
Abstract
The bacteriophage-derived ɸC31 integrase system represents an attractive tool for site-directed recombination in mammalian cells. Its integration reaction is based on recombination between the attachment site attB within an episomal substrate plasmid and either the bacteriophage-derived wild-type attachment site attP or pseudo-attP attachment sites (attP′) present in the mammalian genome. In the present study we aimed at increasing the safety and efficiency of ɸC31 integrase-mediated recombination by mutating the DNA-binding domain located at the C terminus. Using an alanine mutagenesis approach, we generated 22 ɸC31 point mutants that were screened for activities in mammalian cells. Intramolecular excision assays based on recombination between attB and attP revealed five mutants with 2-fold enhanced excision activity. Importantly, we also identified mutants showing enhanced recombination activities between attB and three previously described attP′ sites detected in the mammalian genome, indicating that there may be enhanced specificity for these hot spots. Several mutants showed, in mammalian cells, integration activities that increased in a cell line-dependent manner. The combination of beneficial mutations in addition to optimization of the integrase plasmid dose enhanced integration efficiencies up to 5.5-fold. We also identified three ɸC31 integrase mutants that were recombination defective in all applied assays, suggesting that these amino acid residues are essential for the functionality of ɸC31 integrase in mammalian cells. In summary, we identified critical amino acid residues within the ɸC31 DNA-binding domain. With respect to site-directed recombination and genome engineering these findings have important implications for improved ɸC31 protein design.
Introduction
Several tools for targeted integration at a potentially safe harbor were generated. For instance, zinc finger nucleases (Porteus and Carroll, 2005; Cathomen and Joung, 2008), adeno-associated virus Rep68/78 protein-mediated site-specific integration on chromosome 19 (Recchia et al., 1999; Satoh et al., 2000), and Sleeping Beauty (SB) transposase–E2C zinc finger fusion proteins (Ivics et al., 2007; Yant et al., 2007) as promising tools for targeted integration were developed. As another approach for targeted integration in eukaryotic cells the ɸC31 integrase (ɸC31) derived from a Streptomyces lividans phage was introduced (Groth et al., 2000).
ɸC31 integrase belongs to the superfamily of site-specific recombinases. It is a member of the large serine recombinase subfamily; it is estimated that 30 members belong to this group (Smith et al., 2002). It possesses a conserved N-terminal domain providing catalytic activity for DNA cleavage, strand exchange, and protein–protein interactions (Yuan et al., 2008). The large C-terminal domain, although less well understood, is required for DNA binding (Rowley et al., 2008). Unfortunately, the crystal structure of the ɸC31 integrase is not yet available. However, to further understand the molecular mechanism of ɸC31-mediated recombination in bacteria and to identify critical amino acid residues within the ɸC31 integrase-coding sequences, mutations were introduced into the N-terminal domain (Rowley and Smith, 2008; Keravala et al., 2009) and the C-terminal domain (Rowley et al., 2008; McEwan et al., 2009; Liu et al., 2010).
In nature, ɸC31 integrase mediates site-specific recombination at the wild-type ɸC31 DNA-binding site attB present in the bacterial genome and attP contained in the phage genome (Thorpe and Smith, 1998). With respect to functionality in mammalian cells, it was shown that pseudo-attP sites (attP′) for targeted integration exist in the mammalian genome (Thyagarajan et al., 2001). In a typical approach for achieving ɸC31-mediated somatic integration in mammalian cells, the substrate plasmid containing the gene of interest and the ɸC31 integrase-specific attB attachment site is cotransfected with the ɸC31-encoding plasmid into mammalian cells. After recombination the majority of integration events occur at a limited number of attP′ sites (Chalberg et al., 2006; Ehrhardt et al., 2006).
After ɸC31 integrase-mediated recombination had been shown to be functional in mammalian cells (Groth et al., 2000), several disease-related studies were performed. ɸC31 integrase has successfully been applied in numerous preclinical ex vivo and in vivo gene therapy studies (Olivares et al., 2002; Ortiz-Urda et al., 2002; Ehrhardt et al., 2005, 2007). Furthermore, the ɸC31 integrase methodology represents a platform for genetic engineering of eukaryotic genomes and transgenesis (Belteki et al., 2003; Blaas et al., 2007; Raymond and Soriano, 2007; Thyagarajan et al., 2008; Allen and Weeks, 2009).
However, the ɸC31 integrase system also has its limitations because aberrant events such as chromosomal rearrangements, chromosomal instability as deletions and insertions, and changes at a molecular level occur at a frequency of about 15% (Ehrhardt et al., 2006; Liu et al., 2006). Therefore, several groups aimed to improve the features of the ɸC31 integrase protein.
One study obtained mutants introduced into the N-terminal catalytic domain by alanine scanning mutagenesis, error-prone PCR, and bacterial mutator strains (Keravala et al., 2009). This approach resulted in ɸC31 integrase mutants with moderate (up to 1.4-fold) increased integration efficiencies into attP′ sites present in the genome of mammalian cells and stabilized transgene expression in murine liver. Furthermore, DNA-shuffling approaches were performed (Sclimenti et al., 2001), the simian virus 40 (SV40) large T-antigen nuclear localization signal (NLS) for increased nuclear uptake was fused to the ɸC31 integrase protein (Kalderon et al., 1984; Andreas et al., 2002), and a synthesized codon-optimized ɸC31 integrase was established (Raymond and Soriano, 2007). As a strategy for posttranslational regulation a steroid-induced ɸC31 integrase system was established (Sharma et al., 2008).
In the present study we mutated the DNA-binding domain at the C terminus by an alanine mutagenesis approach. Using this methodology we increased ɸC31-mediated integration and recombination activities up to 2.1- and 5.5-fold at wild-type attP and attP′ sites, respectively. In the future these mutants could be useful for targeted integration in gene therapeutic studies and genome engineering.
Materials and Methods
Plasmid constructs
The wild-type ɸC31 integrase expression plasmid pCSI (wtɸC31) and the plasmid pCSmI (iɸC31), encoding wild-type and the recombination-deficient version of the ɸC31 integrase (Groth et al., 2000; Ehrhardt et al., 2005), were a generous gift from M.P. Calos (Stanford University, Stanford, CA). The mutated integrase gene represents an integration-defective version of the ɸC31 integrase and contains the point mutation S12F (Thorpe and Smith, 1998). The ɸC31 integrase substrate plasmid p7, which was used for colony-forming assays, contains the attB attachment site and a neomycin resistance gene. Plasmid p7 was described in our earlier study (Ehrhardt et al., 2005).
The reporter plasmid pattPpolyAattBLuciferase (pLucCR) was a generous gift from C. Rudolph (Ludwig Maximilians University Munich, Munich, Germany). It contains a cytomegalovirus (CMV) promoter, native attP and attB sites flanking a poly(A) site (termination site), and a luciferase reporter gene downstream of the termination site. For this reporter plasmid luciferase expression is dependent on excision of the termination signal by ɸC31-mediated recombination between attB and attP.
Furthermore, the reporter plasmid pLucCR was used as a template construct to exchange the wild-type attP site with three previously described pseudo-attP sites (attP′) on chromosomes 2 (hs2), 12 (hs12), and 19 (hs19) (Chalberg et al., 2006; Ehrhardt et al., 2006). For DNA amplification of these hot spots, a PCR was performed with genomic DNA isolated from HEK293 cells and HCT116 cells. To generate the respective fragments with the attP′ site and surrounding genomic DNA of up to 430 bp in length, the following primers were used (AgeI and StuI restriction enzyme recognition sites are underlined): forward AgeI hs2, 5′-
The vector pCMV-attB-BGH-polyA-attP-EGFP (named pEGFP-att in the present study), with a neomycin resistance marker, was a generous gift from W. Jia (Chen et al., 2006). Similar to pLucCR, this plasmid contains a termination signal bovine growth hormone (BGH) poly(A) signal flanked by attB and attP attachment sites upstream of the enhanced green fluorescent protein (EGFP)-coding sequence.
