
Abstract
Destruction of cancer cells by genetically modified viral and nonviral vectors has been the aim of many research programs. The ability to target cytotoxic gene therapies to the cells of interest is an essential prerequisite, and the treatment has always had the potential to provide better and more long-lasting therapy than existing chemotherapies. However, the potency of these infectious agents requires effective testing systems, in which hypotheses can be explored both in vitro and in vivo before the establishment of clinical trials in humans. The real prospect of off-target effects should be eliminated in the preclinical stage, if current prejudices against such therapies are to be overcome. In this review we have set out, using adenoviral vectors as a commonly used example, to discuss some of the key parameters required to develop more effective testing, and to critically assess the current cellular models for the development and testing of prostate cancer biotherapy. Only by developing models that more closely mirror human tissues will we be able to translate literature publications into clinical trials and hence into acceptable alternative treatments for the most commonly diagnosed cancer in humans.
Introduction
Validity of Cell Lines That Represent Prostatic Disease
Prostate cancer has been poorly served by the establishment of cell lines. In general, new agents for prostate cancer are tested mainly in three cell types; PC3, LNCaP, and DU145. A review of the gene therapy literature suggests there are 2650 PubMed references that include the terms prostate cancer and gene therapy. Of those, 562 employ only LNCaP cells; PC3 cells are used in 153 and DU145 cells in 179. The remainder employ a mixture of all three. In reality, only relatively few published studies make it through to the clinic. The Journal of Gene Medicine database (March 2009;
The LNCaP, PC3, and DU145 cell lines were derived almost 30 years ago from therapy-resistant metastatic cancers (Table 1). The TSU-Pr1 cell line was also considered to be from prostate, but was subsequently shown to be derived from the human bladder cancer cell line T24 (van Bokhoven et al., 2001). What is frequently lacking from these models is any representation of untreated low Gleason grade tumors within the prostate (Gleason, 1966), which have a high degree of normal prostate differentiation, or less structured, untreated but “undifferentiated” higher Gleason grade tumors. As screening and diagnostic techniques improve for prostate cancer (Schröder et al., 2009), “clinical” prostate cancers will be downgraded to lower Gleason grades, which will comprise the primary target for new therapies. At present, however, most biotherapies are used as a “last resort” after failure of hormone therapy, radiotherapy, and chemotherapy.
Abbreviations: ATCC, American Type Culture Collection; HPV, human papilloma virus; hTERT, human telomerase reverse transcriptase; ND, not determined; SV40, simian virus 40.
Can be induced to express androgen receptor and luminal markers by culture in three dimensions and/or androgens.
It is common in studies of new agents for prostate cancer to demonstrate their efficacy on one or more cancer cell lines, but only rarely do the experiments offer any estimation of damage to the normal prostate, as the prostate is considered a nonessential organ. Nonmalignant cell lines (Maitland et al., 2001; Peehl, 2005) are listed in Table 1 along with many of the existing cancer cell lines. Primary cell cultures (see later), grown in a serum-free or low-serum medium, are frequently used as a comparator with the established cancer cell lines. These primary cultures are of a basal cell type, unless induced to differentiate by growth in a medium supplemented with serum, calcium, and androgens (Collins et al., 2005). Similarly, most nonmalignant epithelial cell lines from prostate are also basal in phenotype, and express cytokeratin-5/14, but not the androgen receptor.
Thus, when studying vector-mediated gene transfer efficiencies in prostate cell cultures, one must be careful which parameter is under study, for example, between androgen-sensitive and androgen-insensitive cancers. In this case the comparison is frequently between LNCaP and PC3/DU145. This is more precisely a comparison of luminal cells and basal cells, and a better comparison would be LNCaP with one of the many derived androgen receptor-negative subclones such as C4-2 (Wu et al., 1994). The comparison of the more recently established wild-type androgen receptor-expressing PC346C with one of its androgen receptor-negative subclones provides an even more clinically relevant alternative (Marques et al., 2005, 2006).
