
Abstract
Adoptive cellular therapy provides the promise of a potentially powerful general treatment for cancer. Although this is a complex and challenging field, there have been major advances in basic and translational research resulting in clinical trial activity that is now beginning to confirm this promise. However, these trials are also identifying new challenges and this review focuses on these clinical issues. For tumors such as melanoma, in which tumor-specific T cells can be readily identified and isolated, the adoptive transfer of “tumor-infiltrating lymphocytes” (TILs) already appears to offer significant patient benefit and this approach now warrants further development. Genetically engineered T cells offer a means to endow peripheral blood T cells with antitumor activity and in principle these techniques could allow the treatment of a wide range of cancers. Genetic engineering also offers the means to endow T cells with new properties and enhanced functions. There have been clear proof-of-principle trials showing responses with T cell receptor (TCR)-engineered T cells and this can be built on with further development. By contrast, other trials have produced significant toxicity related to expression of target antigen on normal tissue, particularly with enhanced receptors. The challenge ahead lies in understanding how to achieve the balance between targeted antitumor immune responses while avoiding toxicity associated with on-target destruction of antigen-expressing normal tissues. Cellular therapy of cancer will need continued preclinical evaluation as well as carefully designed clinical trials addressing the various issues. For these challenges to be fully assessed, and for progression to a widely used, effective and safe therapy, development as cooperative groups is an appropriate way forward.
Introduction
The concept of adoptive immunotherapy forms a more drastic approach to alter the established balance between cancer cells and immune cells. First, by using immune cells with cancer specificity grown outside of the patient and infused in larger numbers immune regulation during the expansion of these cells is circumvented. Second, during this in vitro growth phase, the properties—and in particular the recognition potential—of these cells may be modified through genetic engineering. The earliest approaches of adoptive immunotherapy used unselected cells activated with IL-2 and produced modest successes that did not prove to be beneficial over IL-2 alone in randomized trials (Rosenberg et al., 1993). More recent approaches have used T cells possessing tumor specificity generated by selecting naturally occurring antigen-specific T cells or generated by gene modification from polyclonal nonspecific T cells. Preceding articles in this review series have comprehensively reviewed the various technologies for producing gene-modified cells and this will not be reviewed in detail here (Dotti et al., 2009; Schmitt et al., 2009). This article reviews the clinical progress in cellular therapy and proposes coordinated efforts to develop this patient-specific approach to treatment as a routine cancer therapy.
Adoptive T Cell Therapy in Melanoma
Much of the current clinical work in adoptive T cell therapy has been undertaken in melanoma by Rosenberg and colleagues at the National Cancer Institute (NCI, Bethesda, MD). The reasons to target melanoma include the relatively frequent finding of tumor-specific T cells in patients with melanoma, the molecular characterization of their T cell receptors, the large number of tumor-specific antigens discovered in melanoma, and the fact that metastatic melanoma has proven resistant to other forms of treatment, with standard chemotherapy having little clinical effect (reviewed by Kirkwood et al., 2008). Limited efficacy for IL-2 in metastatic melanoma treatment trials, in conjunction with adoptive T cell transfer, was first reported in 1988 (Rosenberg et al., 1988) with further updates in 1994 (Rosenberg et al., 1994). Cells grown from melanoma tumors (“tumor-infiltrating lymphocytes”) were cultured and expanded ex vivo in IL-2 and returned to the patients, either with or without cyclophosphamide. All patients received high-dose IL-2 after cell infusion. Overall, the response rate was good at 34%, but the duration was often short (median, 4 months for partial responses; Rosenberg et al., 1994). A key correlate of response was, however, the short time in culture/short doubling time, and in vitro antitumor reactivity also seemed beneficial (Schwartzentruber et al., 1994); these are markers of “young” cells.
In a series of major trials following these initial observations a variety of improvements were explored. The outcome of these trials may be summarized as follows: Cloning cells to select individual reactive clones resulted in decreased in vivo T cell survival and a reduced clinical response rate (Dudley et al., 2001). Using cells that were only partially selected for specificity but introducing more intensive preconditioning resulted in better T cell survival (indeed, in many cases permitted significant in vivo expansion) enhancing both response rate and duration of response (Dudley et al., 2002). Key correlates of response were, however, long telomere lengths and high surface expression of the CD27 molecule, both markers of young cells (Zhou et al., 2005; Huang et al., 2006). Using intense preconditioning resulted in high response rates (more than 70%) with proven durability in some cases (Dudley et al., 2008).
