
Abstract
Although the precise pathophysiological mechanism of muscle damage in dystrophin-deficient muscle remains disputed, calcium appears to be a critical mediator of the dystrophic process. Duchenne muscular dystrophy patients and mouse models of dystrophin deficiency exhibit extensive abnormalities of calcium homeostasis, which we hypothesized would be mitigated by increased expression of the sarcoplasmic reticulum calcium pump. Neonatal adeno-associated virus gene transfer of sarcoplasmic reticulum ATPase 1a to the mdx diaphragm decreased centrally located nuclei and resulted in reduced susceptibility to eccentric contraction-induced damage at 6 months of age. As the diaphragm is the mouse muscle most representative of human disease, these results provide impetus for further investigation of therapeutic strategies aimed at enhanced cytosolic calcium removal.
Introduction
Extensive evidence suggests calcium homeostasis is dysfunctional in dystrophic muscle. Sarcoplasmic reticulum calcium ATPase (SERCA) activity is most likely not impaired in dystrophic muscle, although disparate results have been reported depending on the muscle group examined and method used (Kargacin and Kargacin, 1996; Khammari et al., 1998; Culligan et al., 2002; Divet and Huchet-Cadiou, 2002; Plant and Lynch, 2003). Numerous abnormalities of SERCA and other calcium-handling proteins have been reported in dystrophin-deficient skeletal muscle. In the mdx mouse, levels of SERCA1a are elevated in the spared intrinsic laryngeal and toe muscles, unchanged in the mildly affected tibialis anterior, and reduced in the moderately affected extensor digitorum longus (EDL) muscle (Culligan et al., 2002; Dowling et al., 2002, 2003; Ferretti et al., 2009). Similarly, calsequestrin, a high-capacity intraluminal calcium-binding protein, is elevated in intrinsic laryngeal muscle and reduced in the EDL (Ferretti et al., 2009). Other reports describe decreased levels of calsequestrin-like protein, whose function is unknown but may contribute to reduced calcium-binding capacity in the dystrophic sarcoplasmic reticulum (Leberer et al., 1988; Culligan et al., 2002; Doran et al., 2004).
These observations led us to hypothesize that increased expression of sarcoplasmic reticulum ATPase 1a would reduce muscle damage following eccentric contraction. We used neonatal gene transfer of adeno-associated virus (AAV) pseudotype 2/6 to overexpress the fast skeletal muscle isoform of SERCA in the diaphragm of the dystrophin-deficient mdx mouse model and analyzed muscle function and morphology at 6 months of age. Increased SERCA1a protein levels in the diaphragm increased the proportion of type IIA fibers, reduced the percentage of centrally nucleated fibers, and attenuated the loss of force production following eccentric contractions.
Materials and Methods
The use of mice in these experiments was approved by the University of Pennsylvania Animal Care and Use Committee. The viral construct used in this study consisted of the constitutive chicken β-actin promoter and the murine fast skeletal muscle isoform of SERCA (SERCA1a). AAV pseudotype 2/6 was produced by the University of Pennsylvania Vector Core, and 1E12 genome copies of virus in a total volume of 50 μl was injected into neonatal mice (Bish et al., 2008). At 6 months of age, muscle function was measured, and tissues were harvested for further analysis. Immunoblotting, muscle histology, and functional assessments were performed as described previously (Morine et al., 2010), with the following modification for the assessment of eccentric contraction-induced force deficit. Following measurement of maximum isometric tetanus, the diaphragm was subjected to a series of five eccentric contractions with a 5-min rest between contractions. Muscles were stimulated for a total of 700 msec and stretched 10% of optimum length in the final 200 msec of stimulation. The two-tailed unpaired Student's t test was used to compare means between groups; data are reported as means ± SD.