It was the aim of the GIANT (Gene Therapy: An Integrated Approach to Neoplastic Treatment) program (
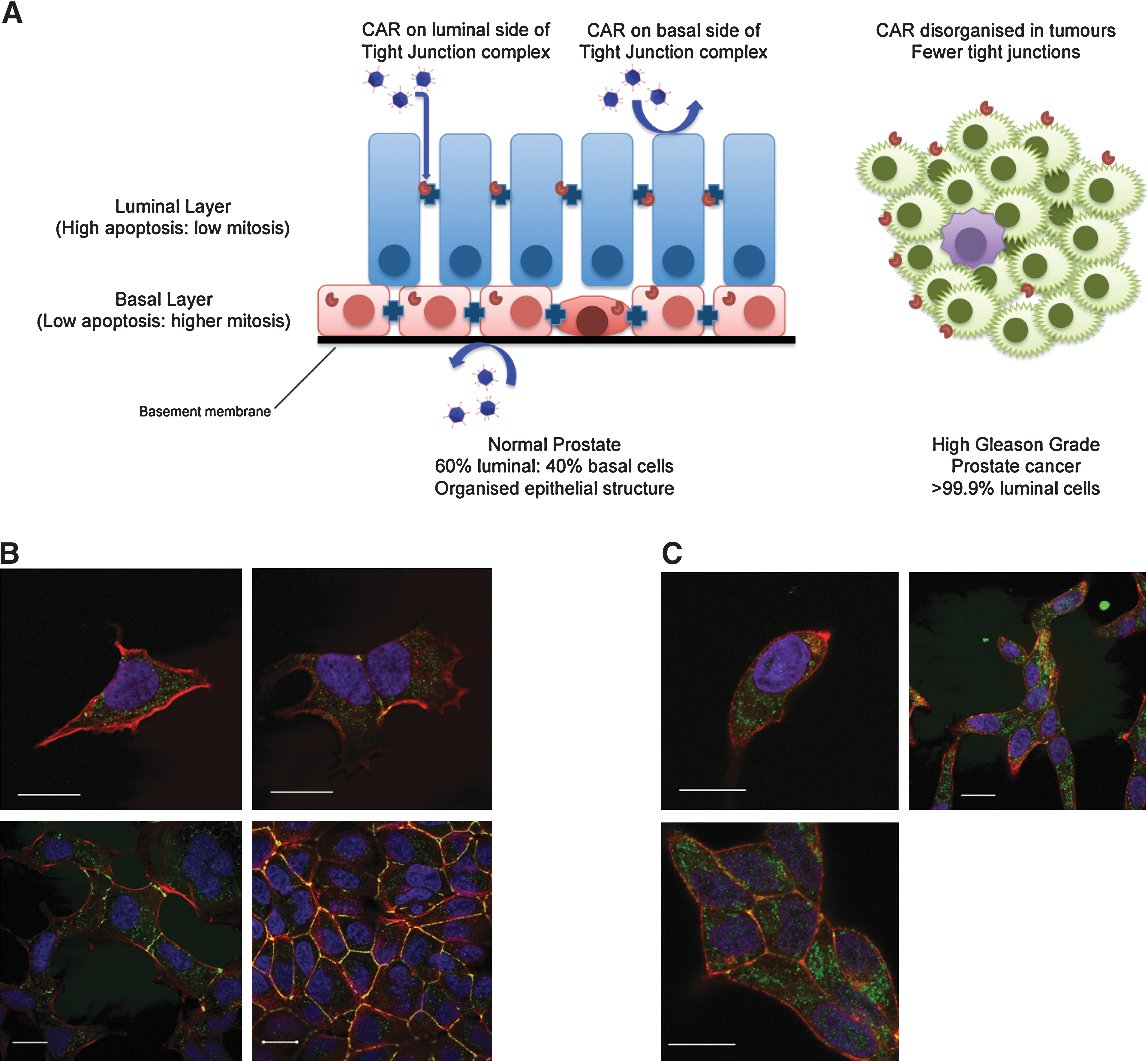
A model for virus binding and penetration in prostate epithelium. (
Toward Better Preclinical Systems: Vector Adhesion in Two-Dimensional Cell Cultures
Initial testing of infectious agents in cell cultures is often optimized for viral replication or infectivity, for example, attachment of viruses to the cell surface. Many viruses use adhesion molecules as receptors and adenovirus is no exception (reviewed in Greber and Gastaldelli, 2007). For example, the main adenovirus receptor, the coxsackievirus–adenovirus receptor (CAR), forms homodimers between contacting cells and serves as a cell adhesion molecule (van Raaij et al., 2000), but adenoviruses also require a secondary receptor in the form of integrins for high-efficiency transduction. Of these, αvβ3 is probably the key integrin involved in virus uptake (Wickham et al., 1993; Nemerow, 2000). CAR protein is also expressed in mobile membrane lipid rafts (Ashbourne Excoffon et al., 2003) and coimmunoprecipitates with ZO-1, a tight junction protein (Cohen et al., 2001).