These trials along with supporting preclinical data provide the current paradigm for the use of adoptive cell transfer, in which infusion of cells is combined with preconditioning chemotherapy (Fig. 1). In these trials the importance of T cell characteristics was confirmed and the benefit of young, rapidly growing cells held up from the initial trials. This has been further confirmed in trials using young TILs, wherein cells are not selected for specificity at all. Remarkably, this produces excellent response rates—about 50%—either with all cells (Besser et al., 2009, 2010; Markel et al., 2009) or with CD8-selected TILs (M.E. Dudley, personal communication). This approach is practical and deserves further testing as a treatment for melanoma. A key question that remains unanswered concerns which T cell reactivities in TILs are involved in the observed clinical responses. If the presence of certain T cell reactivities can be correlated with clinical course, for instance, through the use of high-throughput T cell immunomonitoring (Hadrup et al., 2009), this may allow the future development of more defined T cell products that are enriched for these specificities.
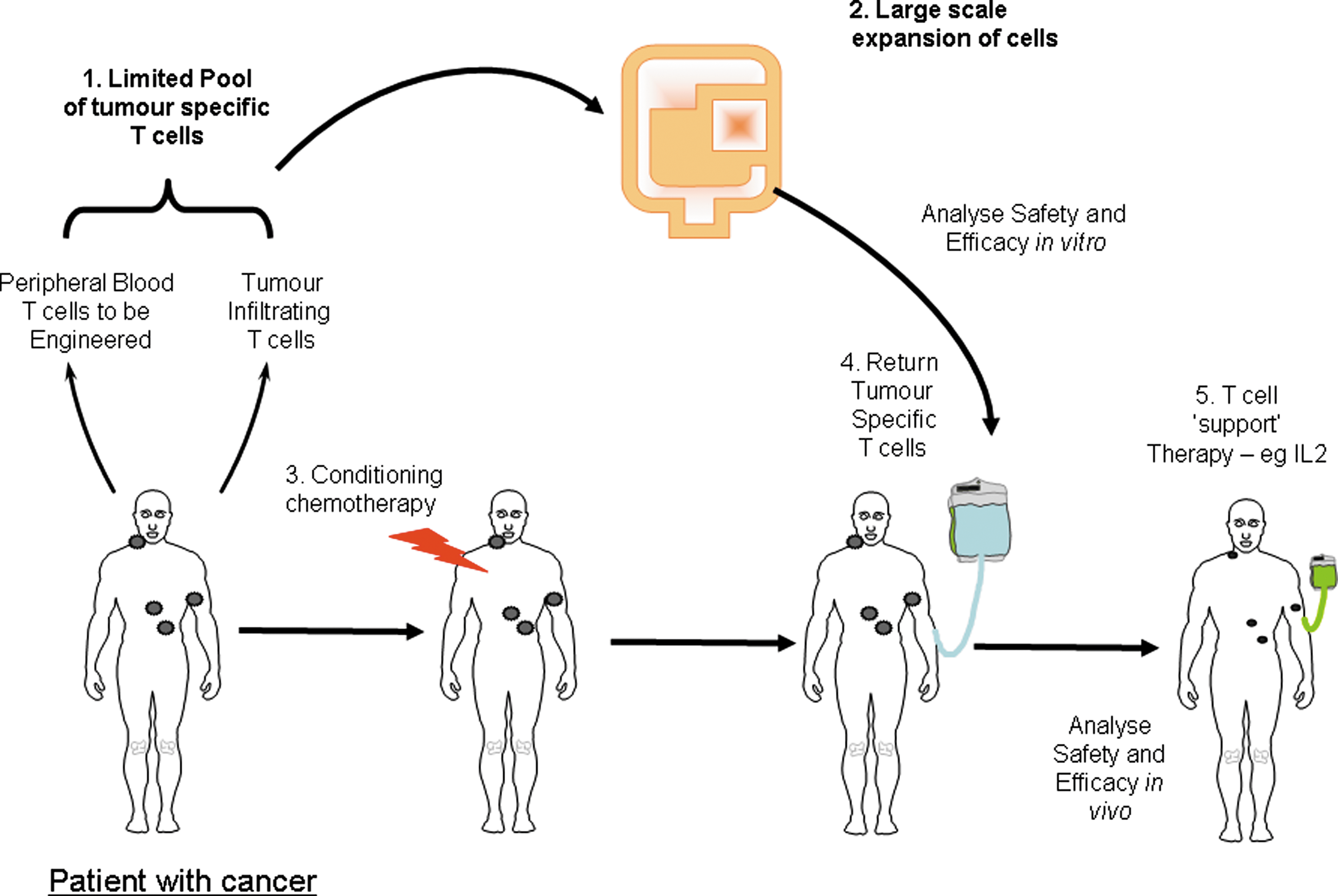
Overview of the current approaches to adoptive cell therapy for cancer. IL2, interleukin-2.
