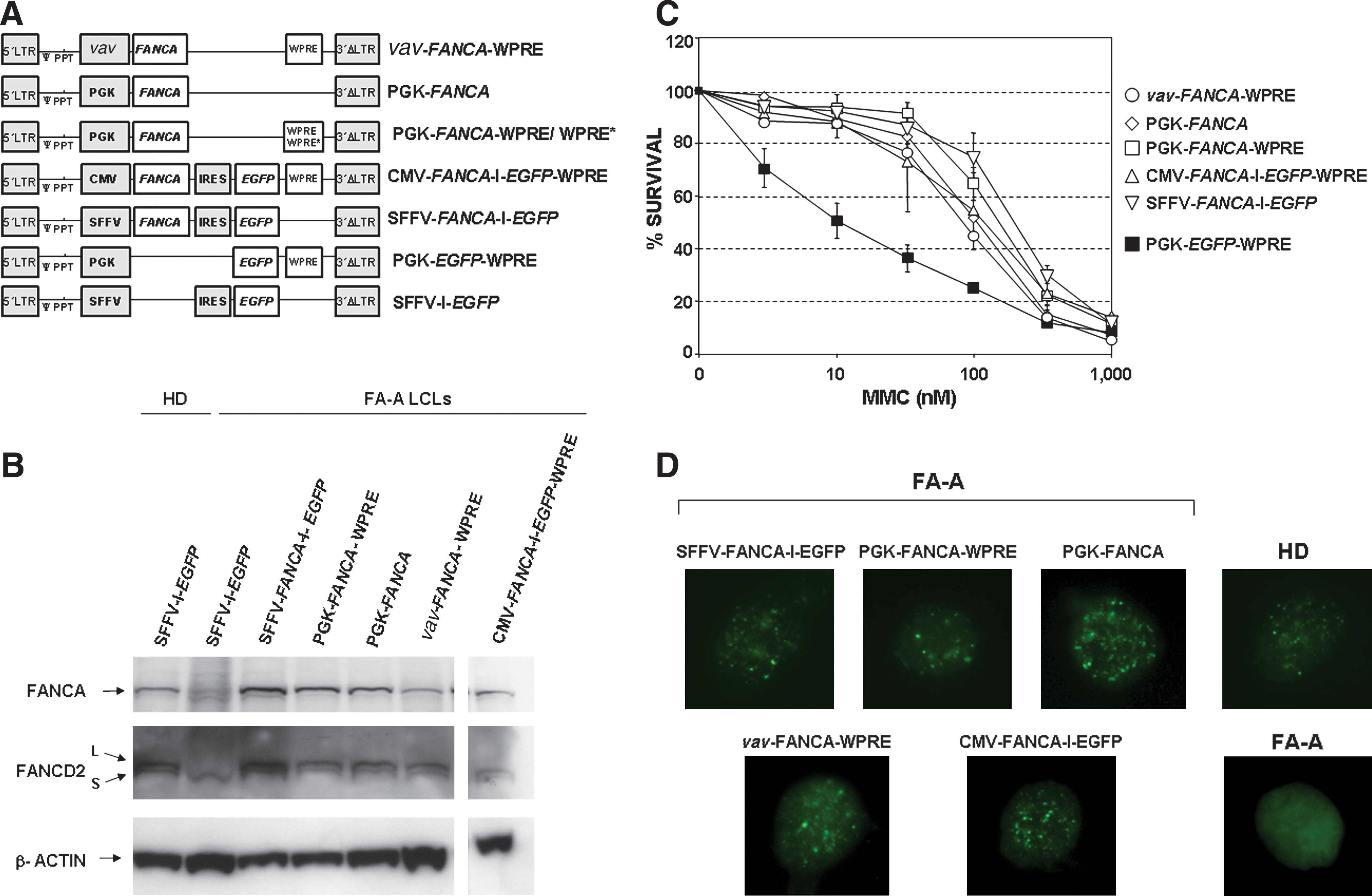
Functional analysis of lentiviral vectors expressing FANCA under the control of different internal promoters. (
To determine the level of FANCA mRNA that was conferred by each vector, transduced FA-A LCLs were selected with 30 nM MMC for 5 days. After the selection process, transduced FA-A LCLs contained 0.81 to 3.04 copies of the respective LV per cell (Table 1). Unselected FA-A LCLs transduced with EGFP LVs and untransduced LCLs from a healthy donor (HD) were used as controls.
