
Abstract
β-Thalassemia is a severe inherited anemia caused by insufficient production of β-globin chains. Allogeneic hematopoietic stem cell (HSC) transplantation is currently the only cure, and is limited by donor availability and regimen-related toxicity and mortality. Gene therapy is a promising therapeutic tool for all thalassemic patients lacking a compatible donor and potentially provides transfusion independence in the absence of transplant-related complications, such as graft rejection and graft-versus-host disease. The issue of HSC procurement is critical in this setting because of the specific features of thalassemic syndromes, which include bone marrow (BM) expansion, ineffective erythropoiesis, and splenomegaly. Little is known about the efficiency of CD34+ cell yield from steady-state BM harvests from thalassemic patients. We have collected data on safety and cell yield from 20 pediatric patients with β-thalassemia who underwent autologous BM harvest before allogeneic HSC transplantation, and from 49 age-matched sibling donors who also underwent BM harvest. The procedure was safe, as no significant adverse events occurred. In terms of cell yield, no difference was found between patients and normal donors in the number of CD34+ cells and total nucleated cells harvested. Most importantly, no difference was found in the proportion of myeloid and erythroid progenitors, suggesting a similar repopulating capacity. On the basis of these results, we conclude that steady-state BM can be used as a safe and efficient source of HSC for gene therapy of β-thalassemia.
Introduction
β-
At present, the only curative approach is represented by allogeneic hematopoietic stem cell transplantation (HSCT). However, this therapeutic option is limited, as only one of four patients has a fully compatible bone marrow (BM) donor. HSCT is also associated with significant morbidities and mortality due to treatment-related toxicity, graft-versus-host disease, and graft rejection (Lucarelli and Gaziev, 2008). Moreover, the use of immunosuppressive drugs can lead to organ toxicity and infectious complications.
A gene therapy approach based on autologous transplantation of genetically corrected hematopoietic stem cells (HSCs) could overcome major HSCT limitations, as it is potentially applicable to all patients and, in the absence of immunological barriers, could provide hematopoietic reconstitution by means of gene-corrected autologous HSCs (Sadelain et al., 2007; Cavazzana-Calvo et al., 2010). We developed a novel β-globin-expressing lentiviral vector, GLOBE, that has proven effective in correcting the major β-thalassemia phenotype both in the murine model (Miccio et al., 2008) and in human thalassemic cells (Roselli et al., 2010).
The issue of procurement of a sufficient number of HSCs for gene therapy remains critical and the source a matter of debate. Current human gene therapy clinical trials use either steady-state BM or peripheral blood stem cells (PBSCs) mobilized by granulocyte colony-stimulating factor (G-CSF). The amount of gene-modified autologous CD34+ cells required to achieve cure is still unknown but clearly depends on the diagnosis and conditioning regimen. In the adenosine deaminase severe combined immunodeficiency (ADA-SCID) trial (Aiuti et al., 2009) the infusion of a mean dose of 8.2 × 106 BM-derived CD34+ cells/kg of body weight (28.6% transduced colony-forming units) resulted in immune reconstitution and normalization of T cell function in 9 of 10 treated patients. In the X-linked SCID (SCID-X1) trial (Hacein-Bey-Abina et al., 2010) a median dose of 4 × 106 BM-derived CD34+γ-chain+ cells/kg (range, 1 × 106/kg to 22 × 106/kg) resulted in reconstitution of the T cell pool and protection from infections. In a patient with BE/B0 thalassemia (Cavazzana-Calvo et al., 2010), a dose of 3.9 × 106 gene-modified BM-derived CD34+ cells/kg (0.6 vector per cell) was successful in conferring transfusion independency. Two adults with X-linked chronic granulomatous disease had sustained engraftment of functionally corrected cells after infusion of 5.1 × 106 and 3.6 × 106 peripheral blood-derived CD34+gp91+ cells/kg, respectively (Ott et al., 2006). Finally, two patients affected by X-linked adrenoleukodystrophy had polyclonal reconstitution and arrest of progressive cerebral demyelination after infusion of, respectively, 4.6 × 106 (50% ALD+) and 7.2 × 106 (33% ALD+) peripheral blood-mobilized CD34+ cells per kilogram of body weight (Cartier et al., 2009).