The donor plasmid pBS-attB-hFIX, used for injection into mouse liver, has been introduced elsewhere (Miao et al., 2000; Ehrhardt et al., 2005). It contains the native attB attachment site and a human coagulation factor IX (hFIX) expression cassette with the hFIX cDNA and part of the first intron of the hFIX gene (hFIX minigene). Expression is controlled by the liver-specific α1-antitrypsin promoter (hAAT-p) and two liver-specific enhancers.
All constructs used in this study including the molecular setup of reporter constructs are schematically represented in Fig. 1.
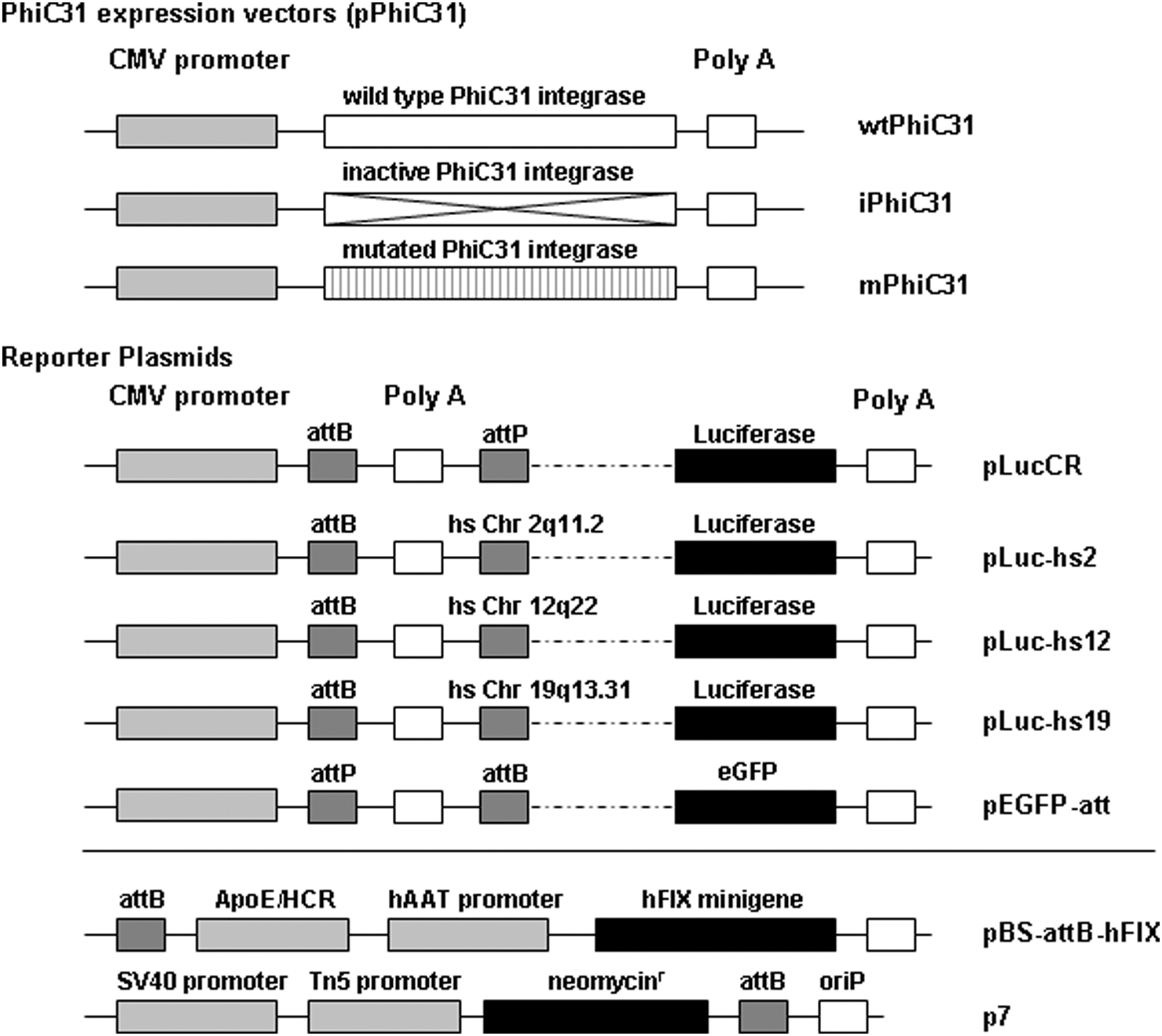
All DNA constructs used in this study. Top: The first three constructs schematically show all φC31 integrase-encoding plasmids (pφC31). Wild-type (wtφC31), inactivated (iφC31), and mutated (mφC31) φC31 integrase-coding sequences were expressed under the control of the cytomegalovirus (CMV) promoter. Middle: The reporter plasmids pLucCR, pLuc-hs2, pLuc-hs12, pLuc-hs19, and pEGFP-attB express luciferase or enhanced green fluorescent protein (eGFP) under the control of the CMV promoter. For plasmids pLucCR and pEGFP-attB reporter gene expression is dependent on φC31 integrase-mediated recombination between native attB and attP integrase recognition sites. For plasmids pLuc-hs2, pLuc-hs12, and pLuc-hs19 luciferase expression is dependent on recombination between attB and the previously described hot spots on human chromosomes 2 (hs2), 12 (hs12), and 19 (hs19). All reporter constructs contain a bovine growth hormone polyadenylation signal (Poly A) between the φC31 recognition sites. Bottom: The φC31 integrase substrate plasmid pBS-attB-hFIX (Ehrhardt et al., 2005; Miao et al., 2000) expresses a human coagulation factor IX (hFIX) minigene containing the hFIX cDNA and part of the first intron of the hFIX gene under the control of the human α1-antitrypsin promoter (hAAT) and the hepatic locus control region from the apolipoprotein E gene locus (ApoE/HCR). The plasmid p7 (Ehrhardt et al., 2005; Miao et al., 2000) used for colony-forming assays contains a neomycin resistance gene (neomycinr), the simian virus 40 (SV40) promoter, the bacterial transposon promoter Tn5, and a bacterial origin of replication (oriP).
Site-directed mutagenesis of the C terminus of ɸC31 integrase
ɸC31 integrase mutants (mɸC31) were obtained by a two-step overlapping PCR approach. The pCSI vector carrying the ɸC31 integrase gene was used as a template for each obtained mutant. Alanine substitutions were introduced into the coding sequence of the ɸC31 integrase, using mutagenic primers (length, 21 bp) with the nucleotide mutation in the center. Primers for all generated mutants are summarized in Supplementary Table 1 (see
Primers BamHIforw-outer and BstEIIrev-outer were used for the final PCR. This PCR product with the mutated DNA-binding domain was cloned into the pTOPO vector (Invitrogen), excised by BamHI and BstEI restriction enzyme digest, and subsequently cloned into the BamH and BstEI sites of the plasmid pCS + NotI. Plasmid pCS + NotI was produced by excising the BamHI and BstEI fragment at the C terminus of the ɸC31 integrase-coding sequence from pCMV-Int and adding a NotI linker instead. This linker was produced by annealing the oligonucleotides BamHINotIBstEIIfor (GAT CC
Double mutants were constructed, respectively, using the mutated ɸC31 integrase sequence as template. Mutations were confirmed by DNA sequencing.
Cell culture
HeLa cells and HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (PAA Laboratories, Pasching, Austria) supplemented with penicillin–streptomycin (PAA Laboratories) and 10% fetal bovine serum (FBS; Invitrogen). The human hepatoma-derived cell line Huh7 was maintained in DMEM supplemented with
Luciferase reporter assay for detection of ɸC31 integrase excision activities from episomal DNA
To evaluate ɸC31 integrase-mediated intramolecular excision from a substrate plasmid, luciferase assays were performed in HEK293 cells. For Dual-Luciferase assays a triple transfection protocol with the respective integrase-encoding plasmid (wtɸC31, iɸC31, or mɸC31), the firefly luciferase-encoding plasmid pLucCR, and an internal transfection control (Renilla luciferase-encoding plasmids pRL-TK or phRL-O) was applied.