Abbreviations: CMV, cytomegalovirus; EGFP, enhanced green fluorescent protein; FA-A, Fanconi anemia complementation group A; HD, healthy donor; MMC, mitomycin C; PGK, phosphoglycerate kinase; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element; WPRE*, mutated WPRE.
Analyses of samples transduced with FANCA-expressing LVs were always conducted in cells selected with 30 nM mitomycin C.
To estimate levels of FANCA mRNA and FANCA protein per copy of FANCA, healthy donor LCLs were considered to have two copies of genomic FANCA.
Total FANCA mRNA levels, as well as relative FANCA mRNA levels per LV copy number, were determined in LCLs transduced with the various LVs (Table 1). Compared with FANCA mRNA levels observed in HD LCLs, similar levels of FANCA mRNA levels per copy were observed in FA-A cells transduced with vav-FANCA- and PGK-FANCA LVs. CMV-FANCA LV and, more significantly, SFFV-FANCA LV conferred supraphysiological levels of FANCA mRNA per copy (3.6- and 5.6-fold, respectively). PGK-FANCA LVs harboring WPRE or the mutated WPRE* sequence (Schambach et al., 2006) increased FANCA mRNA levels 2.3- to 2.6-fold compared with PGK LVs without WPRE. Similar levels of FANCA expression were observed in LCLs that had been transduced with PGK-FANCA-WPRE* LV and maintained in culture for 4 weeks (data not shown).
To test whether differences in FANCA mRNA expression were confirmed at the protein level, Western blot analyses were conducted with the samples shown in Table 1 (Fig. 1B). As for the FANCA mRNA determinations, FANCA protein values were related not only to protein loadings, but also to the proviral copy number determined in each transduced FA LCL. Compared with FANCA protein levels per copy as determined in HD LCLs, essentially normal levels of FANCA per copy were conferred by all tested FANCA LVs, except for SFFV-FANCA LV (see Table 1). In this case, relative FANCA protein levels per copy were 3.4-fold higher than levels determined in HD LCLs. Western blots restained with anti-FANCD2 showed that, whereas FA-A LCLs transduced with the control vector were not able to monoubiquitinate FANCD2, FA-A LCLs transduced with either type of FANCA LV expressed both the nonubiquitinated and monoubiquitinated forms of FANCD2, consistent with a functional FA pathway in these cells (Fig. 1B).
To analyze possible differences in the therapeutic efficacy of the various FANCA-expressing LVs, the efficiency with which each vector corrected the MMC hypersensitivity of FA cells was determined. To this end, FA-A LCLs were transduced with the various LVs (Fig. 1A) and then exposed to increasing concentrations of MMC. Thereafter, the viability of transduced cells was determined as indicated in Materials and Methods. As shown in Fig. 1C, all tested FANCA LVs were equally efficient in reverting the hypersensitivity of FA-A LCLs. Similarly, all vectors promoted the generation of nuclear FANCD2 foci in MMC-treated cells (Fig. 1D), consistent with a functional FA pathway in FA-A cells transduced with either FANCA LV.
Because vectors containing the PGK promoter have been extensively used, and several reports have confirmed the stability (Follenzi et al., 2000) and low genotoxic properties of LVs carrying this promoter (Montini et al., 2006, 2009; Modlich et al., 2009), further experiments were conducted in FA-A LCLs transduced with PGK-FANCA LVs, free from any WPRE or harboring the WPRE or WPRE* sequence. As shown in Fig. 2A, all three vectors conferred the same reversion in the hypersensitivity of FA-A LCLs to MMC.