In normal prostate tissues, CAR is located at the lateral junctions between luminal cells, and at the apical border, whereas in basal cells mostly cytoplasmic expression is observed (Rauen et al., 2002). Prostate cancer is mainly luminal in phenotype, which suggests it should express more CAR, as application of hormones to rat seminal vesicle and prostate epithelium results in a reorganization and differentiation-regulated increase in junction formation (Ortiz and Cavicchia, 1990; Mitra et al., 2006). However, in sections of prostate cancer, the expression of CAR is lost at the cell junctions and also decreases at the protein level (Li et al., 1999; Okegawa, 2000). Therefore the viral vectors may find alterative means of entry, for example, adenovirus may enter through direct contact of the penton base with integrins or other receptors. The loss of CAR was confirmed in our laboratory, where we could not detect expression at the apical border of primary prostate cancer samples (K. Chambers, unpublished data). Although the common productive cell line for adenoviral culture, HEK-293, has extensive intracellular CAR (Fig. 1B), in prostate cancer cell lines, such as LNCaP, CAR is present at lateral junctions but is expressed mainly in the cytoplasm of single cells without adhesions (Fig. 1C; and K. Chambers, unpublished data). CAR is, however, still detectable in subconfluent prostate cancer cell cultures even after disruption of the junctions for flow cytometric analysis (Pandha et al., 2003).
Because of the expression of CAR at the tight junctions, the availability of the receptor from the luminal surface is blocked by their proximity to or incorporation into tight intracellular junctions (Balda and Matter, 1998; Cohen et al., 2001). Access and spread can be governed by restricted access to receptor complexes, as the mean distance between cells in tight and gap junctions is 1.5 and 4 nm, respectively, whereas the full diameter of many nonviral particles is at least 50 nm and that of the rigid adenoviral particle is 100 nm.
With prostate cancers, there is also the question of the cell surface available to be infected in a structured and polarized epithelium (present in most lower Gleason grade tumors). In vivo, it is likely that infection will occur through the luminal surface (see Fig. 1A) rather than through basal contact via the basement membrane, which adenoviruses can fail to penetrate, although genetic modification of the virus by replacement of the E3 gene to encode the relaxin-degradative enzyme can improve penetration (Kim et al., 2006). Other strategies to improve infectivity via CAR include internalization of tight junction proteins such as occludin to expose the tight junction receptor CAR, without affecting the distribution of other tight junctional proteins (Coyne et al., 2007), and increased exposure of CAR through actin remodeling (Coyne and Bergelson, 2005).
In tissues, the damage induced by direct injection results in effective infection restricted to a few millimeters around needle tracks (Patel et al., 2009). Therefore it will probably never be possible in vivo to saturate a tumor with prodrug-activating cells after gene transduction by intratumoral injection of even high titers of the most efficient vectors, without facilitating intratumoral spread. It was for this reason that an oncolytic approach was taken for the initial trials in GIANT (Cheng et al., 2006), where intracellular spread occurs after infected cell lysis.
However, in immunocompetent patients such spread is a matter of timing between the triggering of a potent immune response against adenoviruses and the ability of the virus to reinfect multiple times. Although the immune response may be restrictive in the spread of the virus (Parato et al., 2005), there is evidence to suggest that patients who respond immunologically to a therapeutic viral infection also mount a powerful immune response against tumor cell antigens. The strong adjuvant effect of high levels of viral antigens is thought to be responsible, resulting in regression of not only the primary tumor, but also of distant metastases (Freytag et al., 2002). This type of outcome emphasizes the need to use virotherapies in immunocompetent patients to achieve maximal therapeutic efficacy. Predictably, in most phase 1 (in which safety assessment is the object) but more surprisingly in phase 2 (in which efficacy is sought) clinical trials of biotherapies, this is seldom the case.
Primary Tissue Infections
An excellent model, which takes into account differential patient susceptibility to infection, is direct infection of needle biopsies from fresh prostate cancer tissues, in which tissue architecture and viability can be preserved in culture for a period of up to 2 weeks in a basal medium. Such tests provide, on a patient-to-patient basis, a measurement of susceptibility not only to gene therapy agents but also to many common chemotherapies. However, ethical restrictions in obtaining fresh tissues, or the risk of compromising pathological diagnosis, can compromise the quantities of tissue available to do such studies in a rational and quantitative way.
A good alternative would be to establish and test the transduction of fragments of tissue as primary xenografts in immunocompromised mice. Short-term “culture” of human tissues is possible in xenografts, with maintenance of both tissue architecture and hormone responsiveness at subcutaneous, subrenal capsule, and orthotopic mouse prostate sites. Although the production of such xenografts can be labor intensive, and they are limited in size and scope for the testing of agents, they do offer the best measurement of therapeutic effectiveness as a patient-specific medicine, the “gold standard” for sophisticated and targeted biotherapies. Within the GIANT program we have used xenograft models, established for many years (Table 2).
Abbreviations: AD, androgen dependent; AI, androgen independent; AR, androgen receptor; LN, lymph node metastasis; Met, metastasis; N/A, not applicable; PC, prostate cancer; PSA, prostate-specific antigen; TURP, transurethral resection of the prostate.
Reproduced with permission from van Weerden et al. (2009).