Engineered T Cells: Specificity
The ability to genetically modify T cells allows one to take normal peripheral blood T cells and endow them with tumor specificity, thereby potentially allowing T cell therapy to be applied to cancer types for which specific TILs cannot be routinely obtained. Two basic approaches have been developed to engineer T cells (Fig. 1): first, the gene transfer of natural T cell receptor (TCR)-based receptors; and second, the gene transfer of chimeric receptors typically comprising an antibody fragment linked to a T cell signal-transducing domain. Both these approaches have already been comprehensively reviewed earlier in this series (Dotti et al., 2009; Schmitt et al., 2009). Although in their relative infancy, clinical trials of engineered T cells are producing important results that will guide future development. In particular, the results obtained to date have highlighted both the potential power and the potential toxicity of the approach, and these findings will guide the future development of engineered T cell therapy.
Clinical Results with TCR-Based Engineered T cells
Using TCR gene transfer, the NCI group of Rosenberg has seen consistent evidence of clinical activity but has also seen some significant “on-target” toxicity (Morgan et al., 2006; Johnson et al., 2009). These trials have also helped to define appropriate cell growth conditions for clinical efficacy. All trials used preconditioning chemotherapy consisting of cyclophosphamide and fludarabine and also gave high-dose IL-2 after cell infusion. The initial trial used cells grown long term in IL-2 alone (>14 days). These cells did not survive long term and no clinically beneficial effects were seen. When cells were cultured for a shorter time (about 7 days) or reexpanded in a further rapid expansion protocol (Dudley et al., 2003) the cells persisted in larger numbers and even expanded in vivo, and produced clinical effects, with tumor shrinkage in 15–30% of patients. Other trials targeting different melanoma antigens (several constructs targeting MART1/HLA-A2 and gp100/HLA-A2) have since been reported. Use of TCRs with higher affinity has resulted in pathology that is consistent with “on-target” toxicity (damage to pigmented cells in the eye and ear) (Johnson et al., 2009). Although—at least thus far—manageable for melanoma, this observation suggests that great care should be taken in the selection of target antigens. In addition to the potential for on-target toxicity, the concern has been raised that toxicity may occur through mispairing of endogenously present and genetically introduced TCR chains (Schumacher, 2002). Although this has not been reported so far in clinical trials, animal models do show the potential for this occurrence, and the implementation of engineering strategies that can reduce TCR mispairing is likely to be important when clinical protocols are developed that generate more “fit” gene-modified cells (Bendle et al., 2010). Various molecular strategies have been developed by the ATTACK (Adoptive Engineered T-Cell Targeting to Activate Cancer Killing) Consortium and others to address TCR mispairing (Cohen et al., 2006; Kuball et al., 2007; Thomas et al., 2007; Sebestyen et al., 2008), which should be considered for implementation in future clinical TCR gene therapy trials (see Govers et al., 2010, for a review). At present, there are several ongoing trials targeting melanoma and other tumors. Although the approach appears to be viable, on a practical level it is likely to be restricted to patients with common HLA types.
