
Abstract
The recent development of induced pluripotent stem cells (iPSCs) by ectopic expression of defined reprogramming factors offers enormous therapeutic opportunity. To deliver these factors, murine leukemia virus (MLV)-based vectors have been broadly used in the setting of hematopoietic stem cell transplantation. However, MLV vectors have been implicated in malignancy induced by insertional mutagenesis, whereas lentiviral vectors have not. Furthermore, the infectivity of MLV vectors is limited to dividing cells, whereas lentiviral vectors can also transduce nondividing cells. One important characteristic of MLV vectors is a self-silencing property of the promoter element in pluripotent stem cells, allowing temporal transgene expression in a nonpluripotent state before iPSC derivation. Here we test iPSC generation using a novel chimeric vector carrying a mutant MLV promoter internal to a lentiviral vector backbone, thereby containing the useful properties of both types of vectors. Transgene expression of this chimeric vector was highly efficient compared with that of MLV vectors and was silenced specifically in human embryonic stem cells. Human fetal fibroblasts transduced with the vector encoding each factor were efficiently reprogrammed into a pluripotent state, and these iPSCs had potential to differentiate into a variety of cell types. To explore the possibility of iPSCs for gene therapy, we established iPSC clones expressing a short hairpin RNA (shRNA) targeting chemokine receptor 5 (CCR5), the main coreceptor for HIV-1. Using a reporter construct for CCR5 expression, we confirmed that CCR5 shRNA was expressed and specifically knocked down the reporter expression in iPSCs. These data indicate that our chimeric lentiviral vector is a valuable tool for generation of iPSCs and the combination with vectors encoding transgenes allows for rapid establishment of desired genetically engineered iPSC lines.
Introduction
A number of delivery methods have been used to express the reprogramming factors in cells (for review, see Maherali and Hochedlinger, 2008). However, the most efficient means of generating iPSCs for experimental investigation is through the use of murine leukemia virus (MLV) vectors that stably integrate the proviral DNA into the host genome (for review, see Amabile and Meissner, 2009). An MLV-based vector was the first to be used for an iPSC derivation in mouse and human fibroblasts by Yamanaka and colleagues (Takahashi and Yamanaka, 2006; Takahashi et al., 2007). The importance of using vectors based on MLV lies in the fact that the promoter element within the MLV long-terminal repeat (LTR) is silenced in ESCs (Wolf and Goff, 2007; Wolf et al., 2008a,b). The self-silencing property allows the temporal expression of the factors during reprogramming, but then abrogates the expression of reprogramming factors to maintain cells in an "ESC-like" state (Niwa et al., 2000; Mikkelsen et al., 2008) and to proceed to efficient differentiation (Brambrink et al., 2008). Human immunodeficiency virus type-1 (HIV-1)–based lentiviral vectors have also been used for the transduction of reprogramming factors as well as MLV vectors because of their greater efficiency to infect human cells, including nondividing cells (Blelloch et al., 2007; Yu et al., 2007; Papapetrou et al., 2009). However the internal promoters are generally not efficiently silenced in a pluripotent state (Lois et al., 2002; Pfeifer et al., 2002; Gropp et al., 2003; Ma et al., 2003; Hong et al., 2007; Xia et al., 2007), making the constitutive gene expression and resulting in an impairment of the proper differentiation of iPSCs (Niwa et al., 2000; Brambrink et al., 2008; Mikkelsen et al., 2008).
One potential issue in the use of MLV vectors is their association with malignant transformation in a few cases of patients treated for X-linked severe combined immunodeficiency (Hacein-Bey-Abina et al., 2003; McCormack et al., 2003) and for X-linked chronic granulomatous disease (Ott et al., 2006). This is due in part to insertional mutagenesis as a result of integration adjacent to cellular oncogenes. In fact, MLV is well-known to induce disease naturally in mice by insertional mutagenesis (Mikkers and Berns, 2003). In contrast, lentiviral vectors have not shown evidence of insertional mutagenesis in natural HIV-1 infection or in animal model systems of lentiviral vector gene transfer. Indeed, direct comparison of the two vectors in mice genetically prone to cancer demonstrates that MLV vectors, but not lentiviral vectors, induce cancer (Montini et al., 2006, 2009).
We previously described a series of chimeric vectors containing MLV promoters internal to a lentiviral vector backbone (Kung et al., 2000). These vectors had the advantages of lentiviral vectors for efficient transduction of human cells and expression based on the properties of the internal MLV promoter. One of these vectors was constructed using a mutant MLV promoter (RhMLV) isolated from a rhesus macaque T-cell tumor that had much higher expression in human cells compared with the wild-type MLV promoter (Vanin et al., 1994; Kung et al., 2000). We considered whether this vector would be suitable for iPSC formation because of high lentiviral transduction efficiency and high levels of expression driven by this mutant MLV promoter.
Here, we test the properties of the chimeric MLV/lentiviral vector and demonstrate that it can be used to generate iPSCs. Furthermore, for future HIV-1 genetic therapy we show proof of concept that the iPSC can be genetically modified with other lentiviral vectors through cotransduction of somatic cells prior to reprogramming.
Materials and Methods
Construction of lentiviral vector
For iPSC generation, we used an FG12-based lentiviral vector encoding EGFP (An et al., 2007). The ubiqutin C promoter in FG12 was substituted with the RhMLV promoter (FRh11) derived from the LTR region of the MLV vector in the serum of one rhesus macaque monkey that developed T-cell lymphoma following autologous transplantation (Vanin et al., 1994; Kung et al., 2000). cDNAs encoding human OCT4 (Plasmid 17217), SOX2 (Plasmid 17218), KLF4 (Plasmid 17219), cMYC (Plasmid 17220), NANOG (Plasmid 18115), and LIN28 (Plasmid 16350) were purchased from Addgene (Addgene Inc., Cambridge, MA) and substituted with EGFP in FRh11.
Virus production and titration
Lentiviral vector stocks were generated using a vector plasmid, a packaging plasmid pCMV R8.2 ΔVpr, and a VSV-G envelope protein-coding plasmid by calcium phosphate-mediated transient transfection as previously described (Kamata et al., 2009). After 48 and 72 hr, lentiviral vector particles were harvested, concentrated by ultracentrifugation, resuspended in a 150-fold lower volume of Hanks' balanced salt solutions, and stored at −80°C. The viral titer was measured by anti-p24 Gag ELISA, and the infectious titer was determined in 293T cells by infecting with FRh11 in the presence of 8 μg/ml polybrene.