Primary Epithelial Cell Cultures
As an interim solution, in vitro cultures of primary prostate cancers can be employed. Primary cells have a limited life span in culture, especially in medium containing high fetal calf serum concentrations, which is used to promote rapid growth of not only the primary epithelial cultures, but also the fibroblasts. These culture conditions also stimulate the terminal differentiation of basal epithelial cells. Normal prostate luminal epithelium has a limited ability to persist in vitro and in vivo, whereas it is the basal epithelium that is responsible for driving organ regeneration after castration (Isaacs and Kyprianou, 1987) and has a much higher frequency of mitosis compared with the luminal epithelium. Apoptotic rates are higher (De Marzo et al., 1998) and mitoses are barely detectable in normal luminal cells (Fig. 1), which can be considered the end-stage of a differentiation process, in which cells are shed into the lumen and often appear in the bloodstream as apoptotic or preapoptotic bodies. This is also true in tumors, where circulating tumor cells are mostly of a differentiated phenotype (PSA+, CK18+), many of which are not viable (Riethdorf and Pantel, 2008). It is perhaps due to the high apoptotic rate in the luminal fraction that successful primary cultures of normal human prostate are of a basal cell type. We have not yet solved the problem of both differentiation and apoptosis within these cultures, which is frequently linked to the presence of high amounts of prostate stroma copurified from the dissociated tissues (Cunha, 1984; Hall et al., 2002). To eliminate or reduce the stromal content, cholera toxin can be used along with other epithelial growth-promoting factors within the cell culture medium (Chung et al., 1988; Marques et al., 2005). Tissue dissociation and cell fractionation, for example, by means of immunomagnetic beads, effectively removes all prostate stroma and additional purification by promoting adherence to basement membrane or collagen provides further reduction of both luminal and stromal cells (Richardson et al., 2004).
To at least partly restore luminal differentiation, reconstructions of the multilayered and multicellular prostatic epithelium can be achieved in two dimensions by allowing the epithelial cells to grow out of monolayer and form a bilayer. Induction of luminal differentiation is further enhanced by addition of prostate androgen-responsive stroma and medium supplements such as dihydrotestosterone and calcium, resulting in a polarization of the epithelial layers. At this point the characteristic patterns of expression of cell surface receptors are found, as gene expression switches from basal to luminal cytokeratin expression in the upper (luminal) layer (Swift et al., 2010).
Three Dimensions: Better Than Two?
Whereas two-dimensional cell culture presents a single surface for virus infection, tissues exist in vivo as multiple layers bordered by tight junctions and desmosomes, and containing a multitude of different structurally differentiated cell types, which will inhibit vector penetration. One way to model this is to generate three-dimensional structures in cell culture. To assess the effects of differentiation, three-dimensional cultures can be induced to both polarize and differentiate by suspension in an appropriate matrix, the stimulus to polarize being provided by signals from prostate stromal cells (Lang et al., 2001).
Adenoviruses have a limited ability to extravasate from blood vessels, unless the capillaries are severely damaged or are irregular in development, such as is the case with intratumoral capillaries (Wang and Yuan, 2006). When adenovirus was administered via the intravenous route in mice xenografted with a human prostate tumor, infection was observed mainly in endothelial cells of healthy vessels in the tumor. Few infected tumor cells were detectable after intravenous administration. However, when virus was administered intratumorally in the same model, many infected tumor cells were detected, although the distribution pattern remained concentrated around the needle track (R. Kraaij, unpublished). Complex three-dimensional structures in vitro can also address the issue of penetration.
In another tissue, that is, infections of the eye, adenoviruses were also unable to penetrate the outer layers of epithelium to infect the basal epithelium, whereas enveloped viruses of less rigid structure, such as baculovirus (mean diameter, 60 nm), could do so (Kinnunen et al., 2009). This suggests that particle diameter is not the only parameter to take into account in determining tissue penetration.
To model tissue penetration in the GIANT program, three-dimensional spheroids grown from a number of different prostate cell lines were employed. Different morphologies of spheroid were derived from the different cell types (Fig. 2). Some were rigid and heavily structured, whereas others were much more loosely associated, in which the requirement to form structures and to communicate with other cells had been gradually lost either as a result of oncogenic changes, or through long-term culture. In such full three-dimensional models (Lang et al., 2001) it became apparent that most vectors, including adenoviruses, had difficulties in penetrating basement membranes such as Matrigel, which reflects restrictions on their spread in tissues in vivo as well.
