
Abstract
Glioblastoma multiforme (GBM) is the most aggressive human brain tumor, and is highly resistant to chemo- and radiotherapy. Selectively replicating oncolytic viruses represent a novel approach for the treatment of neoplastic diseases. Coxsackievirus–adenovirus receptor (CAR) is the primary receptor for adenoviruses, and loss or reduction of CAR greatly decreases adenoviral entry. Understanding the mechanisms regulating CAR expression and localization will contribute to increase the efficacy of oncolytic adenoviruses. Two glioma cell lines (U343MG and U373MG) were infected with the oncolytic adenovirus dl922-947. U373MG cells were more susceptible to cell death after viral infection, compared with U343MG cells. The enhanced sensitivity was paralleled by increased adenoviral entry and CAR mRNA and protein levels in U373MG cells. In addition, U373MG cells displayed a decreased ERK1/2 (extracellular signal-regulated kinase-1/2) nuclear-to-cytosolic ratio, compared with U343MG cells. Intracellular content of PED/PEA-15, an ERK1/2-interacting protein, was also augmented in these cells. Both ERK2 overexpression and genetic silencing of PED/PEA-15 by antisense oligonucleotides increased ERK nuclear accumulation and reduced CAR expression and adenoviral entry. Our data indicate that dl922-947 could represent an useful tool for the treatment of GBM and that PED/PEA-15 modulates CAR expression and adenoviral entry, by sequestering ERK1/2.
Introduction
Treatment normally includes tumor resection, radiation, and chemotherapy; however, GBM cells are largely resistant to chemo- and radiotherapy. Consequently, only a small minority of patients with GBM achieve long-term survival (Furnari et al., 2007; Brandes et al., 2008). Novel treatment strategies are therefore required in order to increase the therapeutic options.
Selectively replicating oncolytic viruses represent a novel platform for the treatment of neoplastic diseases and several studies have been performed showing the feasibility of this therapeutic strategy in patients with glioblastoma (Haseley et al., 2009).
dl922-947 is a selectively replicating oncolytic adenoviral mutant bearing a 24-bp deletion in E1A-conserved region-2 (CR2), necessary for binding and inactivation of the pRb family of proteins (Heise et al., 2000); dl922-947 mutant is unable to induce progression from G1 into the S phase of normal cells, but replicates with high efficiency in cells with an abnormal G1–S checkpoint.
The G1–S checkpoint is critical for cell growth progression (Sherr, 2000) and is abnormal in GBM (Solomon et al., 2008); therefore mutant E1A adenoviruses have been proposed for the therapy of gliomas and are now in preclinical development as antiglioma therapy (Vecil and Lang, 2003).
The efficacy of adenoviral vectors as therapeutic agents depends on the ability of neoplastic cells to bind and internalize adenoviruses. Adenoviral infection involves two distinct virus–cell interactions. First, cell surface attachment is mediated by binding of the viral fiber protein to the cellular coxsackievirus–adenovirus receptor (CAR) (Bergelson et al., 1997; Tomko et al., 2000). CAR is a 46-kDa integral membrane protein and variant isoforms, which differ only at the C terminus and that most likely result from alternative splicing, have also been identified in mice, humans, and rats (Fechner et al., 1999; Coyne and Bergelson, 2005).
Internalization, via receptor-mediated endocytosis, involves interactions between the viral penton protein and cellular integrins, such as αvβ3, αvβ5, α5β1, and α3β1, that act as coreceptors (Nemerow, 2000). Low or absent expression of CAR is seen in many primary tumor tissues (Rein et al., 2006) and low expression of CAR has been observed in grade IV gliomas (Fuxe et al., 2003).
However, the molecular mechanisms by which CAR expression is regulated have been only partially elucidated. Interestingly, inhibition of ERK (extracellular signal-regulated kinase)/MAPK (mitogen-activated protein kinase) pathway has been reported to upregulate CAR expression (Anders et al., 2003).
Here we show that, in two glioblastoma cell lines (Hao et al., 2001; Xiao et al., 2002), the expression of PED/PEA (phosphoprotein enriched in diabetes/phosphoprotein enriched in astrocytes)-15, a protein that binds ERK and prevents its nuclear accumulation (Formstecher et al., 2001; Hill et al., 2002; Renault et al., 2003; Whitehurst et al., 2004; Renganathan et al., 2005), correlates with CAR mRNA levels as well as with the sensitivity to adenoviral infection and dl922-947 killing activity. Indeed, silencing of PED/PEA-15 promotes ERK nuclear translocation and simultaneously reduces CAR expression and adenoviral entry into glioblastoma cells.
Materials and Methods
Cell lines, plasmids, and transfections
Glioma cell lines U343MG and U373MG were purchased from the American Type Culture Collection (Manassas, VA). All cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 IU/ml), streptomycin (100 IU/ml), and 2%
The pcDNA3 vector carrying PED/PEA-15 cDNA was obtained as previously described (Condorelli et al., 1998).
Sequences of scramble and antisense oligonucleotides (Sigma-Aldrich, St. Louis, MO) are as follows: AS-PED/PEA-15 human: 5′-TGACGCCTCTGGAGCTGAGA-3′ Scr-PED/PEA-15 human: 5′-GGCAATTTCGAGCGGCACGT-3′
The plasmid pcDNA3-HA carrying ERK2 cDNA (pcDNA3-HA-pMAPK2) was kindly provided by M. Chiariello (Istituto di Endocrinologia e Oncologia Sperimentale, CNR, Naples, Italy).