MLV vector stocks were generated by using a vector plasmid described above and packaging plasmid pCL-Ampho (Imgenex, San Diego, CA) by calcium phosphate-mediated transient transfection. After 48 hr, MLV vector particles were harvested, concentrated 100-fold by Amicon Ultra-100 (Millipore, Temecula, CA), and stored at −80°C. The infectious titer was determined in 293T cells by infecting with MLV vector encoding EGFP (pMX-GFP, Cell Biolabs, Inc., San Diego, CA) in the presence of 8 μg/ml polybrene. Reporter gene expression was monitored by flow cytometry.
Cell culture
293T and NTERA-2 cells were maintained with Dulbecco's modified Eagle medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Omega Scientific, Tarzana, CA) and 2 mM GlutaMAX (Invitrogen). All cells were incubated at 37°C and 5% CO2.
hESCs (H1 clone) and human iPSCs (hiPSCs) were maintained in mTeSR1 (StemCell Technologies, Inc., Vancouver, Canada) on hESC-qualified Matrigel (BD Biosciences, San Jose, CA) coated plates. Differentiated colonies were removed daily through aspiration, and medium was replaced on a daily basis. Cells were passed upon confluency (typically 7–10 days), using 1 mg/ml dispase (StemCell Technologies). All work with hESCs and hiPSCs was approved by the UCLA Embryonic Stem Cell Research Oversight Committee.
Human fetal fibroblasts (HFFs) isolated from the skin of a 16-week-old fetus were expanded with DMEM supplemented with 10% FBS and 2 mM GlutaMAX (fibroblast medium) as reported previously (Normand and Karasek, 1995).
Induction of hiPSCs
The day before lentiviral vector transduction, HFFs (passage 1–3) were seeded at 5 × 104 cells per well of six-well plates and infected with the vectors encoding each reprogramming factor (OCT4, SOX2, KLF4, cMYC, NANOG, and LIN28) at 300 ng (around multiplicity of infection [MOI] of 3–5) of p24 per each virus. Cells were cultured for 3 days in fibroblast medium and replated at 5 × 104 cells per 60-mm dish on an irradiated mouse embryonic fibroblast (MEF) feeder layer. On the next day, the medium was replaced with Knockout DMEM (Invitrogen) supplemented with 20% Knockout Serum Replacer (KSR, Invitrogen), 2 mM GlutaMAX (Invitrogen), 0.1 mM nonessential amino acids (Invitrogen), 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO), 50 ng/ml recombinant human basic fibroblast growth factor (Invitrogen) (hiPSC medium), and also added 0.5 mM valproic acid (Sigma-Aldrich) and 10 μM Y27632 (Tocris Bioscience, Ellisville, MO) for the first 14 days (Huangfu et al., 2008; Park et al., 2008). The medium was changed on a daily basis. On days 21–25, iPSC colonies were identified based on ESC-like morphology as described previously (Takahashi et al., 2007) and picked out into wells of 48-well plates coated with Matrigel and expanded in mTeSR medium. Reprogramming efficiency was calculated as the number of iPSC colonies formed per number of seeded HFFs infected with reprogramming factors.
Embryoid body (EB) formation
Size-controlled EBs (3,000 cells/EB) were formed using AggreWell 400 plates (StemCell Technologies) following the manufacturer's protocol. In brief, hESCs and hiPSCs were incubated with 10 μM Y27632 (ROCK) inhibitor for 24 hr before EB formation. Cells were harvested with Accutase (Innovative Cell Technologies, San Diego, CA) as a single-cell suspension and used for EB formation. EBs were harvested into ultra low attachment plates (Corning, Corning, NY) and maintained in mTeSR. Differentiations into a neural lineage (Chambers et al., 2009), cardiomyocyte (Leschik et al., 2008), and hematopoietic progenitor (Subramanian et al., 2009) were performed following reported protocols.
Flow cytometry
For detection of EGFP and mCherry expressions, single-cell suspensions from 293T, NTERA-2, hESC, and hiPSC were prepared using 0.25% trypsin-EDTA and collected in FACS buffer (2% FBS and 0.01% sodium azide in PBS). For detection of hESC-specific markers, cells were adjusted to 100,000 per sample in 100 μl of FACS buffer and then labeled with monoclonal antibodies conjugated with fluorescent dye (SSEA1 and SSEA3: Alexa488 purchased from eBioscience [San Diego, CA]; SSEA4 [eBioscience]; TRA-1-60 and TRA-1-81: PE purchased from BioLegend [San Diego, CA]). Data were collected on a Cytomics FC500 (Beckman Coulter, Fullerton, CA) and analyzed using FCS Express (De Novo Software, Los Angeles, CA).
Alkaline phosphatase (AP) staining and immunocytochemistry
hESC and hiPSC colonies were grown on poly-L-lysine- and Matrigel-coated glass coverslips. Cells were fixed with 1.0% formaldehyde/PBS for 30 min and permeabilized with 0.2% Triton X-100/PBS for 5 min on ice. Cells were then incubated with anti-human Nanog antibody (Abcam Inc., Cambridge, MA), and subsequently with DyLight 488 conjugated donkey anti-rabbit IgG (BioLegend) and 7-aminoactinomycin D (7-AAD) (Invitrogen) for nuclear staining. After washing, cells were visualized with a LEICA DM IRB (Leica Microsystems Inc., Bannockburn, IL) equipped with a SPOT camera and software (Diagnostic Instruments, Sterling Heights, MI).
AP staining was performed with an AP detection kit (Millipore) following the manufacturer's instructions.
RNA extraction and RT-PCR
Total RNAs were extracted from hESCs and hiPSCs using the RNeasy Mini kit following the manufacturer's protocol (QIAGEN, Valencia, CA). The optional DNase I treatment was included, using the RNase-Free DNase Set (QIAGEN). Two hundred fifty nanograms of total RNA was reverse-transcribed using the Omniscript RT kit (QIAGEN) with a 0.5 ng/ml oligo(dT) primer (Invitrogen) in a 20-μl reaction. PCR was performed with the HotMaster Taq DNA polymerase (5 PRIME, Inc., Gaithersburg, MD), using 0.5 μl of cDNA template and primers at a concentration of 3 pmol/μl. Five microliters of PCR products was loaded in a 2% agarose gel containing ethidium bromide. All primer sequences are listed in Table 1.