G-CSF-mobilized PBSCs are an attractive option for patients with thalassemia who are undergoing gene therapy, because of their high yield of CD34+ cells. Although Li and colleagues (1999) reported feasibility of mobilization by G-CSF in thalassemic patients, the apheretic cell yield per kilogram of body weight has not been reported and concerns persist regarding the safety of this procedure in thalassemic patients. In fact, G-CSF-related spleen enlargement (and, occasionally, splenic ruptures) has been described in healthy donors (Falzetti et al., 1999), and these events could occur more frequently in patients affected by β-thalassemia, who often have splenomegaly. Moreover, G-CSF may induce the activation of both coagulation and endothelial cells, which occasionally leads to the development of thrombotic events in healthy donors (LeBlanc et al., 1999; Canales et al., 2002). This provides an additional risk for patients with β-thalassemia, who have a chronic hypercoagulable state (Eldor and Rachmilewitz, 2002; Taher et al., 2008), and could also lead to splenic hemorrhagic infarcts as observed in thalassemic mice treated with G-CSF (Yannaki et al., 2009b). On the other hand, BM harvest is a safe procedure that is routinely performed worldwide in adult and pediatric patients, with a limited risk of morbidities and mortality represented mainly by lumbar back pain and anesthesia-related risks (Styczynski et al., 2008; Halter et al., 2009). Finally, in this clinical setting the repopulating capacity of PBSCs may be inferior to that of BM-derived cells, as G-CSF administration results in the mobilization of more committed and predominantly myeloid stem/progenitor cells (Bender et al., 1991; Fukuda et al., 1994).
We hypothesize that BM could be a possible source of HSCs for gene therapy if a sufficient number of CD34+ cells can be safely harvested from patients with β-thalassemia. To our knowledge, no data are available in the literature to support this hypothesis. We performed 20 BM harvests from transfusion-dependent patients with β-thalassemia as autologous backup before allogeneic HSCT. We describe the safety and efficacy of these procedures and conclude that adequate numbers of CD34+ cells are harvested in most cases. On the basis of these results, on the safety data of BM harvest from international registries, and on the clinical peculiarities of thalassemic patients, we propose BM as the source of HSCs for a future gene therapy clinical application.
Patients and Methods
Patients and healthy donors
We prospectively collected data from 20 pediatric patients affected by transfusion-dependent β-thalassemia (B-thal) undergoing BM harvest at our hospital between 2005 and 2009 as autologous backup to be used in case of graft failure after allogeneic HSCT. Affected children were referred to our hospital from Mediterranean and Middle Eastern countries sponsored by the Mediterranean Institute of Hematology (
BFU-E, erythroid burst-forming unit; CFU-GM, granulocyte-macrophage colony-forming unit; F, female; M, male; NA, not available; TNC, total nucleated cells; Vol, volume.
B-thal, β-thalassemia; F, female; M, male; ND, normal donor; TD, trait donor; TNC, total nucleated cells; Vol, volume.
Results are given as medians (range in parentheses).
Bone marrow harvest
All BM harvests were performed under general anesthesia. Written informed consent was obtained from the donors' parents or legal guardians in accordance with Italian law. BM collection was carried out via sequential single-hole 3- to 5-ml aspirations taken from the anterior and posterior iliac crests. The target cell dose was >2 × 108 total nucleated cells (TNCs)/kg. The maximal safe volume collected was set at 30 ml/kg of donor weight. Mid-harvest nucleated cell count was used to set the total volume to be harvested. At the end of the procedure, local 0.75% ropivacaine (Naropin, 4 mg/kg) was injected into the aspiration sites. Postoperative pain and antiinfection prophylaxis were managed by intravenous tramadol, paracetamol, and prophylactic ceftriaxone according to local protocols. On the basis of the volume yielded, the preharvest hemoglobin value, and the hemodynamic stability of the subject, irradiated allogeneic red blood cells were transfused during or after the procedure. Patients and donors were expected to be discharged 2 days after the procedure. All grafts were evaluated for total BM volume collected, TNC dose, and number of CD34+ cells. TNCs and CD34+ cells were determined by flow cytometry for expression of CD45 (TNCs) and CD34. BM volumes and cell yields are reported as absolute numbers and per kilogram of body weight, referring to the weight of the individual undergoing harvest. The ability of individual early progenitors to give rise to erythroid colonies (burst-forming unit-erythroid, BFU-E) or colony-forming units granulocyte-macrophage (CFU-GM) was studied by colony-forming unit (CFU) assay. CFU assay was performed starting from both fresh and cryopreserved samples. TNCs were plated in duplicate in 35-mm tissue culture dishes at 2 × 104 or 103 cells/ml, respectively, in semisolid methylcellulose medium supplemented with stem cell factor (SCF), GM-CSF, interleukin-3, and erythropoietin (MethoCult GFH84434 and MethoCult H4534; StemCell Technologies, Vancouver, BC, Canada). Cultures were incubated at 37°C in a humidified atmosphere with 5% CO2 and scored after 14 days of incubation. CFU-GM were analyzed and scored from fresh BM samples. Subsequently, the CFU assay was repeated from cryopreserved samples in order to score both BFU-E and CFU-GM. Absolute numbers represent CFU-GM colonies derived from fresh samples, and percentages represent CFU-GM and BFU-E colonies derived from cryopreserved samples. In nine cases, sufficient amounts of CD34+ purified cells were available to perform CFU-GM and BFU-E assays.