The firefly luciferase-encoding reporter plasmid (pLucCR) contains the ɸC31 attachment sites attB and attP flanking the bovine growth hormone polyadenylation signal (termination signal) upstream of the luciferase reporter gene. In this assay a precondition for luciferase expression is excision of the termination signal by ɸC31-mediated recombination. The transfection ratio on a molecular weight basis (wtɸC31/iɸC31/mɸC31:pLucCR:pRL-TK/phRL-O) used was 10:9:1, with a total amount of 1200 ng of plasmid DNA per individual transfection reaction in one 24-well plate (experimental setup 1). For experimental setup 2, including the attP′ sites from chromosomes 19, 12, and 2, we used a weight ratio of 40:10:1, respectively.
Cells were plated in 24-well plates and plasmid transfections were performed with FuGENE 6 (Roche Applied Science, Indianapolis, IN). For measurements we used a Dual-Luciferase reporter assay system from Promega (Madison, WI). Forty-eight hours posttransfection cells were washed once with Dulbecco's phosphate-buffered saline (DPBS) and thereafter lysed with 100 μl of 1× lysis buffer (25 mM Tris-HCl, 0.1% Triton X-100; pH 7.8). Cells were incubated for 10 min at room temperature, lysed cells were frozen in liquid nitrogen and thawed, and cell debris was removed by centrifugation. The supernatant was either stored at −80°C until use or used immediately. For quantification of firefly and Renilla luciferase expression, 10 μl of the lysate was used and measured in a luminometer (Berthold Detection Systems, Pforzheim, Germany) according to the manufacturer's manual (Promega).
EGFP reporter assay for detection of ɸC31 excision activities from chromosomal DNA
To generate stable cell lines with conditional EGFP expression, HEK293 cells were stably transfected with the plasmid pCMV-attB-BGH-polyA-attP-EGFP, using FuGENE 6. This plasmid was a generous gift from W. Jia (Chen et al., 2006). It contains a neomycin resistance marker for the generation of stable cell lines and a termination signal (bovine growth hormone polyadenylation signal) flanked by attB and attP attachment sites upstream of the EGFP-coding sequence and downstream of a CMV promoter. For this plasmid, expression of EGFP depends on excision of the termination signal by ɸC31 integrase-mediated recombination.
After transfection cells were maintained in medium supplemented with G418 (500 μg/ml), and resulting cell clones were amplified. To test induction of EGFP expression dependent on the activity of ɸC31 integrase, 11 established cell clones were transiently transfected with either wtɸC31- or iɸC31-encoding plasmids. The single clone 28 showed the highest ratio of wtɸC31: iɸC31 activated fluorescence activity and was therefore selected for further analysis.
The integrase-mediated recombination activity of ɸC31 integrase mutants was measured in cell clone 28. We cotransfected ɸC31 integrase-encoding plasmids (wtɸC31, iɸC31, and mɸC31) and a plasmid encoding mOrange, a mutant fluorescent protein derived from the tetrameric Discosoma species red fluorescent protein DsRed, at a molar ratio of 2:1 (ɸC31 integrase:mOrange). This corresponds to 1333 ng of integrase plasmid and 666 ng of mOrange plasmid. The mOrange-expressing plasmid was used to normalize transfection efficiencies. Two days posttransfection cells were harvested by washing once with DPBS, centrifuged at 2000 rpm for 5 min at 4°C, and stored on ice. Up to 10,000 cell counts per sample were analyzed by flow cytometry and the subdivided fractions were measured as nonfluorescent cells, EGFP-expressing cells, or EGFP- and mOrange-expressing cells. The relative EGFP expression level was recorded as a measure of the excision activity of the individual integrase derivatives.
Colony-forming assays to determine integration efficiencies
To determine the ɸC31 integrase-mediated integration efficiencies of (circularized) substrate plasmids, colony-forming assays (Ehrhardt et al., 2005) were performed in HeLa, Huh7, HEK293, HCT, and Hep1A cells. Cells were plated in 6-well plates and transfections were performed with FuGENE 6 (Roche Applied Science). We cotransfected plasmids encoding either active ɸC31 integrase, integrase point mutants, or inactive ɸC31 integrase together with substrate plasmid p7. The initial molar ratio of integrase-encoding plasmid to substrate plasmid p7 was 0.48:1. For dose-dependent studies we changed the molecular ratio to 0.025:1, 0.1:1, 0.5:1, 1:1, 5:1, and 20:1, respectively.
Experiments were preformed in triplicate. Forty-eight hours posttransfection cells were split into 10-cm dishes at a cell density of 4 × 104 cells/10-cm dish. After 14 days of G418 selection at a constant selection pressure of 500 μg/ml, colonies were stained with methylene blue and visually quantified. The number of blue colony-forming units represented the integration efficiency of the respective integrase.
Animal procedures
C57BL/6 mice were kept and treated according to the regulations of the Government of Upper Bavaria in Germany. For hydrodynamic tail vein injections 6- to 8-week-old female C57BL/6 mice (three to five mice per group) were injected as described previously (Liu et al., 1999). Plasmid DNA was diluted in 1.8 ml of physiological saline solution (0.9%, w/v) and injected via the tail vein over a time period of about 6 sec. Plasmid pBS-attB-hFIX (20 μg) and the respective integrase-encoding plasmids (20 μg) were coinjected at a weight ratio of 1:1. Blood was collected periodically and serum was isolated by centrifugation for 2 min at 10,000 rpm at 5°C. Serum samples were stored at −80°C until use.
Rapid cell cycling of murine liver cells was induced by intraperitoneal administration of 50 μl of carbon tetrachloride (CCl4; Sigma-Aldrich, St. Louis, MO) solution (1:1 dilution in mineral oil).
Blood analyses
Human coagulation factor IX (hFIX) expression levels in murine serum were monitored by sandwich ELISA. A murine monoclonal IgG antibody (Sigma-Aldrich) directed against hFIX at a 1:2000 dilution was used for coating. As the secondary antibody we used an IgG antibody conjugated with horseradish peroxidase (Biozol, Eching, Germany) at a 1:1000 dilution. Peroxidase activity was detected at 492 nm with SigmaFast OPD tablets (Sigma-Aldrich) and samples were measured with a plate reader and Magellan 3 software (Tecan Group, Maennedorf, Switzerland).
For quantification of alanine aminotransferase (ALT) levels in murine serum, we followed the instructions of the ALT/GPT (glutamic-pyruvic transaminase) detection kit (Randox Laboratories, Crumlin, County Antrim, UK). We applied 15 μl of murine serum to each reaction.
Statistical analysis
All data are reported as means ± standard deviation unless otherwise noted. Statistical comparison was made by two-tailed Student t test, and a value of p < 0.05 was considered relevant compared with the respective control group.
Results
Generation of ɸC31 integrase mutants
We aimed at changing the biological properties of ɸC31 integrase by identifying beneficial versions of the protein. The ɸC31 integrase DNA-binding domain was used as a template to introduce point mutations. Site-directed mutants were created by using overlapping oligonucleotides in a two-step PCR approach. Positively and negatively charged amino acids were screened and 22 residues were changed to alanine. Mutations were verified by sequencing. All ɸC31-coding sequences including the mutated versions (mɸC31) and the inactive version were expressed under the control of the cytomegalovirus promoter (Fig. 1). The control plasmids are referred to as wtɸC31 (encoding wild-type ɸC31 integrase) and iɸC31 (encoding inactive ɸC31). The putative ɸC31 integrase DNA-binding domain structure and the original amino acid sequence, in which charged amino acid residues were changed to alanine, are summarized in Fig. 2.