Detection of vector integration by Alu-LTR nested PCR
Lentiviral vector integration analysis was performed by Alu-LTR nested PCR as described previously (Liszewski et al., 2009). Genomic DNA was isolated from hESCs either uninfected or infected with FG12 or FRh11 using DNeasy Blood & Tissue Kit (QIAGEN). Provirus-genomic junction fragments were amplified by PCR with primers for HIV gag (5'-GCTCTCGCACCCATCTCTCTCC-3') and Alu (5'-TCCCAGCTACTGGGGAGGCT-3') (1st PCR) using the JumpStart Taq DNA polymerase (D9307, Sigma-Aldrich) with the following cycles: 1 × 94°C for 2 min, 20 × (94°C for 0.5 min, 50°C for 1 min, 72°C for 1.5 min). First PCR products were purified with the QIAGEN PCR Purification kit and eluted with 50 μl of water. For nested PCR, 5 μl of purified 1st PCR product was amplified with specific primers for HIV-1 LTR; NL9076F (5'-TGGAAGGGCTAATTCACTCC-3') and NL9181R (5'-CCTGGCCCTGGTGTGTAGT-3') with the following cycles: 30 × (94°C for 10 sec, 58°C for 20 sec, 72°C for 10 sec). Five microliters of nested PCR products was loaded in a 2% agarose gel containing ethidium bromide.
Western blotting
Western blotting analysis was performed as described previously (Kamata et al., 2009). To compare the expression levels of reprogramming factors, 293T cells were infected with either lentiviral or MLV vector expressing each reprogramming factor (OCT4, SOX2, KLF4, or cMYC). Cells were lysed with 0.5% SDS containing protease inhibitor cocktail (P8340, Sigma-Aldrich) and quantified with a BCA protein assay reagent (Bio-Rad, Hercules, CA). The same amount of total protein (10 μg) was analyzed on 4–20% Precast SDS-PAGE gel (Lonza, Rockland, ME) and transferred onto Immobilon membranes (Millipore). Membranes were reacted with one of the following antibodies: Oct4, anti-Oct3/4 monoclonal antibody (SC-5279, Santa Cruz); Sox2, anti-Sox2 rabbit polyclonal antibody (AB5603, Millipore); Klf4, anti-GKLF rabbit polyclonal antibody (SC-20691, Santa Cruz); and cMyc, anti-cMyc mouse monoclonal antibody (SC-42, Santa Cruz). Membranes were then treated with secondary antibodies conjugated with horseradish peroxidase (Pierce, Rockford, IL) and visualized by chemiluminescence (ECL Plus, Amersham Biosciences, Piscataway, NJ).
Results
Transgene expression driven by the RhMLV promoter is substantially diminished in human embryonic carcinoma (EC) cells and hESCs
The RhMLV promoter was inserted into a self-inactivating lentiviral vector backbone to generate vector FRh11 (Fig. 1A). In this context, the RhMLV promoter drives expression of the reprogramming factors, but transduction and accompanying events in the viral life cycle are mediated by the lentiviral vector. In principle, this vector should exhibit the relatively higher transduction efficiencies of lentiviral vectors in human cells, as well as the silencing of the MLV promoter, which could have an important role for reprogramming. The parental lentiviral vector, FG12 (An et al., 2007), with ubiquitin C as an internal promoter is used as a control (Fig. 1).
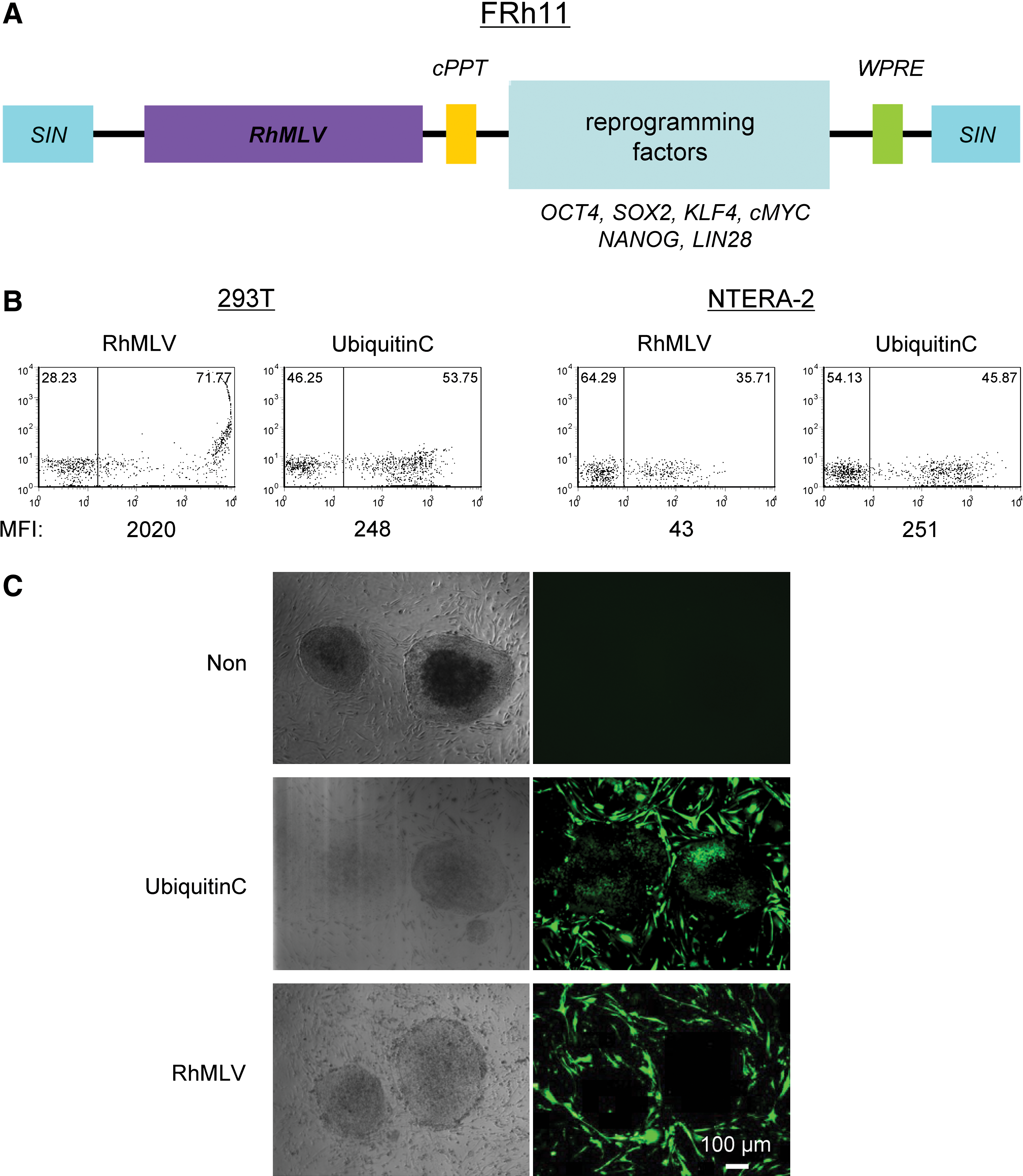
Transgene expression driven by the RhMLV promoter is substantially diminished in human EC cells and ESCs.