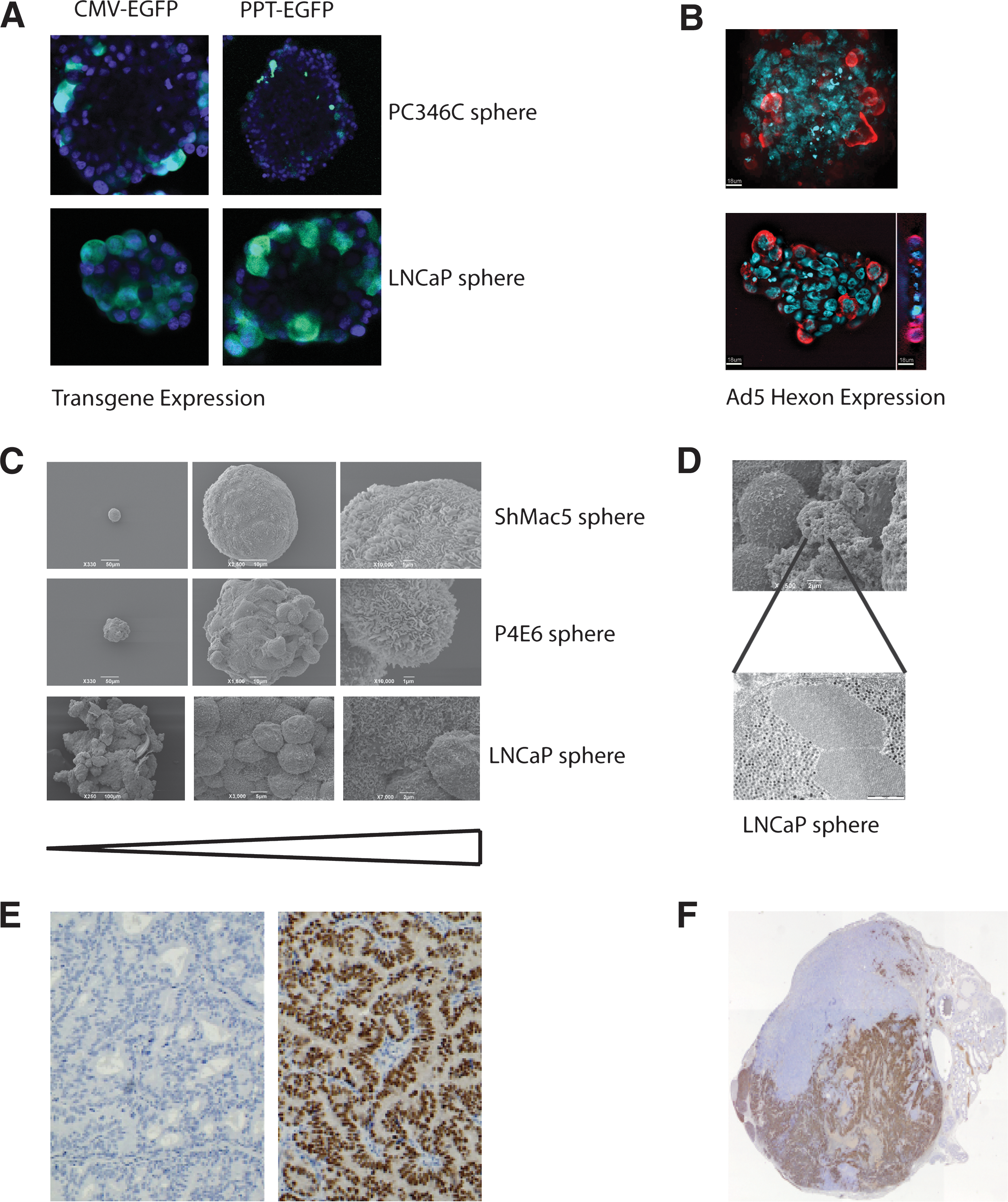
Advanced models of prostate cancer. (
However, even more striking was the inability of adenoviruses to pass through and penetrate organized spheroids as shown in Fig. 2A and B. Furthermore, P4E6 cells (derived from a well-differentiated prostate cancer) form a highly organized spheroid (Fig. 2C), which also contains a stretched layer of thin epithelium to provide structural integrity. Both by immunofluorescence (Fig. 2A and B) and by electron microscopy (Fig. 2D), virus replication could be seen within the outer layer of spheroid epithelium, but spread within the spheroid was almost totally absent. When yields of oncolytic virus in this system were compared with the yields from a replicating monolayer culture and subsequently with xenografts (see later), the three-dimensional culture more closely matched the situation in xenograft models (L. Georgopoulos, H. Evans, and R. Nugent, unpublished data).