Transfection of a pcDNA3 vector carrying PED/PEA-15 cDNA, PED antisense and scramble oligonucleotides, or pcDNA3-HA carrying ERK2 cDNA, was accomplished by the Lipofectamine (Invitrogen, Carlsbad, CA) method as previously described (Condorelli et al., 1998).
Preparation of adenoviruses, infection, and viability assay
dl922-947 is a second-generation adenoviral mutant that has a 24-bp deletion in E1A conserved region-2 (CR2). AdGFP is a nonreplicating E1A-deleted adenovirus encoding green fluorescent protein. Viral stocks were expanded in the human embryonic kidney cell line HEK-293, and purified, as previously reported (Portella et al., 2002).
Stocks were stored at −70°C after the addition of glycerol to a concentration of 50% (v/v). Virus titer was determined by plaque-forming units (pfu) on the HEK-293 cells.
For evaluation of the cytotoxic effects of the dl922-947 virus, 1 × 103 cells were seeded in the wells of 96-well plates, and 24 hr later cells were infected with various multiplicities of infection (MOIs). After 10 days cells were fixed with 10% trichloroacetic acid (TCA) and stained with 0.4% sulforhodamine B in 1% acetic acid (Skehan et al., 1990). The bound dye was solubilized in 200 μl of 10 mM unbuffered Tris solution and the optical density was determined at 490 nm in a microplate reader (Bio-Rad, Munich, Germany). The percent survival rates of treated cells were calculated by assuming the survival rate of untreated cells to be 100%.
For the evaluation of infectivity cells were detached, counted, and plated in 6-well plates at 70% cell density. After 24 hr cells were infected with AdGFP diluted in growth medium at various MOIs; medium was replaced after 2 hr. Cells were washed 24 hr postinfection and then trypsinized and analyzed for GFP expression with a flow cytometer (Dako, Carpinteria, CA) and Summit version 4.3 software (Dako).
Quantitative PCR of dl922-947
To quantify the amount of dl922-947 viral genome, cells were infected with dl922-947 at various MOIs (0.1, 1, and 10 pfu/cell). At 48 hr postinfection, cell supernatant was collected and viral DNA was extracted with a QIAamp DNA mini kit (Qiagen, Valencia, CA) and then quantified by real-time PCR, using assay-specific primer and probe. A real time-based assay was developed with primers 5′-GCCACCGAGACGTACTTCAGCCTG-3′ (upstream primer) and 5′-TTGTACGAGTACGCGGTATCCT-3′ (downstream primer) for amplification of a 143-bp sequence of the viral hexon gene (from bp 99 to 242). For quantification, a standard curve was constructed by assaying serial dilutions of dl922-947 virus ranging from 0.1 to 100 pfu/cell to quantify the input dose.
Detection of cell surface CAR and mRNA quantification
Cells were grown in 6-well plates. After 48 hr cells were detached in phosphate-buffered saline (PBS)–10 mM EDTA, washed with PBS, and then incubated with mouse anti-CAR monoclonal antibody RmcB (Hsu et al., 1988) and a secondary antibody (polyclonal rabbit anti-mouse antibody conjugated to fluorescein isothiocyanate [FITC]; Sigma-Aldrich), and analyzed for CAR expression with a flow cytometer (Dako) and Summit version 4.3 software (Dako). Background emission was subtracted and FITC emission was normalized to control emission.
To block CAR, cells were pretreated with increasing concentrations of the mouse anti-CAR monoclonal antibody RmcB, anti-insulin-like growth factor-1 receptor β subunit (IGF-1R) (Upstate Cell Signaling Solutions/Millipore, Lake Placid, NY), and anti-actin (Santa Cruz Biotechnology, Santa Cruz, CA) antibodies (diluted 1:100, 1:250, and 1:500), for 1 hr at room temperature before addition of virus. Cells were harvested and analyzed as previously described.
To analyze CAR mRNA levels cells were harvested and total RNA was isolated and digested with DNase, using an RNeasy mini kit (Qiagen) according to the manufacturer's recommendations. One microgram of tissue or cell RNA from each sample was reverse transcribed, using SuperScript II reverse transcriptase (Invitrogen). PCR products were analyzed with SYBR green mix (Invitrogen). Reactions were performed with Platinum SYBR green qPCR SuperMix-UDG, using an iCycler iQ multicolor real-time PCR detection system (Bio-Rad). All reactions were performed in triplicate, and β-actin was used as an internal standard.
The primer sequences were as follows: Cxadr forward (5′-ATGAAAAGGAAGTTCATCAACGTA-3′) and Cxadr reverse (5′-AATGATTACTGCCGATGTAGCTT-3′), generating an amplicon of 93 nucleotides scattered among exons 6 and 7; and β-actin forward (5′-GCGTGACATCAAAGAGAAG-3′) and β-actin reverse (5′-ACTGTGTTGGCATAGAGG-3′).