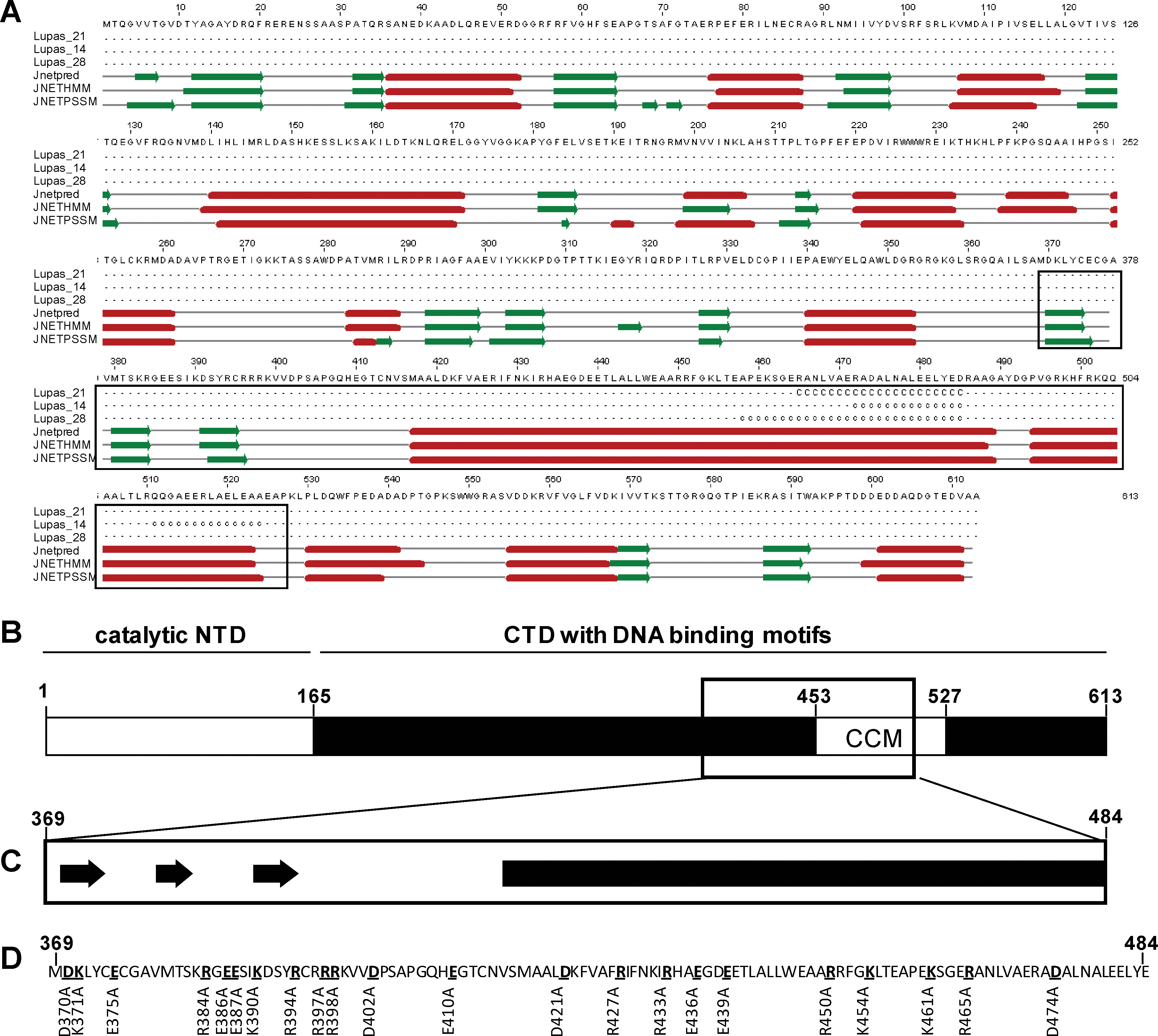
φC31 integrase domain organization. (
According to the Jpred secondary structure prediction server (Cuff and Barton, 2000), the structure of ɸC31 integrase contains several β-sheets and α-helices (Fig. 2A). The C-terminal domain (CTD) contains DNA-binding motifs and the putative DNA-binding domain encompasses amino acid position 369–484. A coiled-coiled motif has been described between amino acid positions 453 and 527 (Fig. 2B and C) (Rowley et al., 2008). Point mutations were introduced into the putative DNA-binding domain spanning amino acid positions 369–484 (Fig. 2D).
Excision activities of ɸC31 mutants in the context of episomal and chromosomal DNA
To determine excision-based recombination efficiencies of ɸC31 integrase mutants we performed an extrachromosomal recombination assay based on transient plasmid transfection. The reporter plasmid pLucCR contains an expression-blocking sequence, flanked by wild-type attB and attP attachment sites between the CMV promoter and the luciferase-coding sequence (Fig. 1). The luciferase gene is turned on after excision of the termination signal because of recombination between the ɸC31 integrase recognition sites attB and attP.
Plasmid pLucCR and the indicated versions of the ɸC31-coding sequence were cotransfected at a molecular weight ratio of 1:0.98 (pLucCR:ɸC31 integrase). Integrase mutants showed excision activities between 4 and 210% compared with wtɸC31, which was set to 100% (Fig. 3A). Five mutants (E375A, E387A, E410A, R427A, and K454A) showed about 2-fold higher excision activity. Eleven mutants (D370A, E386A, K390A, R394A, R398A, D402A, R433A, E439A, R450A, K461A, and D474A) produced results rather similar as wtɸC31, in range of 70 to 130%. Six mutants (K371A, R384A, R397A, D421A, E436A, and R465A) were inactive or showed low activity, ranging from 4 and 30% excision activity. Therefore, this prescreen of generated mutants revealed that recombination efficiencies were altered due to the introduction of point mutations into the DNA-binding domain.
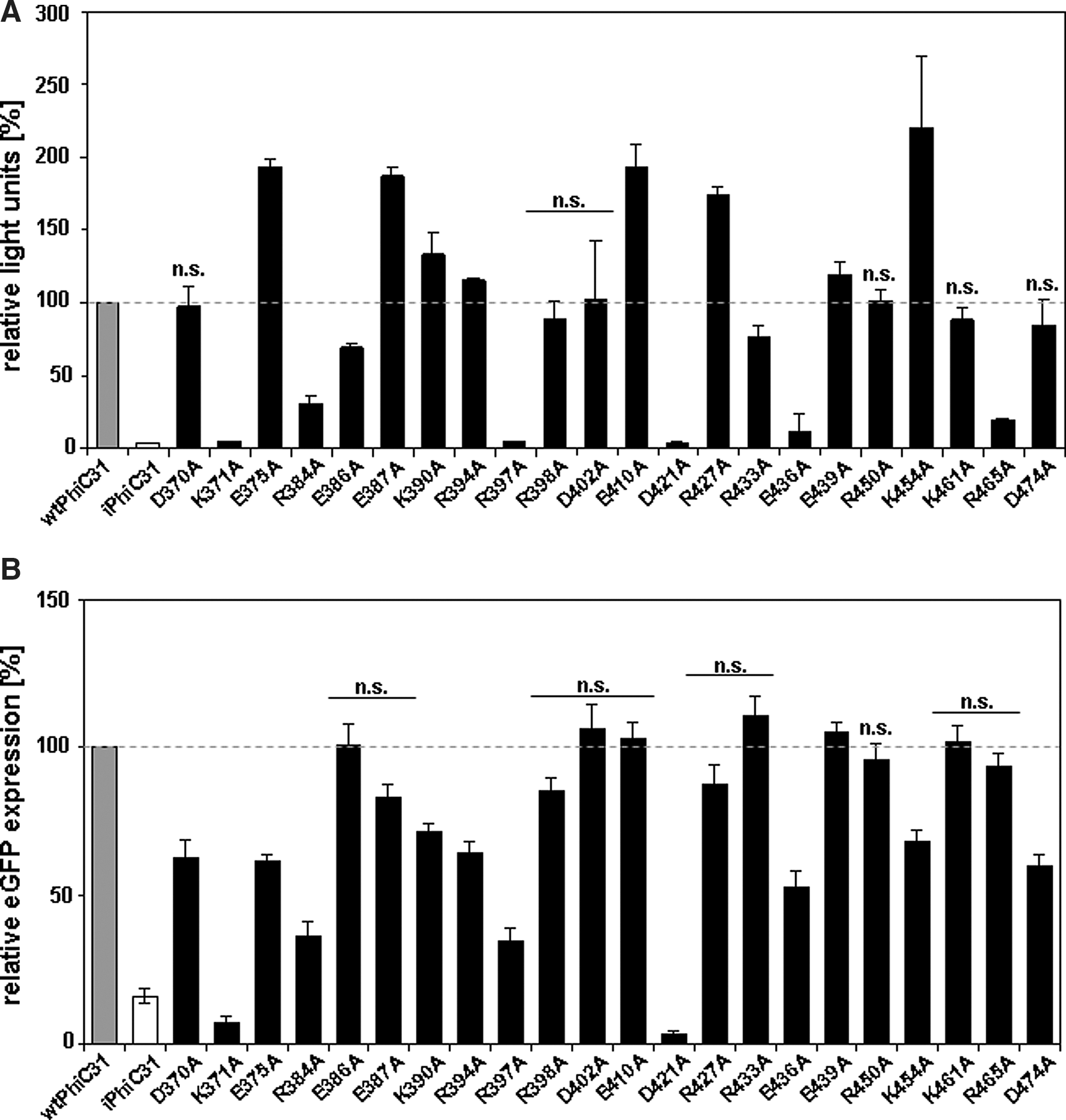
Excision activities of integrase mutants based on recombination between wild-type attachment sites (attP and attB) in HEK293 cells. (
We also aimed at investigating the recombination activity of integrase mutants within the context of chromosomal DNA. Therefore, the reporter plasmid pEGFP-att (Fig. 1) encoding a conditionally active EGFP reporter gene was stably integrated into the genome of HEK293 cells. After selection, 11 single-cell clones were obtained and characterized. In preliminary flow cytometric experiments one cell line (cell line 28) showed the highest induction of EGFP expression after transient transfection of wtɸC31 and subsequent recombination between attB and attP (data not shown). This cell line was used to determine the excision activities of all 22 generated ɸC31 mutants. After transfection of the respective ɸC31 integrase-encoding plasmids (wtɸC31, iɸC31, and mɸC31) into the reporter cell line 28, we measured induction of EGFP expression by flow cytometric analysis.