Cellular Heterogeneity in Prostate Cancer
For most cancer medicines, there has long been the assumption that all the cells in a tumor grow more quickly than their normal counterparts, and that they homogeneously express the tissue-specific mRNA patterns of a prostate tumor. The rapid emergence of therapy-resistant tumors, even for biotherapies, has been cited as proof of the existence of a preexisting therapy-resistant fraction, sometimes termed cancer stem cells or tumor-initiating cells (Diehn et al., 2009). In human prostate cancer, this fraction seems not to respond to androgens, which form the basis of much of the tissue-specific targeting strategies employed in virus targeting (Maitland and Collins, 2008). As suggested in a review (Short and Curiel, 2009), the development of new generations of vectors with the ability to eliminate cancer stem cells, as part of a combined gene therapy approach to shrink tumor bulk, should extend the effectiveness of such therapies.
Contamination of Cell Models
High-throughput testing of infectious agents, relying on both primary cells and cell lines, also presents a number of dangers with respect to the validity of the cells to be employed. It is now mandatory for publication in some journals that all cell cultures be regularly genotyped. The standard applied by the American Tissue Culture Collection (ATCC, Manassas, VA;
Second, cells derived from xenografts or primary cultures passaged on inactivated mouse stromal feeder layers require routine assessment of human cell content and the presence of murine cells and DNA, derived either from murine infiltrates in the xenografts or “breakthrough” of feeder cells resulting from inadequate inactivation, by polymerase chain reaction (PCR) using repetitive species-specific centromeric sequences (Becker et al., 2002).
The last restriction when working with primary tissues, xenografts (in particular), and established cell lines, is the necessity to eliminate mycoplasma contamination, which frequently prevents vector attachment and penetration. A number of excellent commercial PCR-based assays are available for mycoplasma detection.
Ex Vivo Clinically Relevant Testing for Antivector Immunity
One aspect of gene therapy, which cannot be determined in a standard cell culture environment, concerns exposure of the infectious agent to the bloodstream and immune system of the host. The current standard in vivo system, a xenograft of the original cell line used in cell culture in an immunocompromised mouse, suffers 2-fold in this respect.
First, there is no strong humoral or T cell-mediated immune response against the infectious particles, as the commonly used nude mouse is athymic. The nonobese diabetic-severe combined immunodeficient (NOD-SCID) mouse lacks more of the T cell immunity in addition to B cells, but is now being replaced in practical use by even more immunocompromised hosts such as the Rag2/γc double-negative hosts, which most notably also lack most natural killer (NK) cell functions (Le Dévédec et al., 2009). These hosts still retain complement responses, but show different abilities to engraft with human tumor cells. However, all hosts share the inability to mount any immune response to tumor cell lysis products, acknowledged as one of the major influences on tumor destruction (see previously).
Second, infectious particles, in particular adenoviruses, interact in a different way with blood components in murine and human hosts. It has been shown that more than 90% of adenovirus administered to human blood ex vivo binds to human erythrocytes whereas only a negligible fraction (<0.1%) of adenovirus binds to blood cells in freshly isolated murine whole blood (Lyons et al., 2006). Human erythrocytes bind and inactivate adenovirus serotype 5 (Ad5) by expressing both the coxsackievirus–adenovirus receptor (CAR) and the complement receptor-1 (CR1) (Carlisle et al., 2009). Mouse erythrocytes do not express either CAR or CR1. Because complement receptors are important for induction of neutralizing antibodies (Seregin et al., 2009) mouse blood does not mimic the situation in human blood, that is, the effective dose of virus in humans is now calculated to be much lower than that originally predicted from the critical preclinical studies in mice.
As part of the GIANT project, we sought to overcome this by testing vectors in a human ex vivo blood loop system, which had previously been developed for the testing of other biomaterials (Hong et al., 2001). This system is a powerful preclinical model for studies of viral and nonviral vector interactions with all components of whole blood, omitting the need for high-level anticoagulants that can alter the results by chelating the system of positive ions such as Ca2+ and Zn2+. Furthermore, in contrast to the use of sealed small-volume tubes, the loop system also benefits from reproducing shear forces, which are important for platelet function and the continuous flow of the bloodstream.
We have previously shown (Georgopoulos et al., 2009) that a major complement response in blood loops was mounted against a nonhuman virus, baculovirus. Significant binding of IgM and complement components was observed and strong immunoreactivity was characterized by blood clot formation. The importance of the complement responses to baculovirus, against which humans should have no immunological memory, was indicated by clot prevention by use of complement inhibitors such as compstatin (Nilsson et al., 1998). The life span of the vector particles could therefore be extended in serum by coinoculation with specific complement inhibitors.