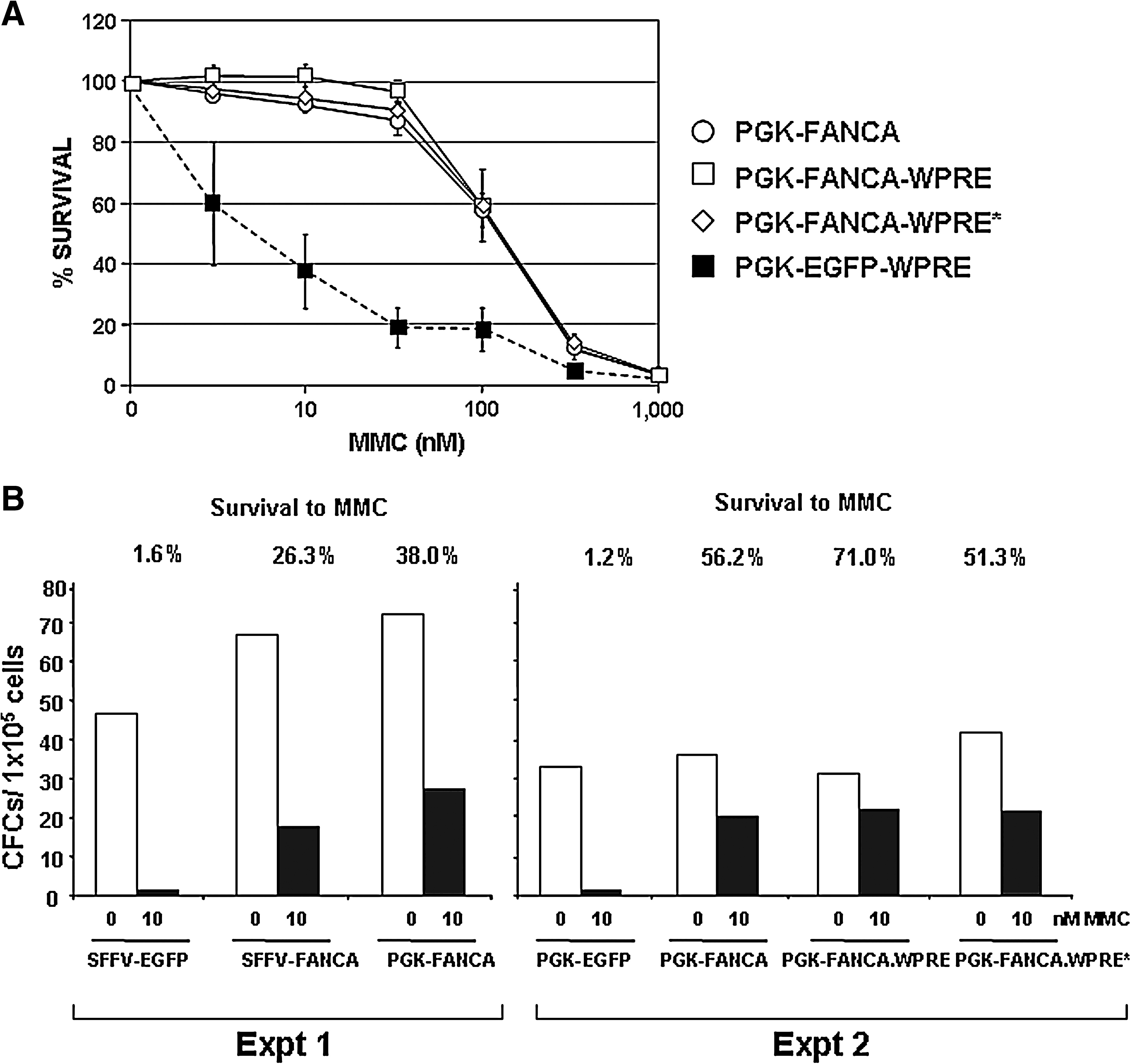
Relevance of the WPRE sequence in the functional properties of lentiviral vectors expressing FANCA under the control of the PGK promoter. (
Finally, to compare the efficacy of SFFV-FANCA- and PGK-FANCA LVs to correct the phenotype of hematopoietic progenitors from patients with FA-A, erythrocyte-depleted BM samples from patients with FA-A were transduced as described (Jacome et al., 2009). To determine the proportion of transduced CFCs, samples were then cultured in methylcellulose, both in the absence of MMC and in the presence of 10 nM MMC. As shown in Fig. 2B, when samples were transduced with EGFP LVs almost no colonies were generated in the presence of MMC. In contrast to this observation, the transduction of FA-A BM cells with SFFV-FANCA- and PGK-FANCA LVs mediated survivals of 26 and 38%, respectively. In a second experiment we compared the efficacy of PGK-FANCA LVs harboring the WPRE and WPRE* sequences, with respect to WPRE-free PGK-FANCA LVs. The number of CFCs present in the BM from this patient with FA-A was about 2-fold lower, compared with numbers corresponding to the previous patient. Significantly, all three LVs mediated a similar level of protection to MMC (Fig. 2B). In this experiment, colonies resistant to MMC were analyzed both for the number of LV copies and for levels of FANCA mRNA, as already conducted with LCLs. Numbers of LV copies per cell were, respectively, 1.84, 1.09, and 1.09 copies per cell, in samples transduced with WPRE-free-, WPRE-, and WPRE*-PGK-FANCA LVs. As happened in FA-A LCLs (see Table 1), levels of FANCA mRNA per LV copy increased 2.7- and 3.0-fold, respectively, when WPRE and WPRE* LVs were used (data not shown), confirming that these sequences not only stabilized the ectopic expression of FANCA in LCLs, but also in primary FA hematopoietic cells derived from corrected progenitors.
Discussion