The conditions used for PCR were 10 min at 95°C and then 45 cycles of 20 sec at 95°C and 1 min at 60°C. To calculate the relative expression levels, we used the 2–
Protein extraction, cell subfractionation, and Western blot analysis
In all experiments, 70% confluent cells were used. Subcellular fractionation was performed by a previously described method (Ruvolo et al., 1998). Briefly, cells were broken in ice-cold hypotonic HEPES buffer (10 mM HEPES [pH 7.4], 5 mM MgCl2, 40 mM KCl, 1 mM phenylmethylsulfonyl fluoride, aprotinin [10 g/ml], leupeptin [10 g/ml]). Broken cells were centrifuged at 200 × g to pellet the nuclei. The resulting supernatants were centrifuged at 10,000 × g to pellet the heavy membrane fraction. The last supernatant represented the cytosolic fraction. The nuclear membranes were isolated by centrifugation of the nuclei through a 2 M sucrose cushion at 150,000 × g. For protein extraction cells were homogenized directly into lysis buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, aprotinin [1 (g/ml], 0.5 mM sodium orthovanadate, 20 mM sodium pyrophosphate). The lysates were clarified by 20 min of centrifugation at 14,000 × g. Protein concentrations were estimated by a Bio-Rad assay, and then proteins were boiled in Laemmli buffer for 5 min before electrophoresis. Proteins were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (10% polyacrylamide) under reducing conditions. After electrophoresis, proteins were transferred to nitrocellulose membranes (Immobilon; Millipore). After blocking with Tris-buffered saline–bovine serum albumin (TBS–BSA), the membrane was incubated with primary antibody: polyclonal rabbit antibody against CAR (SC-15-405, diluted 1:250; Santa Cruz Biotechnology), rabbit anti-PED serum (diluted 1:2000; previously described [Condorelli et al., 1998]), rabbit antibody against ERK1 and ERK2 (diluted 1:1000), rabbit anti-IGF-1R (diluted 1:1000; Upstate Cell Signaling Solutions/Millipore), or rabbit anti-H1 histone (diluted 1:1000; Upstate Cell Signaling Solutions/Millipore), or with the rabbit antibody against actin (diluted 1:2000; Santa Cruz Biotechnology) for an overnight incubation.
Membranes were then incubated with horseradish peroxidase-conjugated secondary antibody (diluted 1:2000) for 45 min (at room temperature) and the reaction was detected with an enhanced chemiluminescence (ECL) system (GE Healthcare Life Sciences, Chalfont St Giles, UK).
Densitometric analysis was performed with Scion Image (Scion, Frederick, MD). All data were expressed as means± SD.
Results
U373MG and U343MG cells show different sensitivities to adenoviral infection
First, we evaluated the antineoplastic activity of the selectively replicating oncolytic adenovirus dl922-947 against U373MG and U343MG glioma cell lines (Fig. 1A).
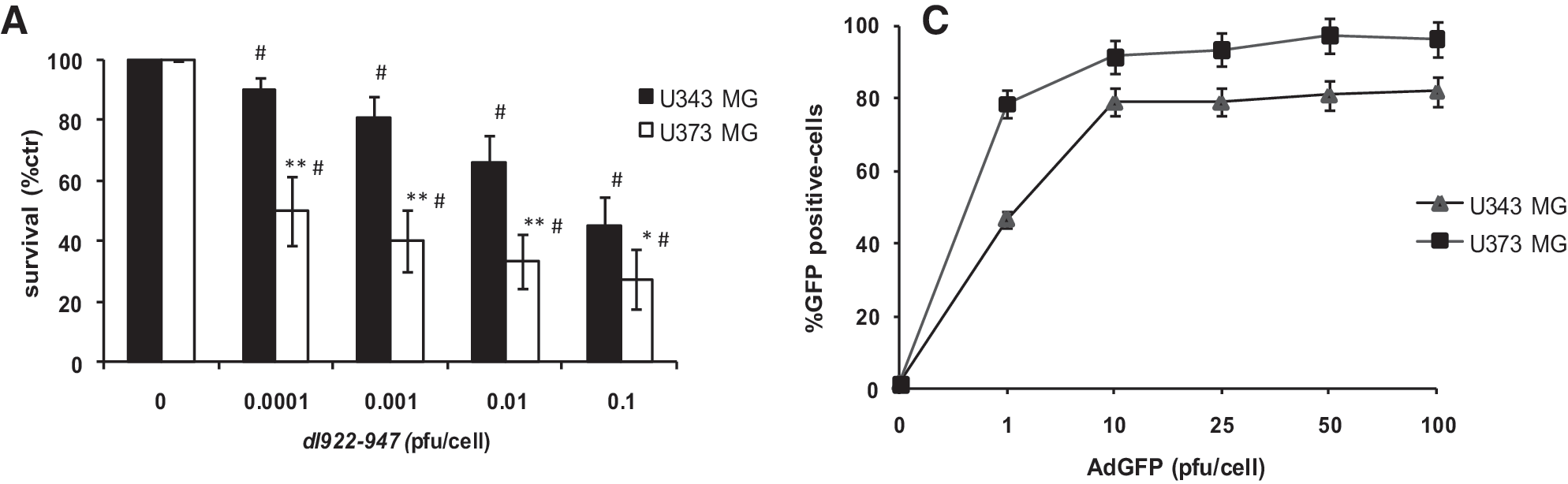
Comparison of the cell-killing activity, replication of dl922-947, and infectivity in U343MG and U373MG glioblastoma cell lines. (
Cells were infected with various MOIs of dl922-947 and cell survival was evaluated after 7 days. The U373MG cell line displayed higher sensitivity to dl922-947, with a 50% inhibitory concentration (IC50) at an MOI of 0.0001 pfu/cell, whereas for U343MG the IC50 was observed at an MOI of 0.1 pfu/cell (Fig. 1A).