Within the GIANT network, we have used the blood loop system to assess the immune response in whole human blood to an Ad5 vector and immunologically “stealthed” PEGylated Ad5. Such PEGylation of Ad5-based vectors has proven efficient in reducing the immune response and increasing the circulation times in mouse models (O'Riordan et al., 1999; Gao et al., 2007) and reducing virus uptake by mouse Kupffer cells (Mok et al., 2005). PEGylation reduced Ad5 adhesion to blood cells, and both complement activation and cytokine release to a certain degree, especially when the neutralizing anti-Ad5 antibody titer was low (Danielsson et al., 2010). However, the reductions were lower than would have been predicted from mouse models (O'Riordan et al., 1999; Mok et al., 2005; Gao et al., 2007; Wortmann et al., 2008). The reason may well be the difference in cell adhesion of Ad5 to human and mouse blood cells and the fact that PEGylation has only a limited protective effect when the neutralizing anti-Ad5 antibody titers are high. This finding simply emphasizes the importance of using a relevant model for evaluating viral vectors.
Toward More Clinically Relevant In Vivo Testing
Current modeling of human prostate cancer in animal models still does not accurately represent the clinical nature of the human disease. A consensus meeting in the United States (Pienta et al., 2008) concluded that newer, more biologically relevant models were still required. Although pure transgenic models have permitted the dissection of basic biological processes underlying prostate cancer, such as the importance of PTEN (phosphatase and tensin homolog) haploinsufficiency (i.e., Di Cristofano et al., 1998), as murine cells are not particularly susceptible to vectors targeting human cancers, and oncolytic adenoviruses do not grow in murine cells, testing systems are usually restricted to human xenografts.
Just as with cell culture lines, until the early 1990s, the limited number of xenograft models for prostate cancer represented predominantly late-stage disease, whereas models of untreated tumors were underrepresented (i.e., radical prostatectomy samples). The more recent establishment of “early-stage” xenograft models and cell lines has largely overcome this limitation (Tables 1 and 2). The latter xenograft models (van Weerden et al., 1996, 2009; Marques, 2006) have become even more relevant as prostate cancer is being diagnosed at an earlier stage. Until more recently, for the testing of gene therapy vectors, the field still remained heavily dependent on the LNCaP, PC3, and DU145 cell lines.
PC3/DU145 Xenografts
PC3 cells form reproducible tumors at most murine sites of inoculation, as subcutaneous, subrenal capsule, and intraprostatic orthotopic grafts (see review by Hoffman, 2007). The tumors are capable of killing the host with an inoculation of about half a million cells within 28 days. This extremely rapid growth from an undifferentiated epithelial cell mass has been used extensively as the target for many prostate-specific therapies. DU145 cells are similarly tumorigenic in immunocompromised animals, although they have been used less frequently (Nemeth et al., 1999).
New variants expressing indicator genes such as luciferase and green fluorescent protein (GFP) have been developed, and more metastatic variants of PC3 have been selected by prolonged passage in nude mice. It could be argued that the lack of structure in the tumors and the rapid growth are not comparable with human prostate cancers, which are much slower growing and display elements of organization and structure even in their least differentiated forms, as described originally by Gleason (1966). One complication with PC3 is that bone tumors, which can be obtained by direct inoculation into bone (Nemeth et al., 1999), are oncolytic rather than osteoblastic, as is commonly found in advanced human cancers (see review by Dotan, 2008).
LNCaP Xenografts
In contrast, the LNCaP cell line expresses androgen receptor (albeit a mutant androgen receptor), which typifies luminal differentiation in well-differentiated common prostate cancers. However, xenografting with LNCaP can be technically difficult, in particular to subcutaneous sites. It is also capable of throwing off many substrains and the C4-2 series originally described by Thalmann and colleagues (1994) clearly has the capacity not only to engraft but also to induce metastasis.
The success of LNCaP xenografts can, however, be substantially improved by coinoculation with prostate stromal cells (Tuxhorn et al., 2002). This again mimicks much more closely the situation in humans.
Other Prostate Epithelial Cell Line Xenografts
The BPH1 cell line, which derived originally from benign prostatic hyperplasia epithelium by introduction of SV40T antigen (Hayward et al., 1995), can be induced to form aggressive tumors by grafting subrenally in the presence of carcinoma-associated fibroblasts (CAFs) from prostate (Hayward et al., 2001). This again emphasizes the importance of the epithelial–stromal interaction in the development of prostate cancers both in vitro and in vivo. The mechanism for this oncogenic transformation of a previously benign cell type simply by the presence of fibroblasts is thought to occur through a complex series of interactions, at least some of which involve transforming growth factor (TGF)-β signaling (Ao et al., 2007). To authentically model lower grades of prostate cancer in the mouse environment will probably require such multicellular grafts.