We found that eight mutants (E386A, D402A, E410A, R433A, E439A, R450A, K461A, and R465A) had similar excision activity compared with wtɸC31 (Fig. 3B). No integrase mutant with significantly higher excision activity compared with wtɸC31 was observed. Interestingly, mutants R371A and D421A were recombination-inactive in both excision-based recombination assays and showed recombination activity similar to that of iɸC31. These critical amino acid residues seem to fail in recombination-based excision in both reporter assays.
A rapid assay for measurement of recombination activities at previously described hot spots
Next, we addressed whether our novel ɸC31 DNA-binding domain mutants display higher specificity with respect to previously described hot spots present in the human genome. Thus, we modified our rapid episomal recombination-based assay, by replacing the native attP site in the reporter plasmid with previously described pseudo-attP (attP′) sites. Three of these ɸC31 integrase hot spots within the human genome, at chromosomal positions 2q11.2, 12q22, and 19q13.31 (Chalberg et al., 2006; Ehrhardt et al., 2006), were chosen. The attP′ site core sequence and flanking genomic DNA was amplified by PCR and inserted into the substrate plasmid pLucCR (Fig. 1). Recombination assays were performed at a high molecular weight ratio of 1:5 (pLucCR:ɸC31 integrase) and a low molecular weight ratio of 1:1 (pLucCR:ɸC31 integrase) for all generated mutants. Figure 4 summarizes the recombination efficiencies between native attB and attP recognition sites and recombination activities between attB and the respective attP′ sites.
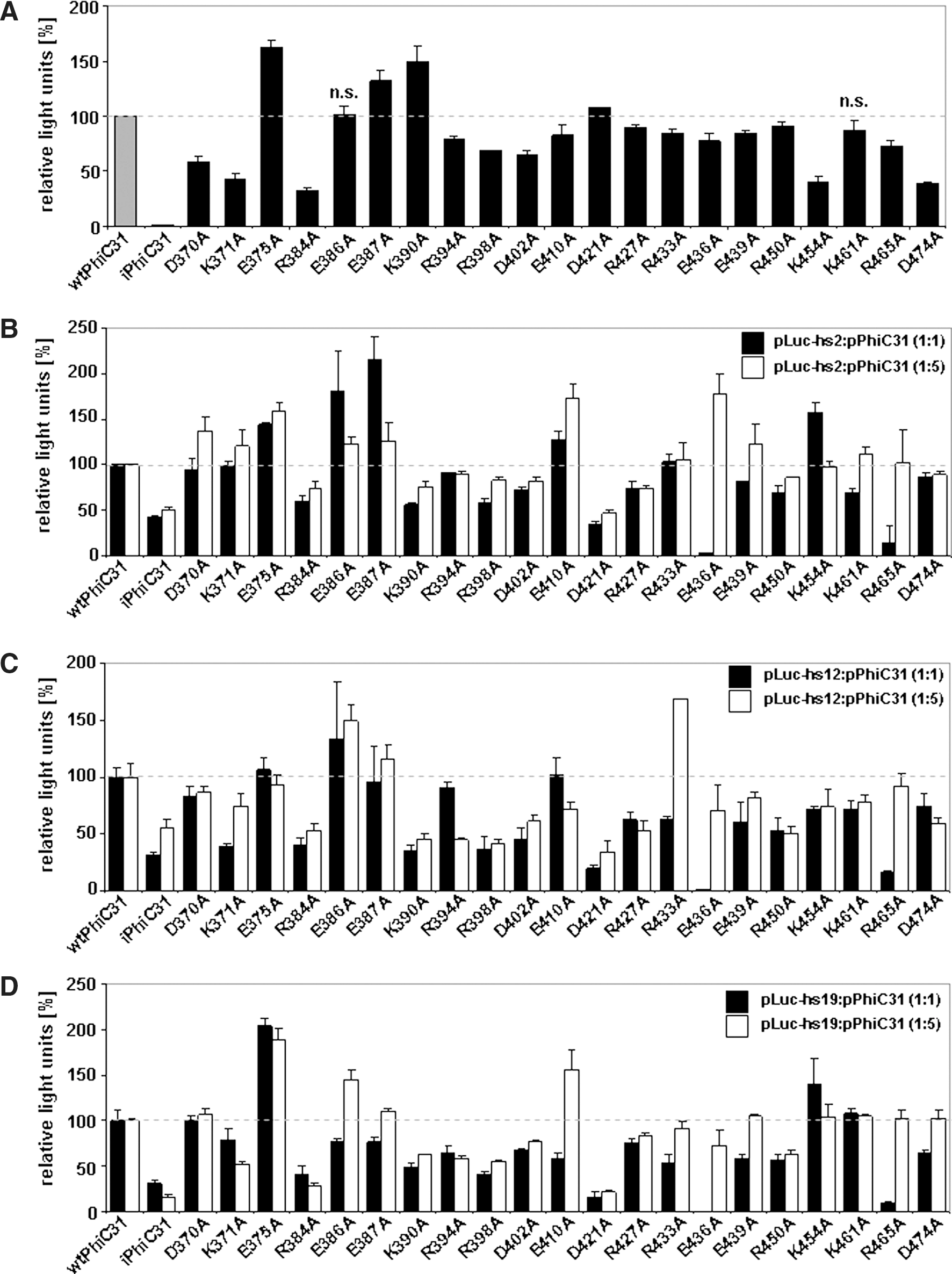
Catalytic activity of integrase mutants after recombination of native attB and attP as well as pseudo-attP sites in HEK293 cells. A Dual-Luciferase assay was performed to measure the activity of wtφC31, iφC31, and mφC31. Relative luciferase activity is indicated as relative light units, expressed as a percentage, corresponding to the excision activity of wild-type integrase (wtφC31) divided by the catalytic activity of integrase mutants. A pLucCR:pInt plasmid ratio of 1:5 was used in Dual-Luciferase assays to detect the excision activity of φC31 integrase after recombining the native attachment sites (
After recombination was induced at native attB and attP sites (Fig. 4A), three mutants (E375A, E387A, and K390A) were identified showing between 1.4- and 1.6-fold improvements in excision activity at a ratio of 1:5 (pLucCR:ɸC31 integrase). The remaining mutants showed between 100 and 40% recombination activity compared with wtɸC31. Recombination assays at the attP′ site derived from chromosome 2q11.2 revealed mutants E386A and E387A and mutants E410A and E436A with an approximately 2-fold increase in recombination activities at a 1:1 ratio and a 1:5 ratio, respectively (Fig. 4B). Mutants R384A and D421A showed activities comparable to iɸC31 at both plasmid ratios. As displayed in Fig. 4C, recombination activities at the attP′ site present on chromosome 12q22 showed improvements for two mutants (E386A and R433A). However, more than two-thirds of the mutants showed lower recombination activity than wtɸC31. Excision activities with respect to the previously described hot spot on chromosome 19q13.31 revealed three mutants (E375A, E386A, and E410A) displaying up to 2-fold improved excision activities (Fig. 4D). For mutants R384A and D421A we observed comparable excision activities as measured for iɸC31.