Statistical analysis
For continuous variables with a symmetric distribution, the results are expressed as medians and ranges. Comparison among patients' and donors' harvest biological characteristics was performed by linear regression analysis. Any other comparison was done by Student two-tailed t test. A p value less than 0.05 was considered statistically significant.
Results and Discussion
Twenty pediatric patients affected by transfusion-dependent β-thalassemia (B-thal), 29 healthy β-thalassemia carrier donors (TDs), and 20 healthy donors (NDs) underwent BM harvest.
Concerning the safety of BM harvest, none of the B-thal patients or healthy donors had any severe adverse event during or after the procedure and all subjects were discharged within 3 days of harvest. The most frequently reported side effect was self-limiting pain at the harvest site, and transient limping or weakness during walking was also occasionally reported. The median hemoglobin value after BM collection was 7.8 g/dl, with an average decrease of 2.3 g/dl compared with the preharvest value. All B-thal patients received allogeneic red blood cell transfusions at a dose of 15 ml/kg during or immediately after the procedure, resulting in restoration of preharvest hemoglobin (Hb) value.
Concerning the efficiency of BM harvest from B-thal patients, a median volume of 25.89 ml/kg (range, 10.58–33.33 ml/kg), corresponding to a total of 599 ml (range, 275–760 ml), was harvested. A median of 3.24 × 108 TNCs/kg (range, 1.07–7.14 TNCs/kg) was collected, corresponding to 13.08 × 106 TNCs/ml BM (range, 5.15–28.56 TNCs/ml BM). Median CD34+ cells harvested were 7.06 × 106 cells/kg (range, 2.78–20.25 cells/kg), corresponding to 0.26 × 106 cells/ml BM (range, 0.10–0.82 cells/ml BM). CD34+ cell yields did not differ depending on severity of B-thal genotype (β0β+ and β+β+ vs. β0β0; p = 0.09). Similarly, no difference was found between CD34+ cell yields from patients in low or high Pesaro risk classes (class I/II vs. class III; p = 0.89) or between splenectomized and nonsplenectomized patients (p = 0.97). Moreover, when CD34+ cell yield was correlated to the degree of iron overload, no significant difference was found (p = 0.8 for serum ferritin and p = 0.96 for liver iron concentration). These data suggest that HSC yield in thalassemic patients is not affected by disease-specific characteristics or iron overload.
Because no data are available in the literature regarding the efficiency of BM harvest in B-thal patients, we compared these yields with those from age- and ethnicity-matched healthy subjects, either NDs (n = 20) or TDs (n = 29) (Table 2). The concentration of CD34+ cells in B-thal BM was similar to that of healthy subjects (median, 0.26 × 106/ml in B-thal, 0.36 × 106/ml in TDs, and 0.24 × 106/ml in NDs; B-thal vs. TDs, p = 0.47; B-thal vs. NDs, p = 0.3) (Fig. 1), as was the total amount collected (median, 7.06 × 106 CD34+ cells/kg in B-thal, 8.57 × 106 CD34+ cells/kg in TDs, 5.5 × 106 CD34+ cells/kg in NDs; B-thal vs. TDs, p = 0.43; B-thal vs. NDs, p = 0.20). As expected, the CD34+ cell yield was positively correlated with the number of TNCs (B-thal, p = 0.0009; TDs, p < 0.0001; NDs, p = 0.02) and negatively correlated with the BM volume collected (B-thal, p = 0.01; TDs, p = 0.0006; NDs, p < 0.0001), suggesting that the efficiency of CD34+ cell yield progressively decreases during harvest, probably due to hemodilution. Regarding individual parameters such as age and weight, no correlation with CD34+ cell yield was observed in B-thal patients (age/yr, p = 0.52; weight/kg, p = 0.3). This differs from what is usually seen in healthy donors (Kao et al., 2009) and from what we observed in our series, where CD34+ cell yield correlated indirectly with donor age (NDs, p = 0.0002; TDs, p 9 0.0001) and weight (NDs, p = 0.005; TDs, p = 0.01). Although we have no explanation for this dissimilarity, reduced body weight secondary to growth retardation of the elderly B-thal subjects in our series and stressed hematopoiesis due to chronic anemia might be contributing factors. The results reported so far suggest that HSC yield in thalassemic patients does not differ from that in healthy donors and that the efficiency of progenitor yield does not decrease with patient age.