The purpose of this study was to develop a therapeutic vector aiming at the efficient and safe gene therapy of patients with FA-A, the most prevalent complementation group in FA (Levitus et al., 2004; Taniguchi and D'Andrea, 2006; Casado et al., 2007; Rosenberg et al., 2008). Although previous studies have demonstrated the efficacy of gammaretroviral and lentiviral vectors to correct the hematopoietic phenotype of FA-A mice (Galimi et al., 2002; Río et al., 2002), no clinical benefits have been reported so far in patients with FA treated by gene therapy (Liu et al., 1999; Walsh et al., 2001; Kelly et al., 2007).
In previous studies we showed that in contrast to what has been observed in samples from healthy donors, hematopoietic progenitors from patients with FA can be efficiently transduced with gammaretroviral vectors in the absence of in vitro prestimulation (Jacome et al., 2006). More recently, to improve the transduction and repopulating properties of FA HSCs, we proposed to transduce unselected hematopoietic samples from these patients over short periods of time, using lentiviral vectors (Jacome et al., 2009). In addition, because of the safer integration pattern of LVs compared with RVs (Schroder et al., 2002; Wu et al., 2003; Mitchell et al., 2004; De Palma et al., 2005; Montini et al., 2006, 2009; González-Murillo et al., 2008), we focused on LVs as therapeutic vectors to correct the phenotype of FA cells.
Because studies have shown that LVs harboring potent internal promoters can also trans-activate neighboring genes (Modlich et al., 2009), we considered that LVs to be used in the clinic should present a compromise between therapeutic efficacy and risks to trans-activate neighboring genes. Our objective was, therefore, to define threshold levels of FANCA expression that could be therapeutic, in order to limit risks of gene trans-activation by the enhancer/promoter that drives the expression of the therapeutic gene.