Interestingly, two mutants were identified (E436A and R465A) as showing enhanced activity at the wild-type attP attachment site after increasing the ɸC31-encoding plasmid dose. These mutants were recombination defective at a 1:1 ratio, but a 5-fold increase in the integrase plasmid dose (1:5 ratio) led to an increase in recombination activities of more than 10-fold (Fig. 4). Importantly, this phenomenon was observed not only for native attB and attP recombination activities; when recombination was performed at the attP′ sites at chromosomal positions 2q11.2, 12q22, and 19q13.31 the same effect was observed (Fig. 4B–D). This indicated that the activity of mutants E436A and R465A may be dose dependent.
Altered somatic integration efficiencies of ɸC31 mutants in mammalian cells
For characterization of ɸC31 integrase mutants we first performed intramolecular plasmid-based assays addressing recombination efficiencies at three particular hot spots (Fig. 4). Next, we wanted to analyze integration efficiencies in the context of the complete mammalian genome containing all potential hot spots for ɸC31 integrase-mediated integration. Although the EGFP-based reporter assay (Fig. 3B) investigated recombination efficiencies in the context of chromosomal DNA, it addressed solely recombination at native attachment sites stably integrated into the host genome. Therefore, we performed colony-forming assays, which allow for selection and quantification of integration events in the context of the entire mammalian genome. In this assay integration efficiency directly correlates with the number of clones surviving selection after transfection.
Integration assays performed in triplicate were carried out by cotransfecting plasmids encoding either wtɸC31, the nonfunctional ɸC31 (iɸC31), or the respective newly constructed ɸC31 mutants and the substrate plasmid p7 (Fig. 1) at defined plasmid ratios. The molar ratio used for initial screening in HeLa cells was 0.48:1 (integrase-encoding plasmid:substrate plasmid p7). The integration efficiencies of the 22 different single amino acid substitution mutants in relation to integration efficiencies of wtɸC31 are summarized in Fig. 5. Five integrase mutants (D370A, E386A, E387A, K461A, and D474A) were observed showing 1.2- to 1.7-fold increased integration efficiencies compared with wtɸC31. Mutants K371A, R384A, R397A, R398A, D421A, E436A, and K454A were defective with respect to integration into the mammalian genome. The integration efficiency of the inactive iɸC31 control group was comparable to the latter inactive mutants identified in this screen. Ten mutants (E375A, K390A, R394A, D402A, E410A, R427A, R433A, E439A, R450A, and R465A) showed integration efficiencies comparable to wtɸC31-mediated integration. In summary, under the described conditions a 1.7-fold improved ɸC31 integrase mutant could be identified.

Overall integration efficiencies of φC31 mutants in HeLa cells. To quantify integration efficiencies colony-forming assays were performed with a φC31 integrase substrate plasmid (p7) carrying a neomycin resistance gene. This assay determines recombination between the native attB attachment site contained in the substrate plasmid p7 and genomic pseudo-attP sites. Relative integration events mediated by φC31 mutants were determined after G418 selection and normalized against the catalytic activity of wtφC31, which was set to 100% (dashed line). (
To further improve integration efficiencies in mammalian cells and to further characterize our novel ɸC31 integrase mutants three approaches were addressed: (1) we generated ɸC31 integrase double mutants combining single mutants with increased integration efficiencies, (2) we aimed at optimizing the ɸC31 plasmid dose, and (3) we screened integration efficiencies of double mutants in cell lines of different human origin because it has been shown in the past that ɸC31 integrase-mediated integration efficiencies are cell line dependent (Ehrhardt et al., 2006; Maucksch et al., 2008).
Optimizing the dose of ɸC31-encoding plasmid for improved somatic integration
It was demonstrated for the SB transposase system that increasing the dose of SB results in inhibition of transposition events (Geurts et al., 2003; Zayed et al., 2004). This phenomenon is also referred to as the overproduction inhibition effect. To study the dose dependency of wtɸC31 and ɸC31 integrase mutants we either increased or decreased the amount of transfected ɸC31-encoding plasmid while keeping the substrate plasmid amount constant. On the basis of results presented in Fig. 5A we chose mutants E387A, K461A, and D474A as examples of hyperactive ɸC31 versions for dose-dependent studies. The molecular weight ratios of substrate plasmid p7 to ɸC31 integrase-encoding plasmids ranged from 1:0.025 to 1:5. As displayed in Fig. 5B, lowering the dose of ɸC31 integrase-encoding plasmid resulted in decreased colony-forming units for all groups. In contrast, we found that increasing the plasmid dose of both ɸC31 integrase point mutants resulted in similar (mutants E387A and D474A) or up to 2.3-fold enhanced integration at the 1:5 ratio (Fig. 5C). Under these conditions the integration efficiency of wtɸC31 was increased up to 1.4-fold. Because there was no decrease in colony-forming units after increasing the ɸC31 integrase plasmid dose, we concluded that there was no evidence of an overproduction inhibition effect as described for SB transposase for the ɸC31 mutants analyzed.
Integration efficiencies of generated ɸC31 integrase double mutants are cell line dependent
To analyze whether there are synergistic effects of beneficial single mutants, double mutants of these particular ɸC31 integrase candidates were generated. On the basis of results presented in Fig. 5, we constructed the two double mutants K461A/D474A and E386A/D474A in HeLa cells, the human hepatoma-derived cell line Huh7, the human embryonic kidney cell line HEK293, and the human colon carcinoma-derived cell line HCT.
Integration efficiencies were directly compared with wtɸC31 activities, which were set to 100%. Double mutants K461A/D474A and E386A/D474A showed up to 5.5-fold improved integration efficiencies in HeLa cells compared with wtɸC31 (Fig. 6B). This clearly demonstrated that the combination of beneficial ɸC31 mutants can result in an additive effect with respect to integration activities. We observed 2.4- and 1.5-fold increased integration efficiencies for the double mutant E386A/D474A in Huh7 cells and HEK293 cells, respectively (Fig. 6A and C). In HCT cells the double mutant K461A/D474A showed slightly higher integration efficiencies (up to 1.5-fold) (Fig. 6D). In total, these results indicated that the efficacy of analyzed ɸC31 integrase derivatives is cell line dependent.