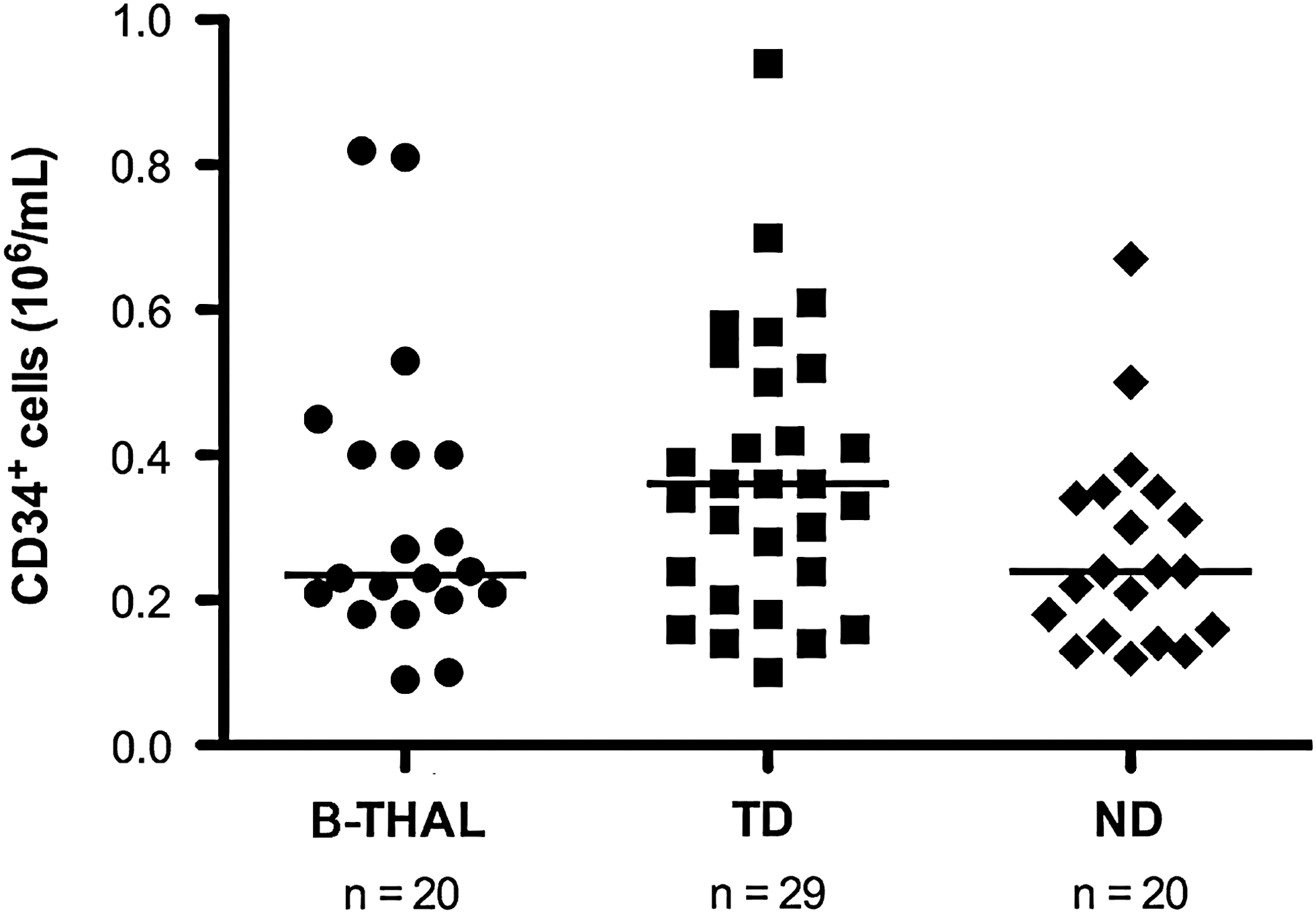
Concentration of CD34+ cells (106/ml) in bone marrow (BM) harvests from patients with β-thalassemia and donors. B-THAL, patients with β-thalassemia; ND, healthy donors; TD, β-thalassemia carrier donors. Horizontal bars represent the median.