In relative terms we observed that vav- and PGK-FANCA LVs conferred physiological levels of FANCA mRNA in FA-A LCLs, whereas CMV and more significantly SFFV promoters, conferred supraphysiological mRNA levels of the transgene, per copy of FANCA. Consistent with other studies (Zufferey et al., 1999; Schambach et al., 2006; Zanta-Boussif et al., 2009), the insertion of the WPRE or WPRE* sequence significantly increased FANCA mRNA levels both in LCLs and in primary hematopoietic cells transduced with PGK-FANCA LVs. Because the wild-type WPRE encodes a C-terminal truncated version of the hepatitis virus X protein, which could mediate a tumorigenic effect (Bouchard and Schneider, 2004; Kingsman et al., 2005), LVs with a mutated WPRE* sequence, lacking any residual open reading frame, are considered more adequate for clinical use (Schambach et al., 2006). Our observations showing similar levels of FANCA protein in transduced FA-A LCLs, except in cells transduced with SFFV-FANCA LV, suggest that FANCA may be posttranscriptionally regulated, as has already been shown for FANCC and FANCD2 (Heinrich et al., 2000; Jacquemont and Taniguchi, 2007).
When FANCA expression levels conferred by the various LVs in FA-A LCLs are compared with those observed in HD LCLs (with two copies of FANCA), we can conclude that the insertion of two LV copies per cell may result in physiological levels of the therapeutic protein, except in the case of SFFV LVs, which would confer supraphysiological levels of FANCA. Achieving this copy number may fit the requirements of a clinical trial of patients with FA, where transduction efficacies of at least 50% may be desired because of the low number of progenitors present in the BM of these patients (Larghero et al., 2002; Jacome et al., 2006, 2009; Kelly et al., 2007). Low transduction levels, which would result in the insertion of only one LV copy per cell, would, on the other hand, mediate FANCA expression levels equivalent to those corresponding to heterozygous FA-A carriers, with no FA phenotype.
The functional studies shown in Figs. 1 and 2 confirmed that FANCA expression levels mediated by relatively weak promoters such as the vav or PGK promoter are as efficient as those conferred by potent promoters such as the SFFV promoter. Although insertion of the WPRE* sequence (Schambach et al., 2006) was not necessary to improve the efficacy of PGK-FANCA LVs, this element may offer a redundant system to maintain, in the long term, therapeutic levels of FANCA in the patient.
Our results, together with previous observations showing the stability (Follenzi et al., 2000) and safety properties of PGK LVs (Montini et al., 2006, 2009; Modlich et al., 2009) and also the efficacy of the mutated WPRE* (Schambach et al., 2006), strongly indicate that the PGK-FANCA-WPRE* LV may constitute an efficient and safe vector for the gene therapy of patients with FA-A.
Footnotes
Acknowledgments
The authors are indebted to the patients with FA and their families for their kind cooperation. The authors also thank Aurora de la Cal, Elena Lopez, Sergio Garcia, and Israel Ormán (CIEMAT/CIBERER) for technical assistance. The authors also thank Genethon for kindly providing the pRRLcpptPGKGFP-WPRE-Alb plasmid for copy number quantification in CFCs. This work was supported by grants from the European Program “Life Sciences, Genomics and Biotechnology for Health” (CONSERT; Ref LSHB-CT-2004-5242), the Centro de Investigación en Red de Enfermedades Raras (CIBER-ER), the Ministerio de Ciencia e Innovación (Projects 2009-7164 and PLE 2009-0100), and Genoma España (FANCOGENE). The authors also thank the Fundación Marcelino Botín for promoting translational research at the División de Hematopoyesis y Terapia Génica of CIEMAT.
Author Disclosure Statement
H.H. may receive royalties based on a license agreement between Indiana University and Takara Shuzo, resulting from the sale of the fibronectin fragment CH296 (RetroNectin). The rest of the authors declare no competing financial interests.