Results and Discussion
Neonatal mdx mice (n = 9 control mdx, n = 9 mdx/SERCA1a) were injected via a subxyphoid approach with 1E12 genome copies of AAV2/6 CB.SERCA1a to target transduction of the diaphragm (Bish et al., 2008). Mdx diaphragm was found to contain approximately one third of the SERCA1a protein content of C57 Bl/6 muscle, whereas gene transfer restored SERCA1a protein levels to 52% of normal (Fig. 1a). Analysis of muscle morphology revealed that increased expression of SERCA1a did not alter fiber size, increased the proportion of type IIA fibers from 54% to 75%, and decreased the proportion of type IIX fibers from 27% to 10% (Fig. 1b and c). Quantification of centrally nucleated fibers, a marker of fibers that had previously undergone degeneration and regeneration, demonstrated a reduced percentage of centrally nucleated fibers in the mdx/SERCA1a diaphragm (Fig. 1d). Dystrophin-deficient muscle fibers are profoundly sensitive to eccentric muscle contractions and exhibit a rapid loss of force production following repetitive stimulation (Petrof et al., 1993). Following a series of five eccentric contractions, the mdx/SERCA1a diaphragm was partially protected from force loss (Fig. 1e). Specific force, or absolute force production normalized to cross-sectional area, was unchanged (9.4 ± 0.7 N/cm2 in control mdx vs. 9.9 ± 1 N/cm2 in mdx/SERCA1a).
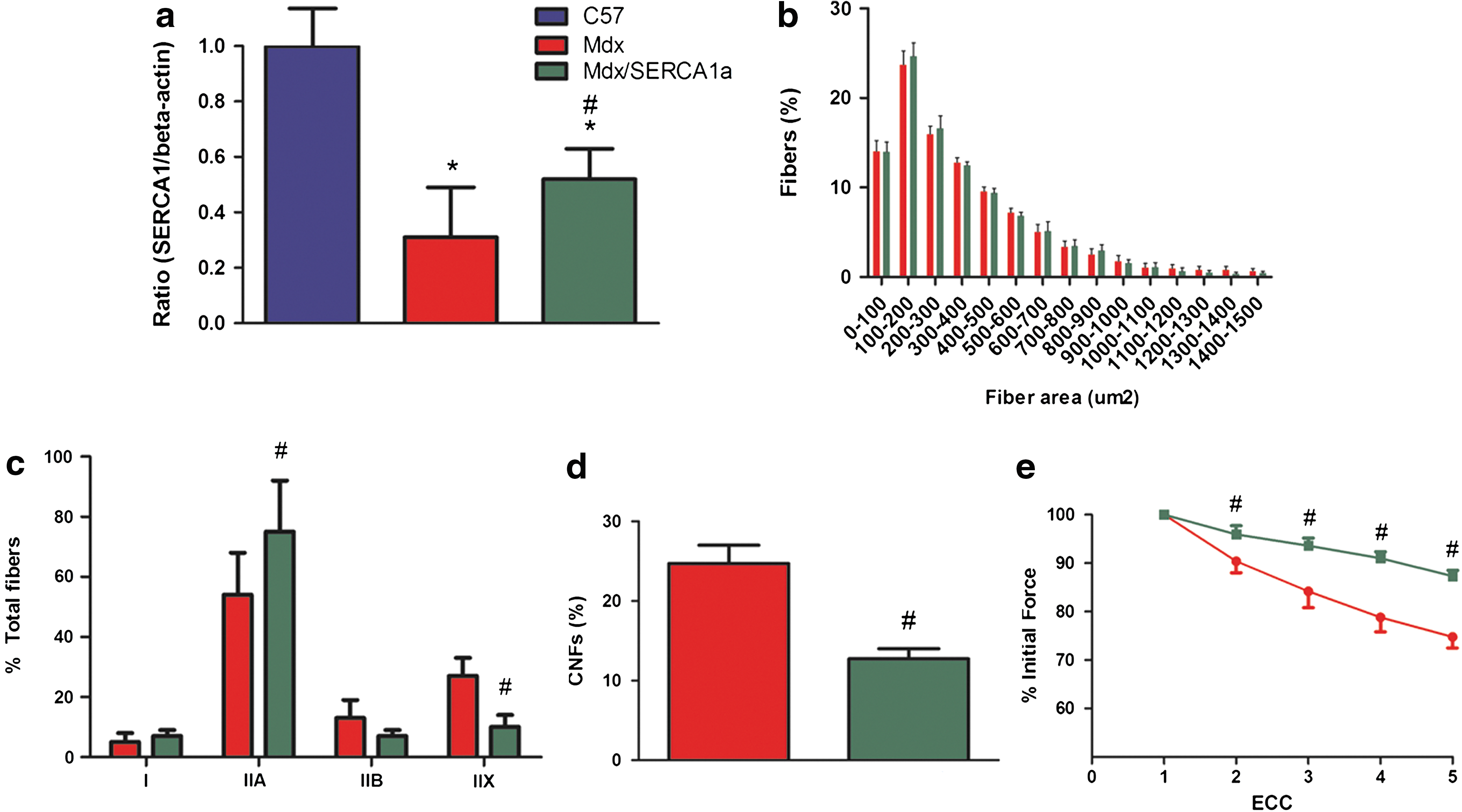
Overexpression of SERCA1a in the mdx diaphragm protects from eccentric contraction–induced loss of force production. (
Here we show that SERCA1a overexpression was sufficient to ameliorate eccentric contraction–induced damage and improve some aspects of disease pathology in the mdx mouse model. The progression of dystrophic pathology is largely accounted for by the loss of muscle fibers following muscle contraction (Lynch, 2004). In dystrophin-deficient muscle, sarcolemmal stabilization achieved by dystrophin replacement or utrophin up-regulation prevents abnormal calcium influx and muscle damage due to contraction and slows or prevents disease progression (Harper et al., 2002; Krag et al., 2004; Liu et al., 2005; Gregorevic et al., 2006, 2008; Odom et al., 2008). Modulation of sarcoplasmic reticulum calcium uptake activity thus represents a novel therapeutic target for muscular dystrophy.