Transduction of 293T cells with the lentiviral vectors showed that both ubiquitin C and RhMLV promoters expressed EGFP as a reporter (Fig. 1B). In contrast, in an EC cell line, NTERA-2, the ubiquitin C promoter expressed EGFP at a similar level assayed by mean fluorescent intensity (MFI) in 293T cells, but the expression from the RhMLV promoter was substantially diminished, consistent with silencing of the RhMLV promoter in NTERA-2 cells. We further tested the expression from the RhMLV promoter in the hESC line, H1 (Fig. 1C). The expression was strongly suppressed in hESCs (Fig. 1C, RhMLV), whereas that from the ubiquitin C promoter was active (Fig. 1C, Ubiquitin C). As a control, mouse fibroblast (iMEF) feeder cells surrounding the hESC colonies expressed EGFP by both ubiquitin C and RhMLV promoters. For semiquantitative analysis of these promoter activities, hESCs were plated on Matrigel and infected with a lentiviral vector encoding EGFP driven by either the ubiquitin C or the RhMLV promoter. The levels of EGFP expression were analyzed by flow cytometry after 2 weeks of culture (Fig. 2). Semiquantitative genomic PCR for integrated proviruses revealed that both vectors were transduced into hESCs at similar levels (Fig. 2B). The expression from the RhMLV promoter, however, was strongly diminished; it was 20-fold lower than that from the ubiquitin C promoter by MFI (Fig. 2A). These results indicate that the transgene expression of the RhMLV promoter in the context of the lentiviral vector is silenced in human EC cells or hESCs, although some residual expression remains.
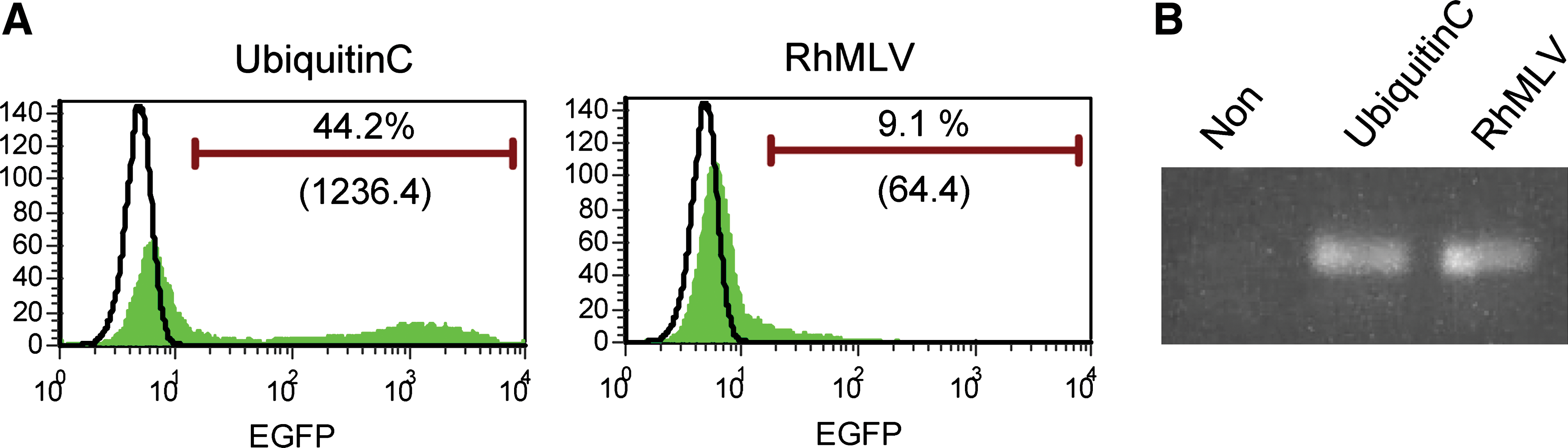
Semiquantitative analysis of transgene expression by the RhMLV and the ubiquitin C promoters in hESCs. hESCs grown on Matrigel under feeder-free conditions were infected with a lentiviral vector encoding EGFP driven by either the ubiquitin C or the RhMLV promoter at an MOI of 10. After 2 weeks of culture, the cells were collected with 0.25% trypsin and analyzed for expression levels of EGFP by flow cytometry
Generation of hiPSCs with the FRh11 vector
We then tested the generation of hiPSCs with FRh11 vectors encoding each reprogramming factor (OCT4, SOX2, KLF4, and cMYC) using fibroblasts isolated from dermal skin of a 16-week-old fetus (HFFs). We first compared the expression levels of each reprogramming factor by the FRh11 vector and the MLV vector, pMXs, used by Yamanaka and colleagues for hiPSC derivation (Takahashi et al., 2007) (Fig. 3B). In transduced 293T cells, the RhMLV promoter expressed each reprogramming factor at levels higher than the MLV vector. We used a combination of each vector individually expressing a single reprogramming factor to transduce HFFs and generate iPSCs following the schedule shown in Fig. 3A. Colonies with the characteristic morphological appearance of the iPSCs were isolated between days 21 and 28 (Fig. 3C). These colonies were easily distinguished from the transformed colonies; hiPSC colonies are characterized by small and tightly packed colonies with smooth borders similar to hESCs (Fig. 3C, Day 21), whereas transformed colonies are mostly larger than those of hiPSCs and configured with heavily granulated cells as reported previously (Takahashi et al., 2007; Yu et al., 2007). In some cases, we used six factors (OCT4, SOX2, KLF4, cMYC, NANOG, and LIN28); in others, we cotransduced with a vector carrying short hairpin RNA (shRNA) for chemokine receptor 5 (CCR5) (sh1005) or the mutant (mutant sh1005) with four factors described above (Table 2). In all cases, the frequency of hiPSC formation was comparable and was similar or slightly higher than that using other vector systems (for review, see Kiskinis and Eggan, 2010). Cells from all of the hiPSC colonies were positive for Nanog (one representative clone is shown in Fig. 4B). Twelve independent hiPSC clones were randomly selected: eight clones from four factors with no therapeutic gene, two clones from four factors with the sh1005 (sh1005 #1 and #10), and two clones from four factors with the mutant sh1005 (mu. sh1005 #1 and #11) from established hiPSC clones; these were further examined for the expression profile of stem-cell specific markers. All 12 clones were positive for SSEA3, SSEA4, TRA-1-60, and TRA-1-81, but negative for SSEA1, similar to hESCs (one representative clone is shown in Fig. 4C). These iPSC clones were differentiated into EBs (one representative clone is shown in Fig. 4D). We further confirmed the expression levels of hESC-specific mRNAs by reverse transcriptase PCR (RT-PCR) analysis (Fig. 5). Twelve selected hiPSC clones were all positive for NANOG, REX1, LIN28, UTF1, DPPA5, hTERT, DNMT3B, OCT4, and SOX2, similar to hESCs. As expected, the expression of factors from the RhMLV promoter was reduced in hiPSC clones. The EBs could be further differentiated into cells with characteristics of hematopoietic progenitors, positive for CD34 and CD43 (Choi et al., 2009) (data not shown), a neural lineage with neural tube-like rosettes (Chambers et al., 2009) (data not shown), and beating cardiomyocytes (Leschik et al., 2008) (data not shown). These results indicate that the chimeric lentiviral vector carrying the RhMLV promoter as an internal promoter is a useful tool for hiPSC generation.