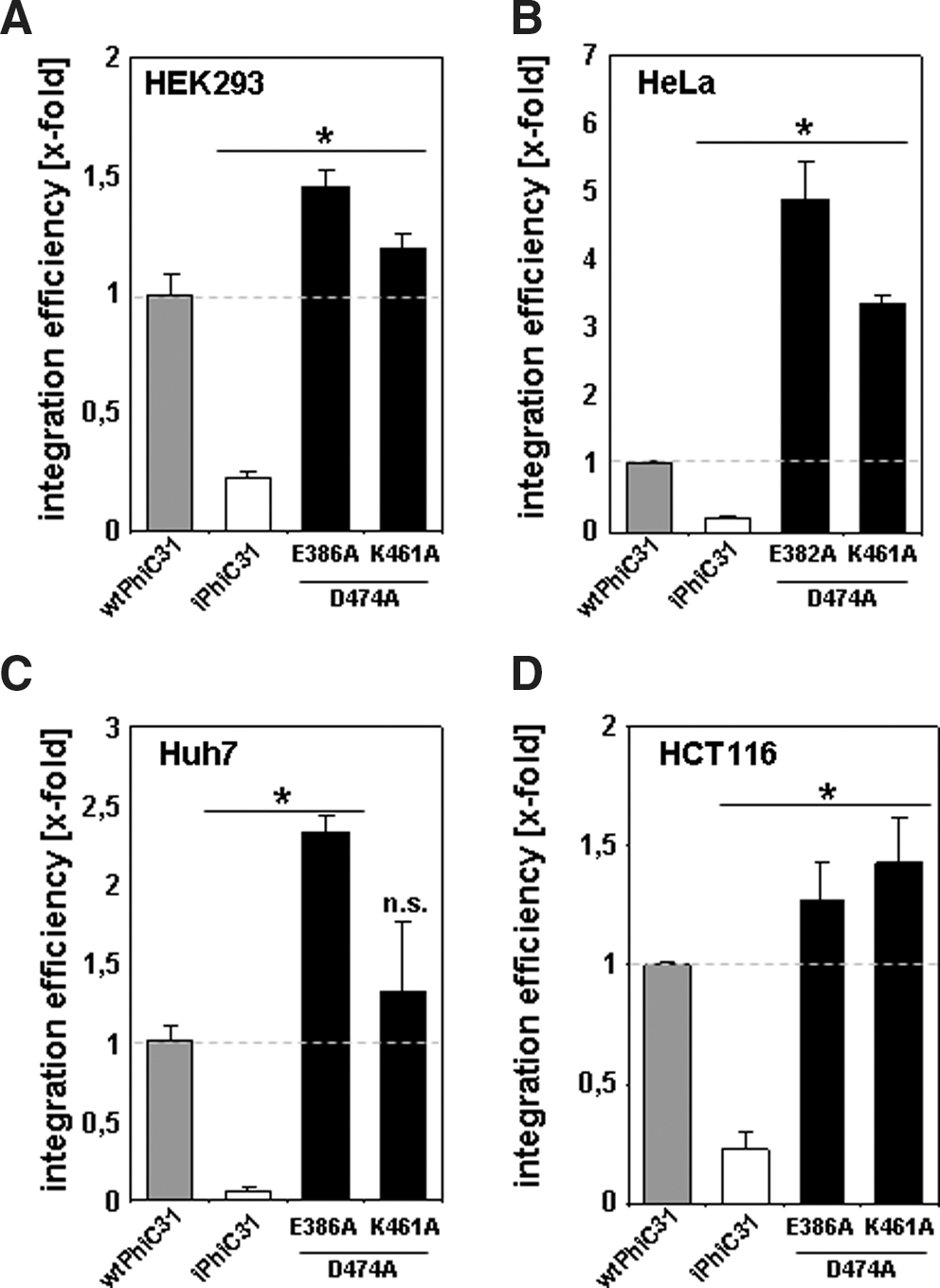
φC31-mediated integration activities are cell line dependent. Colony-forming assays were performed in HeLa cells, the human hepatoma-derived cell line Huh7, the human embryonic kidney cell line HEK293, and the human colon carcinoma-derived cell line HCT116. Integration efficiencies for generated φC31 double mutants E386A/D474A and K461A/D474A in HEK293 cells (
Efficacy of mutants K461A and D474A in mouse liver in vivo
We identified several mutants that showed increased integration efficiencies in vitro (Fig. 5). As a further step we wanted to investigate whether these mutants are also advantageous for in vivo applications. Therefore, we selected the most beneficial mutant, D474A (Fig. 5), and as another example the hyperactive mutant K461A for in vivo studies (see also Figs. 5 and 6A). Control mice either received wtɸC31 or inactive ɸC31 integrase (iɸC31), respectively. A substrate plasmid, pBS-attB-hFIX (Fig. 1), carrying the native attB site and a human coagulation factor IX (hFIX) expression cassette, was coinjected with various versions of the integrase-expressing plasmid into C57BL/6 mice by high-pressure tail vein injection. Blood was collected periodically and serum hFIX levels were determined by ELISA.
In initial experiments we coinjected 2 μg of the hFIX-expressing substrate plasmid and 20 μg of the respective ɸC31 integrase-encoding plasmid. However, initial serum hFIX levels in all groups were low (serum levels were less than 200 ng/ml), indicating that the hFIX-encoding substrate plasmid dose was not sufficient. In the next experiment we coinjected 20 μg of the hFIX-expressing substrate plasmid and 20 μg of the respective ɸC31 integrase-encoding plasmid.
To determine the toxicity profile of wtɸC31 and ɸC31 integrase derivatives, limited toxicity studies were carried out during the first 15 days postinjection. We measured liver enzyme alanine aminotransferase (ALT) present in the serum of treated mice as an indicator of acute toxicity at enhanced levels. We measured elevated liver enzyme levels, which were similar for all groups on day 1 postinjection (Fig. 7A). This observation is most likely a consequence of the harsh hydrodynamic injection conditions. However, 8 and 15 days postinjection ALT levels declined to the normal range (Fig. 7A). Importantly, there was no significant difference in ALT level at any measured time point for all groups.

Human coagulation factor IX expression and in vivo performance of mutants K461A and D474A. C57BL/6 mice were coinjected via hydrodynamic tail vein injection, using 20 μg of φC31 integrase-encoding plasmid and 20 μg of the human coagulation factor IX (hFIX)-encoding substrate plasmid (pBS-attB-hFIX). Rapid hepatocyte proliferation was induced by intraperitoneal injection of carbon tetrachloride (CCl4; injection is indicated by black arrows) on days 49 and 71 postinjection to analyze the stability of transgene expression. (
The time course of hFIX serum concentrations of the four groups is shown in Fig. 7B. For mutants K461A and D474A and for wtɸC31, serum hFIX levels remained stable with levels of up to 3500 ng/ml until day 42 postinjection. Mice that received iɸC31 showed slightly decreased serum hFIX concentrations at this time point. To investigate whether the selected mutants show stabilized transgene expression levels we induced rapid cell cycling in murine liver after intraperitoneally administering carbon tetrachloride (CCl4) on days 49 and 71 postinjection. CCl4 leads to liver necrosis (Weber et al., 2003). To reconstitute normal liver size and function the treated liver undergoes cell cycling and regenerates. This in turn leads to loss of episomal DNA. However, as demonstrated in Fig. 7B, hFIX expression in mice that received either wtɸC31 or ɸC31 integrase mutants remained at a similar level, about 800 ng/ml, over time (Fig. 7B). This suggested that the ɸC31 integrase mutants analyzed did not result in enhanced integration activities in murine liver.
Discussion
In the present study we introduced novel ɸC31 integrase derivatives, with a focus on the putative DNA-binding domain at the C terminus of the ɸC31 protein. Our mutagenesis screening was based on a widespread technique, termed alanine scanning, in which charged amino acids are replaced with alanine. Parts of the putative DNA-binding domain (amino acid positions 369–480) served as a template to generate 22 single mutants. These mutants were screened for activity in mammalian cells. Critical amino acids at the C terminus for recombination activities at wild-type and pseudo-attP (attP′) attachment sides were identified.
In a previously published study the N-terminal catalytic domain of the ɸC31 protein (amino acids 1–150) was mutated by error-prone PCR and alanine scanning (Keravala et al., 2009). This approach uncovered ɸC31 integrase mutants P1, P2, and P3 showing up to 2.3-fold improvement in wild-type attB and attP recombination. Furthermore, mutant P2 resulted in stabilized transgene expression levels in mice after transduction of murine hepatocytes. In the future it may be interesting to combine beneficial mutants at the N terminus with mutants in the putative DNA-binding domain at the C terminus. However, it remains to be investigated whether the mutant P2 generated by Keravala and colleagues (2009) also shows improved integration not only in mouse hepatocytes but also in cells of human origin.