Clinical Results with Chimeric Receptor-Based Engineered T cells
The early trials of chimeric receptor-engineered T cells produced little evidence of efficacy or toxicity. In these trials, cell growth methods were not optimal and no preconditioning was given and, probably because of this, the cells survived for only short periods of time (Kershaw et al., 2006; Lamers et al., 2006a,b). However, even under these suboptimal conditions, it was clear that short-term persistence could already produce significant on-target toxicity in a case in which the tumor-selective antigen was also expressed on important cells that were readily accessible to the infused cells (Lamers et al., 2006a). It is known from labeling experiments that cells localize nonspecifically in lung and liver immediately after infusion (Fisher et al., 1989). It is also known that the G250 antigen, targeted by T cells in an immunotherapy protocol in patients with renal cell carcinoma, is expressed on bile duct cells and it seems this combination of T cell localization and antigen expression on normal tissue was sufficient to cause reversible yet discrete cholangitis and damage to bile duct epithelium. This observation illustrates the potential power of the approach, but also the need to consider the choice of target antigen, along with the site/accessibility of any normal antigen expression as well as its extent.
Introduction of the basic chimeric receptor protein (Gross et al., 1989; Eshhar et al., 1993) into T cells produces only suboptimal cellular activation of cells, as costimulation is not provided. Initial formats of chimeric receptors provided costimulation through a separate chimeric molecule (Alvarez-Vallina and Hawkins, 1996), as occurs naturally when costimulatory receptors are distinct from the TCR. Subsequent second- and third-generation chimeric receptors have combined both TCR activation and costimulation in one molecule (Finney et al., 1998). This design has produced a range of second- and third-generation receptors incorporating one or more costimulatory domains (for review see Dotti et al., 2009; Bridgeman et al., 2010). Such receptors have greater efficacy in preclinical testing (Haynes et al., 2002; Brentjens et al., 2003; Moeller et al., 2004), including increased resistance to the effects of immune-suppressive cytokines (Koehler et al., 2007), and it is plausible that this increased activity will be mirrored in clinical trials. However, as no tumor antigens except viral antigens or unique mutations are truly tumor specific, the potential for on-target toxicity to normal tissue is clear. Many preclinical models are not adequate to assess such toxicity but a report at the European Society of Gene and Cell Therapy (Cheadle et al., 2009) highlighted the potential for both short-term toxicity and long-term toxicity mediated by chimeric receptor-modified T cells. In the short term, large number of T cells encountering antigen-expressing cells can produce a cytokine release syndrome resulting in major toxicity. In the longer term, continued stimulation of engineered T cells with second-generation receptors can result in uncontrolled proliferation with gross lymphocyte accumulation. This fully autologous mouse model (targeting murine CD19 as a model of lymphoma) showed that second-generation receptors were more effective in controlling target cells (normal B cells), but also produced increased toxicity in a dose-dependent manner.
Unfortunately, toxicity has also been seen in the clinic with such receptors. One patient in a trial targeting CD19 with a second-generation chimeric receptor has died of acute toxicity (Brentjens et al., 2010). The precise cause is unclear and infection may have played a role, but there is concern that it may also be T cell related. In a trial targeting Her-2/neu at the NCI with a third-generation receptor a patient suffered sudden respiratory toxicity after T cell infusion, and subsequently died (Morgan et al., 2010). Detailed analysis showed massive cytokine release and T cell infiltration in the lungs. The overall effect was thought to be due to low-level expression of Her2 in the normal lung, where the cells localize after infusion. In both cases the investigators are reducing the number of T cells given before continuing. However, the pharmacodynamics of engineered T cells are clearly much more difficult to predict than for classical drugs or antibodies. These results also highlight the need to further consider the design of engineered T cells to a given antigen and to carefully consider the expression profile of that antigen.
Approaches to Enhance the Efficacy of Engineered T Cells
The chimeric receptor approach remains of interest because of the potentially broad applicability of antibody-based targeting, as it avoids issues of MHC restriction and because of the clear potential of the approach if it can be adequately targeted. The problems encountered suggest that approaches other than the second/third-generation receptors should also be considered and that additional safety features might also be engineered in. In the normal immune system costimulation occurs on a restricted range of target cells—this could be achieved with chimeric receptors if costimulation and TCR activation were targeted separately to different antigens (Alvarez-Vallina and Hawkins, 1996), and this approach would enhance specificity. Other approaches include methods to enhance cell survival independent of costimulation/antigen recognition, and this might also be beneficial.