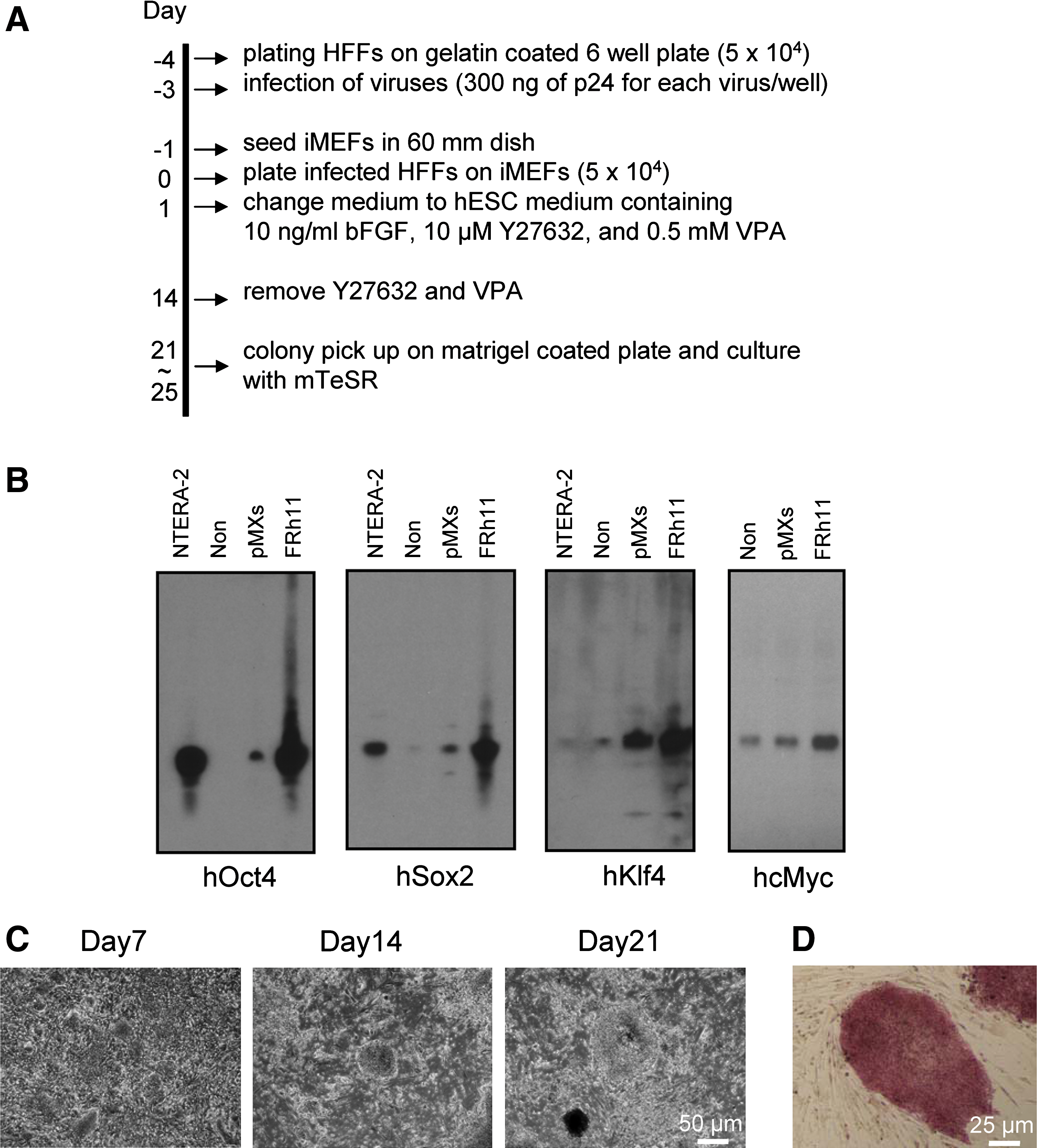
Induction of hiPSCs from HFFs with the FRh11 encoding human reprogramming factors.