Our data indicate that both glioma cell lines are sensitive to dl922-947, although displaying different sensitivities to its oncolytic activity. Genome equivalent copies analysis showed that both cell lines sustain the replication of dl922-947 (Fig. 1B).
To study whether this difference could be due to dissimilar infection efficiency, we used a nonreplicating reporter adenovirus encoding the green fluorescent protein (AdGFP), as previously reported (Watanabe et al., 2006). Cells were infected with various MOIs of AdGFP and 24 hr postinfection the amount of GFP-positive cells was quantified by flow cytometric analysis.
At 1 pfu/cell of AdGFP 80% of U373MG cells showed positivity for GFP expression, whereas a higher viral dose (10 pfu/cell) was required to obtain the same percentage of GFP-positive U343MG cells. Starting from 10 pfu/cell no further increase in GFP-positive cells was observed in either cell line.
These differences in infection efficiency could explain the differences in viral replication, because at 0.1 and 1 pfu/cell a highly significant difference (p < 0.001) in genome copies of dl922-947 was detected in U373MG compared with U343MG.
Conversely, at 10 pfu/cell the differences in genome equivalent copies of dl922-947 were less evident (p < 0.05).
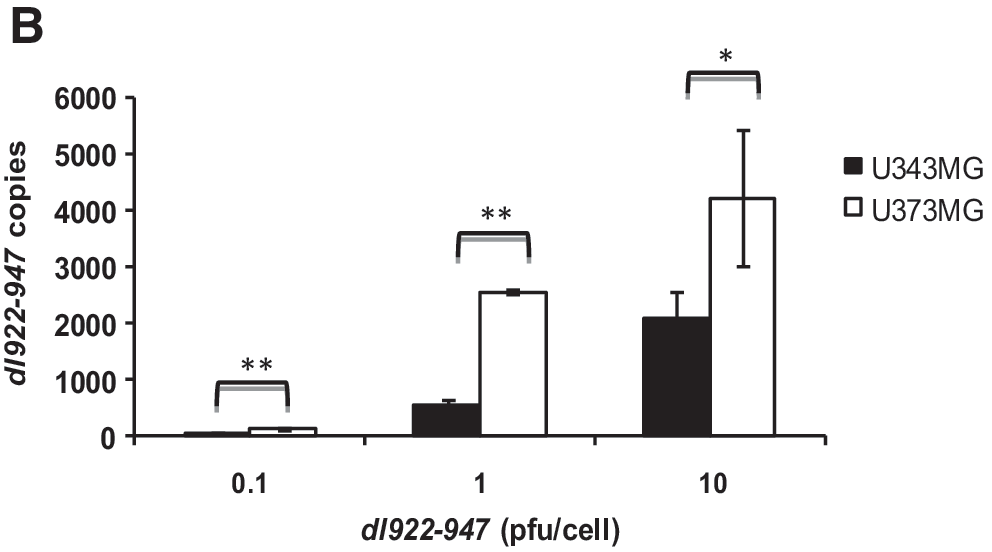
Infectivity of U343MG and U373MG cells is mediated by coxsackievirus–adenovirus receptor
It has been reported that CAR is the main mediator of adenoviral entry (Bergelson et al., 1997; Nemerow, 2000; Tomko et al., 2000; Arnberg, 2009). Therefore, we analyzed CAR expression by Western blot and cytofluorimetric analysis in U343MG and U373MG cell lines. Two CAR-specific bands of 44 and 46 kDa, respectively, were detected in all samples (Fig. 2A), as previously reported (Cohen et al., 2001; Libertini et al., 2007).
Coxsackievirus–adenovirus receptor (CAR) in U343MG and U373MG cells. (
U373MG cells displayed higher total levels (Fig. 2A) and membrane levels (Fig. 2B) of CAR, compared with U343MG cells; this observation parallels the higher viral entry and sensitivity to the oncolytic activity of dl922-947. To evaluate whether CAR may play a direct role in infection efficiency, U343MG and U373MG cells were pretreated for 1 hr with increasing amounts of the blocking anti-CAR monoclonal antibody RmcB and infected with AdGFP at 25 pfu/cell. We also pretreated cells with two control antibodies: anti-IGF-1R and anti-actin. Significant decreases in GFP emission were observed in U373MG cells (p < 0.005) and U343MG cells (p < 0.001), upon treatment with RmcB, but not upon treatment with anti-IGF-1R or anti-actin antibodies (Fig. 2C and D). These data confirm that CAR plays a crucial role in infection efficiency in both cell lines.