To further increase integration efficiency, we investigated the effect of enhancing the integrase plasmid dose on colony formation. Improvements in the integration efficiency of particular single and double mutants could be achieved with a higher plasmid dose (Fig. 6). Because there was no decrease in colony-forming units at high ɸC31 integrase plasmid dose, we concluded that there was no evidence of an overproduction inhibition effect as described for SB transposase. This finding is in contrast to the SB transposase system, because dose dependency studies showed that increasing the dose of SB reduced transposition activity, a phenomenon also referred to as the overproduction inhibition effect (Geurts et al., 2003; Zayed et al., 2004). Although we did not observe such an effect for ɸC31 integrase, it remains unknown whether a large amount of ɸC31 protein molecules within a cell leads to any other negative effects not related to integration activities.
As demonstrated in Fig. 6, we observed various integration efficiencies in different cell lines. This finding is in concordance with earlier studies, because it was shown that ɸC31 integrase efficacy is influenced by the cell line used (Ehrhardt et al., 2006; Maucksch et al., 2008). The study by Maucksch and colleagues suggested that different expression levels of the death domain-associated protein DAXX may be responsible for cell line differences. DAXX protein was shown to be an inhibitor of ɸC31 integrase activity (Chen et al., 2006; Maucksch et al., 2008) and, therefore, cell lines with high expression levels of DAXX may show decreased ɸC31 activity. However, DAXX expression levels in cell lines used in our study remain to be investigated. Another explanation for cell line dependency could be the varying accessibility of attP′ sites present in different cell types or organs. Accessibility, especially around the attP′ sites, may vary in a cell line-dependent manner because of different chromatin structures and transcription patterns. In fact, within mammalian cells, the chromatin represents a highly flexible structure and, according to the needs of the individual cell, chromatin undergoes various structural and epigenetic changes. If the hot spot targeted by ɸC31 integrase displays high transcriptional activity and therefore is present as euchromatin, these DNA sequences could be preferentially targeted. In addition, one may also hypothesize that there are yet unidentified cell type-dependent cofactors of ɸC31 integrase, other than DAXX protein, that could influence ɸC31 integrase activity.
To move further toward gene therapeutic applications, the ɸC31 integrase system was transferred to mice. In contrast to the study by Keravala and colleagues (2009), no beneficial effect on the stability of hFIX expression between wild-type integrase and analyzed mutants was observed. One potential explanation for this finding could be that our chosen mutants for the in vivo study did not show enhanced recombination at the predominant hot spot mpsL1 for ɸC31 integrase-mediated integration into the murine genome (Olivares et al., 2002; Ehrhardt et al., 2005). In our in vitro prescreening experiments we optimized only for recombination in cell lines of human origin with a potentially different integration pattern and varying hot spots compared with mouse hepatocytes. One may also speculate that the accessibility of hot spots within the genome of murine hepatocytes could be different compared with human cell lines because of varying chromatin structures and transcription patterns. In our previous study we found that the hot spot mpsL1 within the murine genome is located within an intron of a predicted gene (Ehrhardt et al., 2005), which may suggest that transcriptional activity occurs. However, experiments analyzing the chromatin structure including the histone code at these particular hot spots within the murine and the human genomes remain to be performed. For instance, chromatin immunoprecipitation to investigate modifications such as acetylation of core histones (histones H2A, H2B, H3, and H4) bound to DNA hot spots for ɸC31-mediated integration may be a suitable method to address this issue.
Several critical amino acid residues within the ɸC31 integrase DNA-binding domain were identified in the present study as having beneficial effects in mammalian cells. In contrast to a previously published study performing mutational analysis of the ɸC31 integrase N-terminal domain (Keravala et al., 2009), we evaluated our mutants directly in mammalian cells; no prescreen in bacteria was performed. However, it remains to be shown whether there is a direct correlation between activity in bacteria and mammalian cells. Other than the DAXX protein, it is not clear at this time whether there are other cellular cofactors in eukaryotic cells that influence ɸC31 activity.
According to secondary structure prediction (Fig. 2) the DNA-binding domain comprises three repetitive β-sheets within positions D370–R394 and a long α-helix comprising positions M417–E484. A putative coiled-coil region (length, about 28 amino acids) with a periodicity of four heptads forming several supercoiled α-helices has also been observed within the binding domain (Rowley et al., 2008; McEwan et al., 2009). In the first β-sheet comprising amino acid sequence 369-MDKLYC-374, mutant K371A was created in this study, showing as low activity as the inactive ɸC31 mutant in the applied assays, indicating that this residue is critical for ɸC31 integrase functionality. Similarly, replacement of the side chain of the positively charged amino acid arginine (R) with the small methyl group of alanine at position 384 (mutant R384A) located between two connecting β-sheets reduced activity dramatically. The dramatic decrease in activity of mutant D421A observed in all three assays suggested that the negatively charged side chain of aspartic acid (D) is also critical for ɸC31 integrase recombination activities.
With respect to integration efficiencies, the most beneficial mutant, D474A, lies within the structural motif of a coiled-coil region between two alanine residues (Fig. 2), in which four α-helices are coiled together in heptamers (Rowley et al., 2008; McEwan et al., 2009). However, the molecular mechanism underlying hyperactivity or inactivity of the respective mutants needs to be further analyzed. For instance, binding assays such as electrophoretic mobility shift assays (EMSAs) may help to understand effects of particular mutant proteins in combination with different attachment sites. Additional analysis of potential attachment sites and thus far unknown binding proteins could be determined and analyzed by DNase I footprinting experiments.
Integration specificity gained great attention because it was shown in the past that integration of the gene of interest at certain sites in the host genome can lead to adverse events. For ɸC31 integrase-mediated integration specificities of up to 15% can be achieved (Chalberg et al., 2006; Ehrhardt et al., 2006). In the present study we identified several ɸC31 integrase mutants by excision assays displaying improved recombination activities between attB and previously described hot spots on chromosomes 2, 12, and 19 (Fig. 4). This may indicate that these sites might represent preferentially targeted recognition sites by the particular integrase mutants within the genomic context as well. However, further studies need to be performed. Although we have begun to analyze integration events of ɸC31 integrase mutants in more detail by applying a plasmid rescue protocol (data not shown), the limited number of identified sites of insertion allows no conclusion with respect to integration specificity.
In the future, determination of the crystal structure would help to apply more rational and likely promising mutagenesis. A large-scale genetic screen involving gene shuffling and exchange of conserved amino acids, as successfully performed with the SB transposase (Mates et al., 2009), will likely result in additional efficiency and specificity mutants. A next step might represent the generation of custom-built recombinases comprising the ɸC31 integrase and a specific DNA-binding domain such as a zinc finger. This approach has already been used for SB transposase and retroviral integrase, which were successfully linked to the zinc finger domain E2C (Tan et al., 2006; Ivics et al., 2007; Yant et al., 2007). Moreover, it may be interesting to take advantage of the inactive ɸC31 mutants identified in this study. Rescue of potentially lost DNA-binding properties by fusion of a zinc finger may be an interesting approach, because now the ɸC31 integrase fusion protein would completely rely on specific DNA binding by the zinc finger and not the natural ɸC31 DNA-binding domain. Notably, in contrast to emerging zinc finger nucleases, which take advantage of the nonspecific DNA cleavage domain FokI, the catalytic domain of ɸC31 integrase responsible for DNA cleavage may be more specific and may result in fewer unwanted side effects. With respect to delivery, it would also be of interest to combine efficient transduction of foreign DNA, using adenoviral vectors with the improved versions of the ɸC31 integrase identified in the present study. Adenovirus-based delivery of the wild-type ɸC31 integrase integration machinery has already been successfully used (Ehrhardt et al., 2007).
In summary, this study describes novel ɸC31 integrase mutants and identified critical amino acid residues within the ɸC31 integrase domain providing DNA binding. We believe that these findings have important implications for rational and improved ɸC31 integrase protein design.
Footnotes
Acknowledgments
This work was supported in part by DFG grants SFB 455 and SPP 1230, EU Framework Programme 7 (Persistent Transgenesis), and the Friedrich Baur Foundation (A.E.). W.Z. was supported by a fellowship from the Chinese Scholarship Council.
Author Disclosure Statement
The authors state that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.