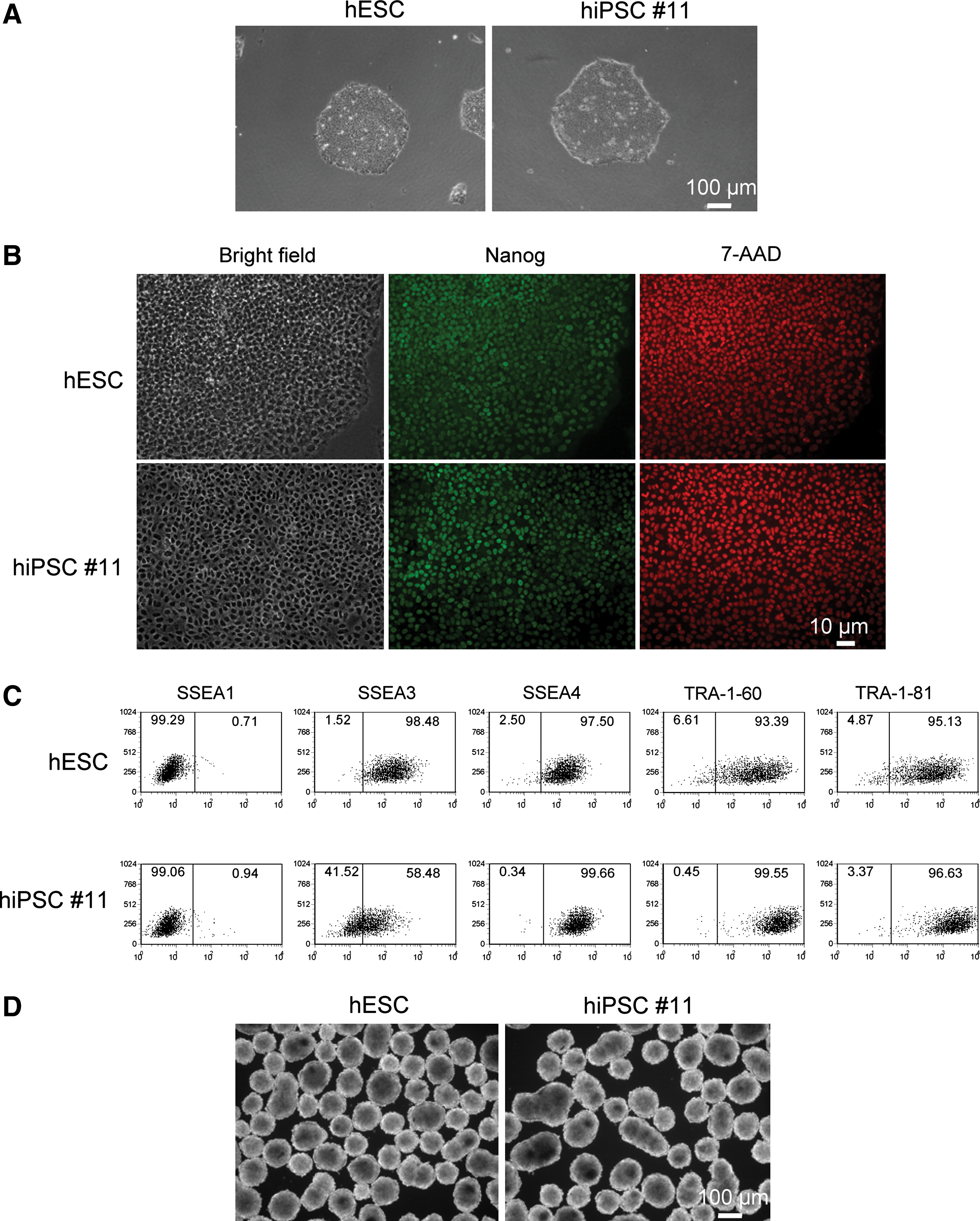
hiPSC clones derived from HFFs by infection of the FRh11 encoding human reprogramming factors share hESC character.
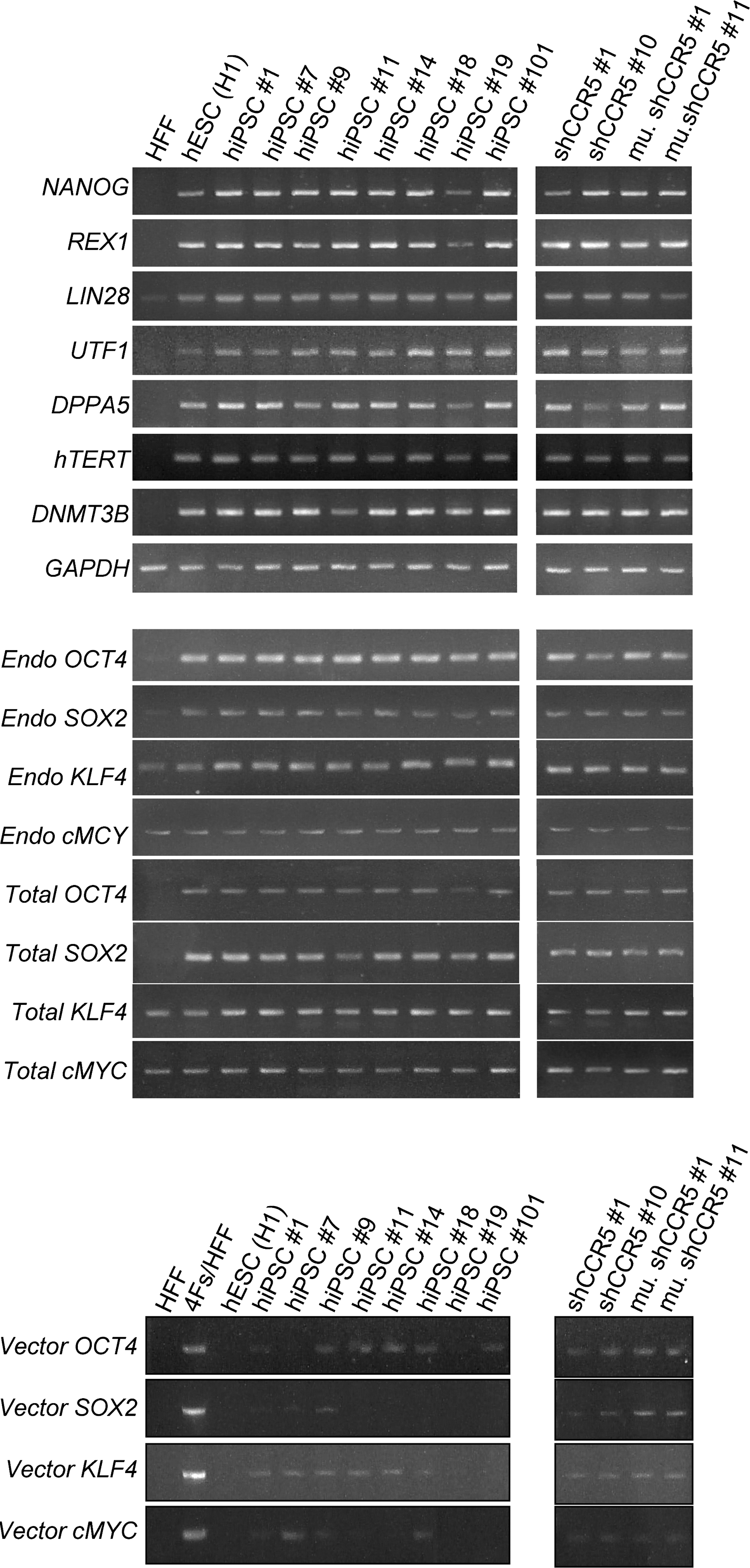
Molecular characterization of hiPSCs transduced with or without the lentiviral vector expressing CCR5 shRNA. Total RNA was isolated using QIAGEN's RNeasy Mini kit from untransduced HFFs, HFFs transduced with four reprogramming factors (Oct4, Sox2, Klf4, and cMyc) (4Fs/HFF), hESCs (H1), and hiPSC clones transduced with the lentiviral vector expressing either CCR5 shRNA (sh1005 #1 and #10) or the mutant sh1005 (mu. sh1005 #1 and #11), and hiPSC clones without transduction of any shRNA expressing lentiviral vector (hiPSC #1, #7, #9, #11, #14, #18, #19, and #101). Total RNA (250 ng) was reverse-transcribed using QIAGEN's Omniscript reverse transcription kit and used as a template in subsequent PCR with 5-PRIME's HotMaster Taq DNA polymerase. PCR products were analyzed on a 2% agarose gel. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. Primers used for NANOG, REX1, LIN28, UTF1, DPPA5, hTERT, DNMT3B, GAPDH, Endo OCT4, Endo SOX2, Endo KLF4, and Endo cMYC specifically detect the transcripts from the endogenous genes. Primers used for Vector OCT4, Vector SOX2, Vector KLF4, and Vector cMYC specifically detect the transcripts from the vectors used for reprogramming. Primers used for Total OCT4, Total SOX2, Total KLF4, and Total cMYC detect the transcripts from both endogenous and vectors. All primer sequences are listed in Table 1.