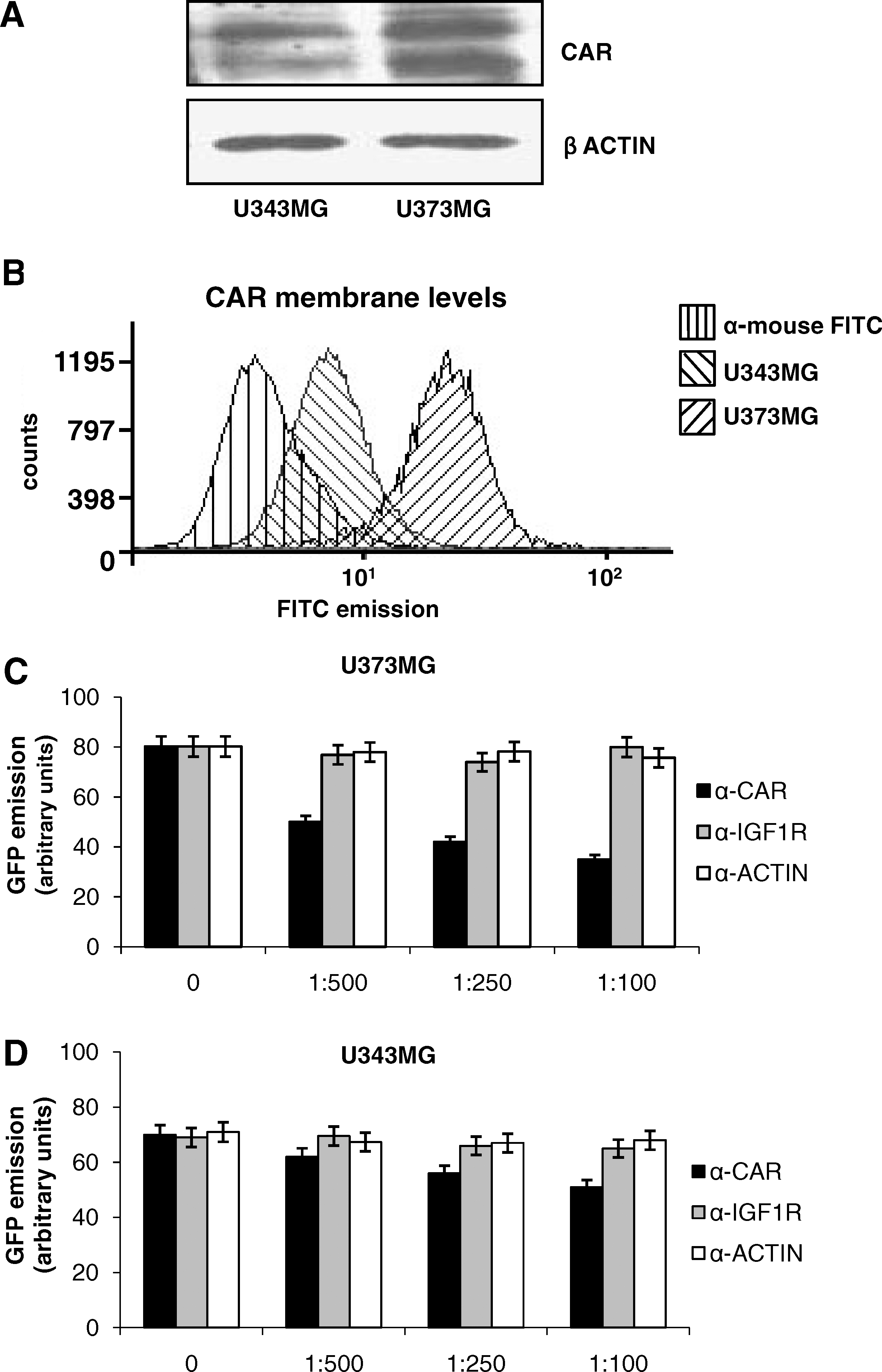
ERK modulates U373MG cell line infectivity by regulating CAR expression
It has been described that ERK signaling regulates CAR levels in cancer cell lines (Anders et al., 2003). Therefore, we evaluated ERK1/2 phosphorylation and subcellular localization in U373MG and U343MG cells. No difference in ERK1/2 phosphorylation was detected in the two cell lines (Fig. 3A). However, in U373MG cells, ERK1/2 cytosolic content was higher than in U343MG cells. Conversely, U343MG cells displayed higher levels of ERK1/2 in the nucleus than did U373MG cells (Fig. 3B), suggesting that ERK1/2 nuclear localization might contribute to downregulate CAR expression.
ERK pathway regulates CAR expression in glioma cells. (
Next, a plasmid carrying ERK2 cDNA (pcDNA3-HA-pERK2) was transiently transfected into U373MG cells, in order to force ERK into the nucleus. Transfection efficiency was evaluated by Western blot (Fig. 3C). ERK2 overexpression was paralleled by increased detection of ERK2 in the nuclei and by a reduction of CAR total and membrane levels (Fig. 3C). CAR mRNA levels, evaluated by reverse transcription (RT) real-time PCR, also showed a reduction of about 80% (Fig. 3D), thus suggesting that an ERK nuclear shift downregulates CAR gene expression. Accordingly, a significant reduction in GFP emission was observed in ERK2-transfected U373MG cells, after infection with AdGFP (Fig. 3E).