PC346C Xenografts
The more recently established PC346C cells reliably form undifferentiated epithelial tumors after subcutaneous or intraprostatic orthotopic inoculation of 1 million cells within 20 days. Importantly, PC346C is androgen sensitive, expresses a wild-type androgen receptor, and secretes large amounts of prostate-specific antigen, which typifies well-differentiated common prostate cancers. Prolonged culture of PC346C cells in the absence of androgen and/or in the presence of the antiandrogen hydroxyflutamide, as well as in vivo passage in castrate recipient mice, has resulted in several castration-resistant variants (Marques et al., 2005, 2006). Furthermore, orthotopic injection of PC346C has provided variant sublines derived from metastatic lesions (lymph node and lung) that are equally tumorigenic (W.M. van Weerden, unpublished data). At present, PC346C variants expressing indicator genes such as luciferase and GFP or RFP fluorescence are being developed, which will permit real-time monitoring of tumor cell fate.
Within the GIANT program we chose to exploit the latter xenografts (for a review see Van Weerden et al., 2009), which have been selected to span a range of pathologies, from androgen dependency through to androgen independency and a number of different morphologies and histological grades (see Table 2).
Tumors of early prostate cancer, as represented by the androgen-responsive PC82, PC295, and PC310 xenografts, are well-differentiated tumors with a cribriform growth pattern that continue to express the prostate antigens and more critically a wild-type androgen receptor (Fig. 2E). In contrast, xenografts that represent later stage disease, such as PC133, PC135, and PC339, are relatively undifferentiated.
Xenografts are propagated by tumor fragment transplantation without additional human stromal cells. Instead, tumors are supported by infiltrating murine prostate stroma, which provides the required vascularization and putative stroma-derived factors that may be required for xenograft growth. These xenografts have growth rates that are variable between the various models, with relatively slow growth rates for the androgen-responsive tumors (tumors reaching a volume of 500 mm3 by 60–70 days after transplantation) and faster growth rates for the less differentiated xenografts (tumors reaching a volume of 500 mm3 by 30–40 days after transplantation). Also, as the tumors continue to secrete prostate-specific antigen (PSA), the principal biomarker for clinical studies of prostate cancer (van Weerden and Schröder, 2008), tumor growth can be readily monitored.
The limitation of the xenograft panel largely lies in the fact that these tumors, except for PC346, can be propagated only subcutaneously, as we have been unable so far to establish permanent cell lines from these xenograft models. Because subcutaneously grown tumors generally do not metastasize, these tumors cannot be used to study metastasis from the primary site. However, PC346C cells that are inoculated into the prostate have been shown to spread to other organs (metastases in the lymph nodes and lung). For preclinical testing, the GIANT program has employed the PC346C tumor, which has the great advantage of being able to be grown in vitro and as a serially transplanted xenograft. The ability of PC346C cells to sustain virus growth has been previously demonstrated (Kraaij et al., 2005), and several prostate-specific oncolytic adenoviral vectors, such as Ad[I/PPT-E1A] (Cheng et al., 2006) and Ad-ZH/3 (Magnusson et al., 2007), have now been preclinically validated using the orthotopic PC346C xenograft model. (Fig. 2F; and R. Kraaij, unpublished).
Future Considerations/Outlook
The potential of gene therapy to provide an alternative treatment for prostate cancer has been discussed since the early 1990s (Sanda et al., 1994). The early promise has not yet been fulfilled, although we now know a great deal more about how to kill the correct cells, and perhaps more importantly how to target potent therapies to the correct cell types. Sadly, the testing strategies have failed to maintain the rapid pace of advance in agent development, which could account in part for the failure to convert our basic knowledge into clinical trials.
The translation from cell lines in culture, and of rapidly growing xenografts of the same cell lines, to a heterogeneous and relatively slowly growing tumor in an immunocompetent patient is rarely considered in vector development strategies. We have attempted to highlight the difficulties in achieving total tumor infection as a result of restrictions in vector spread by using adenoviral vectors as a common example. The restriction of spread from a needle injection in tissues remains limited to at best a few millimeters, requiring precise location of the tumor to be infected (Patel et al., 2009). By treating the vector inoculum as a medicine, requiring formulation just like a small molecule, we can perhaps reduce the in vivo restrictions. For example, blockade of the innate immune response, and incorporation of tissue disruptors such as relaxins (or equivalent small molecules) to open tight junctions and reveal receptors, would aid such spread.
However, the most important consideration must remain patient selection: the stage of prostate cancer to be targeted, and the critical realization that responses in each individual human will vary and also differ from those in mouse models. Increasingly, these responses can now be modeled in the laboratory, restricting unnecessary time and expenditure on premature clinical trials.
Footnotes
Acknowledgments
The authors thank all the collaborators on the EU-funded FP6 Project GIANT for discussions and contributions. The authors particularly thank Michelle Scaife for assistance in manuscript preparation. All original research reported was funded by the GIANT Integrated Project (contract number LSHB-CT-2004-512087). N.J.M. also received invaluable core support from Yorkshire Cancer Research.
Author Disclosure Statement
No competing financial interests exist connected with the information conveyed in this review.