Short hairpin RNA (shRNA) directed to human chemokine receptor CCR5, designated as sh1005, and the 3-nt mismatch mutant (mutant sh1005) (Shimizu et al., 2009b).
Only colonies expressing EGFP were picked and counted.
The average of hiPSC derivation efficiency from four independent trials.
Transduction of a gene-therapeutic vector into the HFFs in combination with vectors encoding each reprogramming factor results in the transgene expression in the hiPSCs
One of our long-term goals is to use hiPSCs for genetic manipulation. To demonstrate proof of concept for genetic therapy, we introduced another gene-therapeutic lentiviral vector into the cells simultaneously with the vectors encoding reprogramming factors. This lentiviral vector expresses a CCR5 shRNA (sh1005) driven by an H1 polymerase III promoter (An et al., 2007; Shimizu et al., 2009). The vector was previously shown to reduce CCR5 expression 5–10-fold when transduced into primary T cells or through transduction of hematopoietic progenitor/stem cells (HPSCs) and in vitro differentiated macrophages (Liang et al., 2010). A mutant with three nucleotide substitutions served as a negative control (Shimizu et al., 2009). The vector encoding either the wild-type CCR5 shRNA (sh1005) or the mutant (mu. sh1005) was cotransduced into HFFs together with vectors encoding each reprogramming factor. Cells transduced with vectors encoding shRNAs were monitored by the expression of EGFP encoded in the vector. As described above, hiPSCs appeared between days 21 and 28. There were no adverse effects on the efficiency of hiPSC induction in the presence of cotransduced vector encoding shRNAs (Table 2). Approximately 60% of total colonies with hESC-like morphology had EGFP expression, and the expression was maintained for over 20 generations, indicating the shRNA did not have adverse effects on the growth of established hiPSCs. Furthermore, hESC-specific mRNAs were expressed in these hiPSC clones comparable to those without shRNAs, suggesting that the cotransduction of the vector encoding shRNA does not affect the induction of hiPSCs (see Fig. 5).
We confirmed that the reprogramming did not affect the shRNA targeting to CCR5 by using a previously described reporter construct containing the mCherry reporter fused to CCR5 target sequences that is sensitive to the activity of CCR5 shRNA (Shimizu et al., 2009). hiPSC lines containing CCR5 shRNA had reduced expression of the mCherry reporter, whereas similarly derived hiPSC lines expressing a mutant CCR5 shRNA maintained mCherry expression (Fig. 6). These results demonstrate that the CCR5 shRNA can be introduced into fibroblasts and its activities maintained after the hiPSC induction.
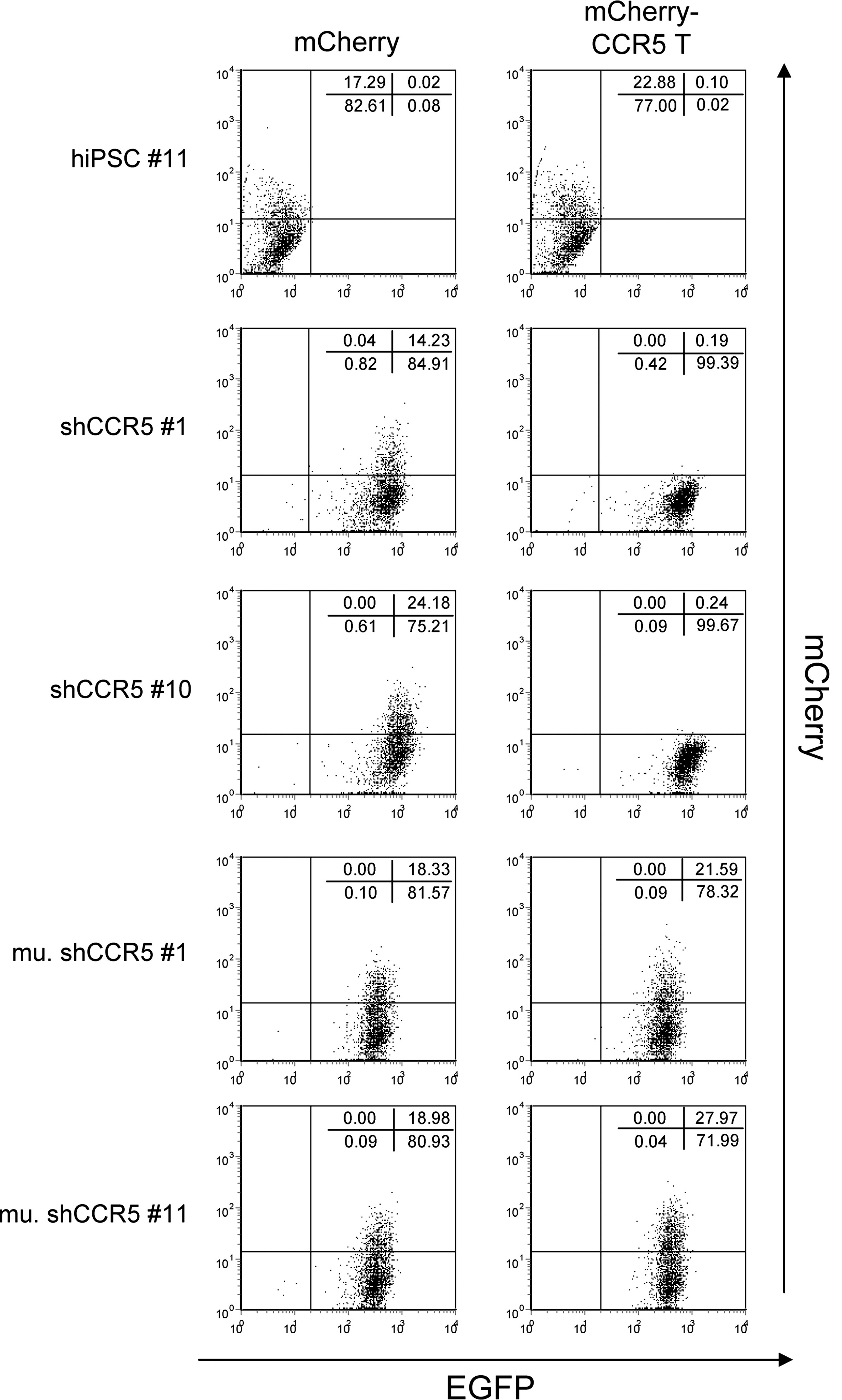
shRNA targeting CCR5 specifically reduces the expression of the chimeric reporter construct of mCherry and CCR5 target sequence in hiPSCs. hiPSC #11, hiPSCs transduced with CCR5 shRNA (shCCR5 #1 and shCCR5 #10), and the mutant CCR5 shRNA (mu. shCCR5 #1 and mu. shCCR5 #11) were infected with the lentiviral vector encoding either mCherry (mCherry) or the chimeric reporter construct of mCherry and CCR5 target sequence (mCherry-CCR5 T) at an MOI of 5. Expression levels of EGFP and mCherry were analyzed by flow cytometry 3 days postinfection. Numbers in each panel represent the percentages in each population.