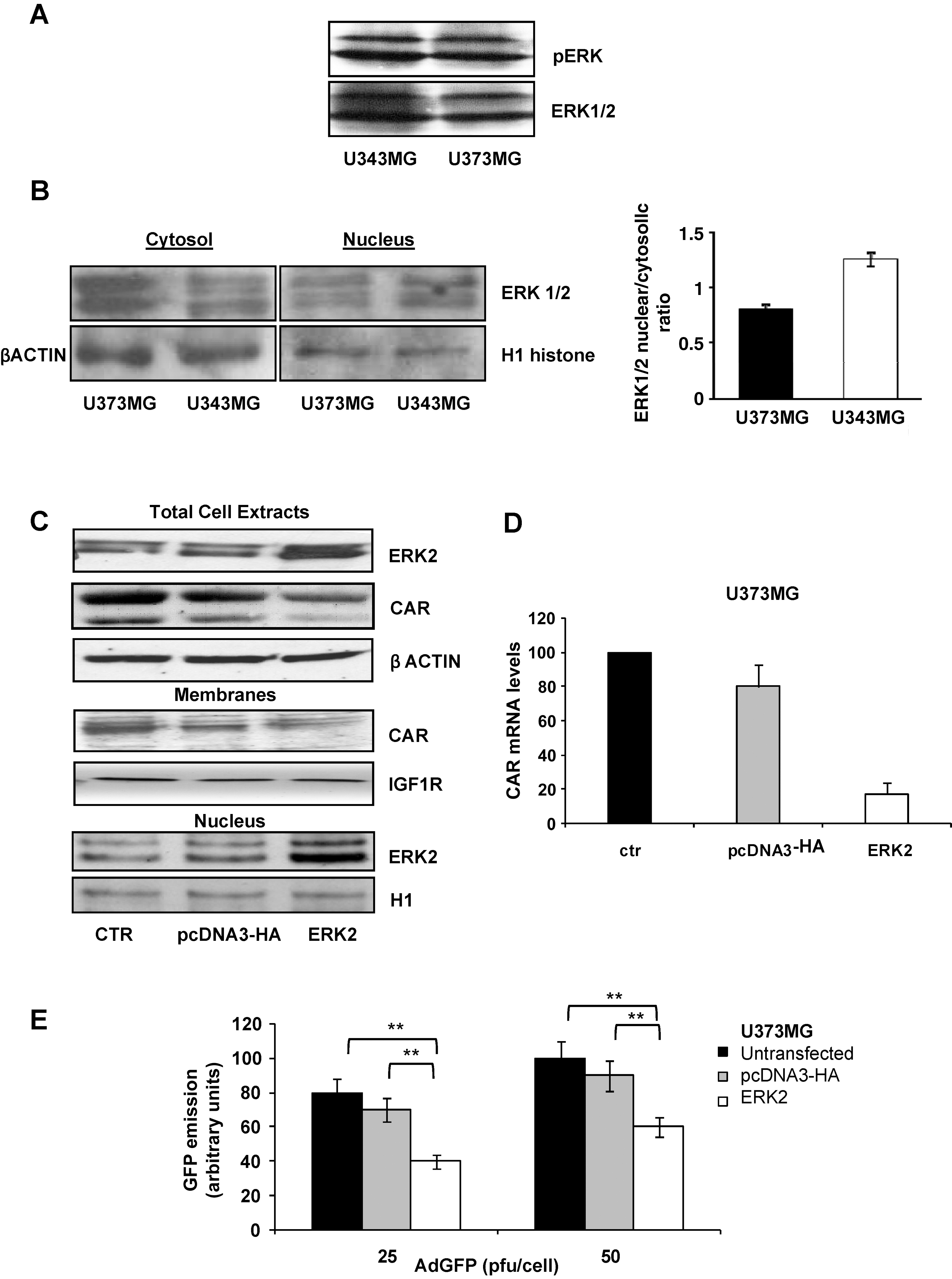
PED/PEA-15 modulates ERK localization and CAR expression
PED/PEA-15 is a death effector domain-containing protein, which is involved in the regulation of apoptotic cell death (Hao et al., 2001; Xiao et al., 2002). PED/PEA-15 is highly expressed in cells of glial origin (Hao et al., 2001; Xiao et al., 2002; Sharif et al., 2004). Moreover, it has been reported that PED/PEA-15 inhibits nuclear translocation and activity of ERK1/2 (Formstecher et al., 2001; Hill et al., 2002; Renault et al., 2003; Whitehurst et al., 2004; Renganathan et al., 2005).
As previously reported (Hao et al., 2001), PED/PEA-15 levels were higher in U373MG cells than in U343MG cells (Fig. 4A). To assess whether PED/PEA-15 may control ERK localization and CAR expression, a PED/PEA-15 antisense oligonucleotide (PED-As) was transfected into U373MG cells, using a scrambled oligonucleotide (PED-Scr) as control, and PED/PEA-15 levels in transfected cells were evaluated by Western blot.

PED/PEA-15 antisense (PED As) transfection. (
After PED-As transfection, a decrease in CAR total levels was observed (Fig. 4B) and the nuclear-to-cytosolic ratio of ERK distribution was shifted toward the nucleus (Fig. 4C).
To further confirm the role of PED/PEA-15 in the expression of CAR, U343MG cells were transfected with a plasmid carrying PED/PEA-15 cDNA, showing an increase in CAR total levels (Fig. 4D).
Downregulation of PED/PEA-15 decreases CAR levels, adenoviral infectivity, and sensitivity to dl922-947 in U373MG cells
Downregulation of PED/PEA-15 was accompanied by a reduction in total (Fig. 4B) and membrane (Fig. 5A) CAR levels, respectively. Transfection of U373MG cells with the scrambled oligonucleotide (PED-Scr) led to a slight reduction (about 30%) of CAR membrane levels, compared with untransfected cells. However, PED-As-transfected cells showed a significant decrease in CAR membrane levels with respect to PED-Scr-transfected cells (p < 0.05).