All harvests were analyzed for routine BM morphology. We found an expected expansion in the erythroid compartment in B-thal patients (erythroid cells, 70%; range, 40–86%) compared with TDs (erythroid cells, 40%; range, 14–66%; p < 0.0001) and NDs (erythroid cells, 35%; range, 14–60%; p < 0.0001). However, this expansion influenced neither the yield of CD45+ and CD34+ cells nor the proportion of myeloid and erythroid progenitors, as shown later.
To assess the clonogenic potential of early progenitors, the proportion of myeloid (CFU-GM) and erythroid (BFU-E) progenitors was calculated from colonies derived from cryopreserved TNCs. Concerning CFU-GM proportion, no difference was observed between the B-thal group (55.95%; range, 40.84–72.88%), NDs (59.62%; range, 41.43–70.11%; p = 0.07), and TDs (59.52%; range, 44.63–75.82%; p = 0.17). Unexpectedly, in spite of the expansion of the erythroid compartment, the BFU-E percentage was not significantly increased in thalassemic individuals (44.05%; range, 27.12–59.16%) compared with NDs (40.38%; range, 29.89–58.57%; p = 0.19) or TD individuals (40.48%; range, 24.18–55.37%; p = 0.09) (Fig. 2A). These results suggest that the erythroid expansion characteristic of thalassemic syndromes is probably caused by more differentiated precursors lacking clonogenic potential.
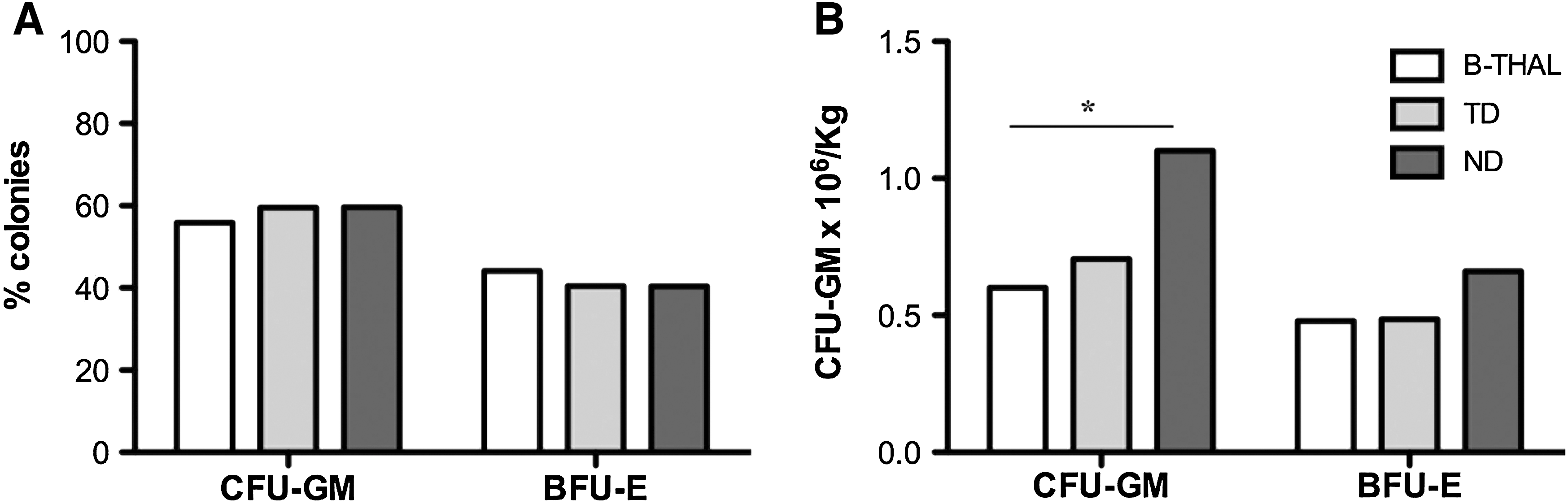
Clonogenic assay performed on BM samples from β-thalassemia patients and donors. (
Conversely, CFU-GM absolute numbers derived from freshly harvested TNCs were halved in B-thal patients compared with healthy donors (0.6 × 106/kg in B-thal vs. 1.1 × 106/kg in NDs; p = 0.03) but similar to thalassemia carrier donors (0.69 × 106/kg in TDs; p = 0.61). No significant difference was observed between splenectomized and nonsplenectomized patients (0.78 × 106/kg vs. 0.54 × 106/kg; p = 0.23). Although CFU-GM in B-thal samples were reduced compared with NDs, the amount of myeloid progenitors was higher than 0.35–0.5 × 106/kg, a cutoff considered predictive of successful engraftment (Miyamoto et al., 2004). Assuming a similar loss of myeloid and erythroid progenitors after thawing (Attarian et al., 1996; Almici et al., 1997; Kim et al., 2007), based on the number of CFU-GM obtained from fresh samples and the CFU-GM and BFU-E proportions obtained from cryopreserved samples, the estimated BFU-E were 0.48 × 106/kg for B-thal, 0.66 × 106/kg for NDs, and 0.49 × 106/kg for TDs. Once more, although the B-thal group showed a reduced number of colonies compared with NDs, the difference was not statistically significant (p = 0.18 and p = 0.68 for NDs and TDs, respectively) (Fig. 2B).