Discussion
Here, we developed the chimeric MLV/lentiviral vector and tested its property for induction of iPSCs from primary HFFs. Transgene expression of this vector was higher than that of the MLV vector and strongly suppressed in hESCs and in hiPSCs. Primary HFFs transduced with this vector encoding each transcription factor were efficiently reprogrammed into hiPSCs within 28 days, as reported by Takahashi et al. (2007). These hiPSCs were virtually indistinguishable from hESCs phenotypically and capable of differentiating into several lineages. Furthermore, we successfully induced genetically modified hiPSCs by cotransducing the lentiviral vector encoding an shRNA targeting to CCR5 with reprogramming factors without any obvious adverse effects on the efficiency of hiPSC induction and the growth. These hiPSCs were also phenotypically indistinguishable from hESCs similar to nonmodified hiPSCs. Moreover, CCR5 shRNA was expressed and effectively suppressed the CCR5 reporter expression in hiPSCs. These results indicate that this novel hybrid vector is a useful tool for iPSC induction having features of both the MLV vector and the lentiviral vector.
Lentiviral vectors have multiple advantages compared with MLV vectors (Kootstra and Verma, 2003): (a) Lentiviral vectors have an ability to transduce both dividing and nondividing cells, whereas MLV vectors require cell division for the integration (Naldini et al., 1996). This feature of the lentiviral vector potentially allows induction of iPSCs from terminally differentiated nondividing cells, like neuron cells and dendritic cells, or quiescent lymphocytes. (b) Lentiviral vectors have lower genotoxicity than MLV vectors (Montini et al., 2006), a critical feature for future clinical purposes. (c) The maximum size of the insertion for MLV vectors is near the size limit of 9 kb, whereas lentiviral vectors can package a much larger insert (Kumar et al., 2001; Kootstra and Verma, 2003). This greater capacity for packaging size may have potential for broader gene therapeutic application. For example, lentiviral vectors can express four reprogramming factors together with multiple therapeutic genes from one vector. Furthermore, the increased length of the packaging size may improve cell-specific expression by inserting cis-element sequences regulating tissue-specific gene expression.
Acquired immunodeficiency syndrome (AIDS) is an incurable disease caused by HIV-1 infection. HIV-1 infects primarily CD4-positive cells such as helper T cells, macrophages, and dendritic cells, and then eventually kills them, resulting in destruction of the immune system (Rosenberg and Fauci, 1989). HIV-1 is mainly classified into two types, R5- and X4-tropic, by the usage of different chemokine receptors CCR5 and CXCR4, respectively, as coreceptors to enter its target cells (Deng et al., 1996; Dragic et al., 1996; Feng et al., 1996). CCR5 is the most physiologically important coreceptor during natural infection, and the reduced expression makes cells resistant to R5-tropic HIV-1 infection (Anderson and Akkina, 2007; Hutter et al., 2009; Liang et al., 2010). Around 1% of Caucasian people bear a 32-bp deletion within the CCR5 gene on both alleles, resulting in resistance to HIV-1 infection (Dean et al., 1996; Liu et al., 1996; Samson et al., 1996). Importantly, these people are apparently phenotypically normal (Smith et al., 1997; Ioannidis et al., 2001; O'Brien and Nelson, 2004), except for an increased risk of symptomatic West Nile virus infection (Glass et al., 2006). shRNA targeting to CCR5 (sh1005) can knock down CCR5 expression in T cells as well as macrophages differentiated from HPSCs in vitro and in vivo, and those cells are resistant to R5 virus infection (Liang et al., 2010; Shimizu et al., 2010). Generation of genetically engineered hiPSCs bearing CCR5 shRNA has potential for cell therapy via transplantation of HPSCs derived from hiPSCs.
Patient-specific iPSCs expressing anti-HIV-1 therapeutic genes have potential for autologous iPSC-based gene therapy without immunologic rejection. Furthermore, the iPSC-based gene therapy could be safer than the current gene therapeutic approach using mobilized HPSCs, because genetically corrected iPSCs allow prescreening of the genomic location of vector integration sites before transplantation into patients, mitigating the possibility of insertional mutagenesis leading to malignant transformation and other adverse effects. Future studies should be helpful for the establishment of the iPSC-based gene therapy approaches to the treatment of AIDS.
Footnotes
Acknowledgments
We are grateful to Si-Hua Mao for technical help. We also greatly appreciate the generous help from Rina Lee for typing and editing this manuscript. This work was supported by grants from the California Institute for Regenerative Medicine (CIRM) (RS1-00172-1), from the NIH (AI055281, AI069350, and AI028697), and from McCarthy Family Foundation.
Author Disclosure Statement
No competing financial interests exist.