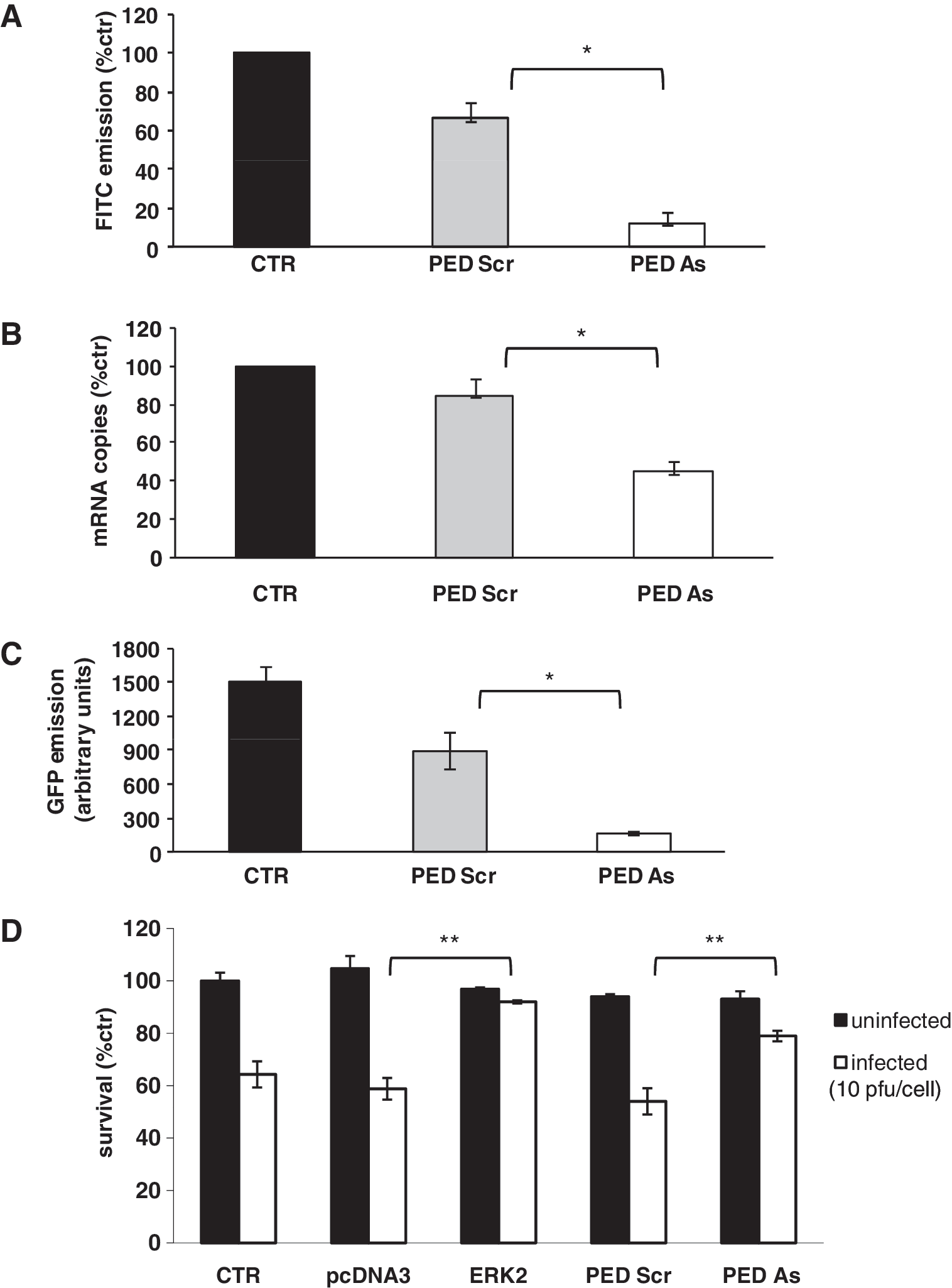
Transfection with PED/PEA-15 antisense (PED As) reduces CAR levels, infectivity, and sensitivity in U373MG cells. (
RT real-time PCR experiments showed an approximately 50% reduction of CAR mRNA levels (p < 0.05) (Fig. 5B) after transfection with PED-As and a significant decrease in GFP emission was observed in PED-As-transfected cells after infection with AdGFP at 25 pfu/cell (p < 0.05) (Fig. 5C).
These data indicate that PED/PEA-15 plays a role in controlling the regulation of CAR expression and in adenoviral infectivity in glioma cells. To confirm that the downregulation of PED/PEA-15 reduces sensitivity to the oncolytic virus dl922-947, U373MG cells were transfected with PED-As or PED-Scr.
U373MG cells were also transfected with pcDNA3-HA pERK2 or pcDNA3-HA plasmid as controls. Forty-eight hours after transfection cells were infected with dl922-947 (10 pfu/cell) and after 48 hr cell survival was analyzed (Fig. 5D). A highly significant (p < 0.001) increase in cell survival after infection was observed in PED As- and ERK2-transfected cells with respect to untransfected or transfected controls (p < 0.001).
Discussion
GBM is surgically incurable in the vast majority of patients (Brandes et al., 2008), with median survival duration of about 9–15 months (Furnari et al., 2007). The protocol of adjuvant therapy, radiation followed by chemotherapy, administered after surgery, has demonstrated only a moderate increase in survival (Argyriou et al., 2009). Therefore novel therapeutic approaches are required. Genetically engineered, conditionally replicating viruses are promising therapeutic agents for cancer and several oncolytic viruses have already been tested in preclinical or clinical studies for the treatment of gliomas (Jiang et al., 2007).