Finally, considering that purified CD34+ cells will be the cellular target for a gene therapy clinical protocol, in nine cases a sufficient amount of cells was available to perform a clonogenic assay. Similar percentages of myeloid and erythroid progenitors were obtained from unseparated BM mononucleated cells and purified CD34+ cells (CFU-GM: 53.86 vs. 58.81%, p = 0.29; BFU-E: 46.14 vs. 41.19%, p = 0.18).
The correlation between the number of CD34+ cells/kg recipient body weight and engraftment is well known. However, no absolute minimal threshold of BM-derived CD34+ cells has been established to guarantee engraftment. The accepted recommendation is to harvest >2 × 108 TNCs/kg or >2 × 106 CD34+ cells/kg for allogeneic HSCTs. Considering that correction of the whole progenitor cell compartment is not essential for amelioration of the thalassemia phenotype, as observed in patients with stable mixed chimerism after HSCT (Andreani et al., 2008), and considering that genetically corrected cells show a selective advantage in vivo in the murine model (Miccio et al., 2008), we hypothesize that the harvest of a double amount of CD34+ cells compared with what is required for allogeneic HSCT should be sufficient for a successful gene therapy approach, in order to balance cell loss due to the transduction procedure. Indeed, the high transduction efficiency of the GLOBE vector (Roselli et al., 2010) should provide an adequate number of genetically corrected stem/progenitor cells. In our opinion, the number of stem/progenitor cells obtained from a single harvest from B-thal patients in this series was nearly always adequate to be employed in a gene therapy clinical protocol.
Although G-CSF-mobilized PBSCs are a well-established source of progenitor cells, G-CSF-induced hyperleukocytosis and vascular activation can result in splenic infarcts and thromboembolic events in both adults and children (Pulsipher et al., 2006). These events may be more frequent or severe in subjects with extramedullary erythropoiesis and splenomegaly, such as B-thal patients. Moreover, even if G-CSF HSC mobilization has been reported in thalassemic patients (Li et al., 1999), kinetics mobilization was suboptimal and CD34+ cell yield was modest, as 11 of 20 patients required more than one apheresis to achieve the minimal target of 1 × 106/kg. In addition, preliminary data by Yannaki and colleagues on G-CSF administration to B-thal patients have shown modest cell yields, especially in splenectomized patients, because of cautious G-CSF dose reduction due to excessive early leukocytosis (Yannaki et al., 2009a). In the future, new mobilizing agents, such as the antagonist of CXCR4, AMD3100 (Pusic and Dipersio, 2010), could prove superior to G-CSF-mobilized PBSCs or steady-state BM as HSC sources for gene therapy. AMD3100 may actually spare patients from hyperleukocytosis and prothrombotic diathesis, and promote a higher yield of activated (and therefore more easily transducible) CD34+ cells (Larochelle et al., 2006) with high engraftment capacity (Hess et al., 2007). Only comparative repopulating experiments will tell the preferable source of HSCs for gene therapy of thalassemia.
In summary, in our experience, BM harvest from B-thal patients is a safe procedure and the yield of HSCs does not differ from what is expected from normal donors. Moreover, CD34+ cell yield in B-thal pediatric patients did not correlate with thalassemia or iron overload-related clinical features, age, or body weight.
As a result of these clinical data, we are currently applying to the Italian authorities for a phase I–II gene therapy clinical trial using BM as the source of HSCs.
Footnotes
Author Disclosure Statement
No competing interests exist.