The prevention of abnormal calcium influx and the activation of downstream effectors in muscular dystrophy have been explored in prior preclinical and clinical studies. In the 1980s, trials of the calcium-channel blockers nifedipine and diltiazem did not attenutate Duchenne muscular dystrophy in humans (Moxley et al., 1987; Bertorini et al., 1988). Although there is in vitro and in vivo evidence from mdx mice that the calcium-channel blockers diltiazem and verapamil can prevent calcium influx (Iwata et al., 2005; Matsumura et al., 2009), these agents predominately act on L-type voltage-dependent calcium channels, and it has not been demonstrated that they act on other pathways of calcium influx implicated in muscular dystrophy, such as stretch-operated and store-operated channels. At doses sufficient to effect clinically meaningful blockade of calcium influx in dystrophic muscle, the untoward cardiovascular side effects of these agents may limit use in humans. Alternatively, selective inhibition of the degradative pathways activated by calcium, such as the calpain and ubiquitin-proteasome system, have shown some promise in preclinical studies (Spencer and Mellgren, 2002; Bonuccelli et al., 2003; Burdi et al., 2006; Briguet et al., 2008).
An important limitation of our study is that transgene overexpression of SERCA1a was lost over time secondary to muscle-fiber turnover. Although we demonstrate that partial restoration of SERCA1a protein content to normal levels provides ∼50% protection from contraction-induced damage, the diaphragm was not fully protected from cumulative muscle damage as specific force was not improved.
Prior evidence indicates that chronic calcium influx can activate the slow/oxidative gene program through the calcineurin/NFAT (nuclear factor of activated T cells) pathway (Chakkalakal et al., 2003; McCullagh et al., 2004; Calabria et al., 2009). Hence, it would be expected that correction of the chronic calcium overload present in dystrophic muscle would prevent the fast to slow transition of myosin heavy-chain expression present in the mdx diaphragm. However, we found that SERCA1a overexpression led to an abundance of type IIA fibers in lieu of type IIX fibers. Type I and IIA fibers express significantly more utrophin than type IIB and IIX fibers (Chakkalakal et al., 2003). In addition to enhanced calcium uptake afforded by SERCA1a overexpression, the fiber-type switch and concomitant increase in utrophin expression may underlie the partial protection from eccentric contraction–induced damage observed in our study. Although increasing extrasynaptic utrophin content is unequivocally beneficial in dystrophin-deficient muscle, it is unclear if inducing the slow gene program in general would be a desirable aim in Duchenne muscular dystrophy patients. Studies of activated calcineurin in the mildly affected mdx mice and the severely affected δ-sarcoglycan-deficient mouse have yielded contrasting results (Chakkalakal et al., 2004; Parsons et al., 2007). Differences in responsiveness to calcineurin inhibition or deletion in these models may reflect underlying differences in pathophysiology or disease severity. Future studies should examine the efficacy of SERCA overexpression in more severe dystrophic models and determine if inducing a fast to slow fiber-type transition is beneficial independent of utrophin up-regulation.
Footnotes
Acknowledgments
This work was supported by a Wellstone Muscular Dystrophy Cooperative Center Grant (U54-AR052646) to H.L.S.
Author Disclosure Statement
No competing financial interests exist.