In the present study, we have observed that dl922-947 is active against two glioblastoma cell lines, U343MG and U373MG, reinforcing the concept that the therapy of glioblastoma could benefit from the use of oncolytic viruses. However, a differential sensitivity to the oncolytic activity of dl922-947 was evidenced in the two cell lines, with U343MG cells being more resistant to the virus.
This difference could potentially be due to factors affecting the viral life cycle (such as attachment, entry, viral gene expression, etc.). It is generally accepted that poor adenoviral entry in neoplastic cells represents the most important obstacle for an effective therapy based on replicating oncolytic adenoviruses (Vähä-Koskela et al., 2007).
Coxsackievirus–adenovirus receptor (CAR) is the primary receptor for adenoviruses, and loss or reduction of CAR greatly decreases adenoviral entry (Bergelson et al., 1997; Nemerow, 2000; Tomko et al., 2000; Rein et al., 2006). Higher levels of CAR expression were observed in U373MG cells, as compared with U343MG cells, and this was paralleled by increased infection efficiency. However, a significant reduction was observed in both cell lines blocking the receptor with an anti-CAR antibody.
Although complete abrogation of adenoviral entry was not obtained, possibly because of residual entry via alternative pathways (Arnberg et al., 2009) or subtotal blockade with the antibody, our data are consistent with the hypothesis that CAR-mediated internalization plays a major role in both cell lines.
It has been demonstrated that disruption of signaling through the Raf/MEK (MAPK/ERK kinase)/ERK pathway by MEK inhibitors (U0126 and PD184352) upregulates CAR expression (Anders et al., 2003). Interestingly, U373MG cells displayed higher ERK1/2 cytosolic localization, compared with U343MG cells, in which ERK1/2 was mostly nuclear. Because nuclear translocation is a crucial step for ERK-mediated regulation of gene expression, we hypothesized that ERK nuclear activity could control CAR expression. Indeed, overexpression of ERK2 in U373MG cells was accompanied by forced nuclear localization and decreased CAR mRNA and protein levels, leading to a reduction in infection efficiency.
PED/PEA-15 is a death effector domain-containing protein involved in the regulation of apoptotic cell death and highly expressed in cells of glial origin (Hao et al., 2001; Xiao et al., 2002). PED/PEA-15 regulates the ERK/MAPK pathway by binding ERK1/2 and preventing its nuclear accumulation and activity (Formstecher et al., 2001; Hill et al., 2002; Renault et al., 2003; Whitehurst et al., 2004; Renganathan et al., 2005). Moreover, abrogation of ERK1/2 binding as a result of point mutations in PED/PEA-15 restores normal ERK1/2 function (Whitehurst et al., 2004). We have hypothesized that PED/PEA-15 could be involved in CAR regulation and adenoviral infectivity by controlling ERK subcellular distribution. Indeed, PED/PEA-15 levels are higher in U373MG cells than in U343MG cells and positively correlate with the relative ERK cytosolic abundance, CAR levels, adenoviral infectivity, and dl922-947 killing capacity.
In U373MG cells genetic silencing of PED/PEA-15 with a specific antisense oligonucleotide enhanced ERK1/2 nuclear distribution and led to a reduction of CAR levels and sensitivity to the oncolytic adenovirus dl922-947. Conversely, overexpression of PED/PEA-15 increased CAR total levels in U343MG cells.
Our data show that PED/PEA-15 levels correlate with infectivity and sensitivity to oncolytic adenoviruses and suggested that, in association with CAR, the evaluation of PED/PEA-15 levels could represent a useful tool to guide patients with glioblastoma toward specific therapeutic options.
It is important to note that PED/PEA-15 is involved in the regulation of apoptotic cell death (Hao et al., 2001; Condorelli et al., 2002; Xiao et al., 2002) and it has been demonstrated that, in glioma cell lines, the apoptotic cascade activated by tumor necrosis factor
These findings indicate that PED/PEA-15 increases the resistance to apoptotic agents, and that PED/PEA-15 expression could predict resistance to apoptosis. It is possible to hypothesize that patients with high PED/PEA-15 levels should not benefit from therapy based on these agents, whereas our data indicate that these patients could benefit from adenovirus-based therapy.
Therefore, PED/PEA-15 expression levels could play a dual predictive role: as a marker of resistance to chemo- and radiotherapy for its antiapoptotic activity and as a marker of sensitivity to adenovirus-based therapies, for its role on CAR expression.
In conclusion, our data show that adenoviral infectivity is mostly CAR-mediated in glioblastoma cells and that PED/PEA-15 upregulates CAR expression, by preventing ERK nuclear translocation. Further studies are required to clearly assess the role of PED/PEA-15 as a predictive marker in glioblastoma.
Footnotes
Acknowledgments
The authors thank Dr. G. Hallden for kindly providing RmcB antibody and Dr. M. Chiariello for the gift of plasmid pcDNA3-HA-pERK2. This study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) and by the Italian Ministry of Instruction, University and Research. S.L. is the recipient of a grant from the Fondazione Italiana per la Ricerca sul Cancro (FIRC).
Author Disclosure Statement
No competing financial interests exist.