Methods to enhance T cell persistence
To generate sufficient numbers of T cells for adoptive transfer, a period of ex vivo culture is required. Clinically applicable methods have been developed that permit the generation of T cells for clinical use (Lamers et al., 2002, 2006b, 2008). These protocols have relied on IL-2 to drive the expansion of T cells; however, studies suggest that other cytokines such as IL-7 (Jaleco et al., 2003) and IL-15/IL-21 (Hinrichs et al., 2008; Pouw et al., 2010a,b) may play important roles in manipulating the final T cell phenotype toward that thought to be more optimal for in vivo activity (Gattinoni et al., 2005; Klebanoff et al., 2005; Hinrichs et al., 2009; Mondino et al., 2010).
Various genetic approaches have also been investigated including the engineering of T cells to incorporate survival signals such as Bcl-XL (Eaton et al., 2002) or the autocrine secretion of IL-2 (Heemskerk et al., 2008); however, the in vivo effects are limited and there are potential toxicities. Other methods have focused on immunological techniques to expand or improve their survival. The combined use of dendritic cell vaccination with chimeric receptor-engineered T cells (Jiang et al., 2006) has demonstrated benefit in animal models. The use of Epstein–Barr virus (EBV)-specific T cells as targets for genetic modification has already been tested clinically and appears to add some benefit in terms of survival of engineered T cells and clinical outcomes (Pule et al., 2008) without any additional toxicity. Other interesting concepts include the use of cytokine secretion by tumors to drive in vivo expansion of tumors (Lo et al., 2008). Such approaches have intrinsic appeal although the risks include the potential for uncontrolled T cell proliferation that may be antigen dependent or independent.
Controlling toxicity
The description of toxicity in clinical trials of engineered T cells highlights the need for careful trial design and also for the possible inclusion of suicide genes. In the past these were problematic, with immune responses generated to suicide genes such as thymidine kinase, although these responses may not abolish the benefit (Traversari et al., 2007). Alternatives include the use of caspase IX, which has shown efficacy in a model of autoimmune toxicity (de Witte et al., 2008) or antibody-based approaches (Kieback et al., 2008), both of which may have benefits. Another strategy is to use RNA-engineered T cells with transient chimeric antigen expression (Birkholz et al., 2009) to explore potential autoreactivity. Although all these approaches appear valid, their efficacy in controlling rapid and severe clinical toxicity, which can occur (Morgan et al., 2010), remains uncertain and their benefit may lie mostly in the prevention of chronic toxicity.
The Way Ahead
Adoptive cell therapy (ACT) with natural or genetically engineered T cells shows significant promise as a strategy for cancer treatment. On the basis of strong preclinical data there are many ongoing and planned clinical trials. Although the field has enormous promise, it is a complex and expensive approach to therapy. Furthermore, there are clear potential toxicities. We organized a clinical workshop (ATTACK Consortium Clinical Workshop, March 2009, Milan, Italy) in order to discuss how the data emerging from this rapidly growing clinical effort may be maximized. On the basis of the output of this workshop, we have formulated the following recommendations, for clinicians and researchers active in this field, as well as for associated funding bodies.
The first recommendation is that tumor-infiltrating lymphocytes for melanoma should be used in multicenter settings and preferably in randomized trials because the present clinical data are compelling and need to be confirmed. Consideration should also be given to exploring this approach in renal cancer and perhaps other cancers. Once established as a routine treatment, this methodology can be further optimized through larger scale trials, and indeed its establishment will facilitate the wider development of adoptive cellular therapy.
Beyond this, the field of adoptive cellular therapy for cancer is in its infancy and small-scale trials in limited numbers of centers are the appropriate approach. The technology of engineered T cells has advanced and has been facilitated by focused consortia such as the ATTACK Consortium (
The complexity of the approach poses challenges for this field that are different from those of standard drug-based therapies: First, at present, no standard in reporting formats or even assay systems to monitor cell therapy-induced immune responses has been established. As a consequence, comparison of clinical trial data is unlikely to be informative. It should be relatively straightforward to solve this issue, provided researchers can agree on the assays that should be considered to be standard. The second, much more fundamental issue, is that the trials that are currently planned differ in such a large number of parameters that, even when reporting standards are implemented, it will be highly problematic to identify which particular aspects of a protocol are critical for its effectiveness (or ineffectiveness). To illustrate this issue, clinical trials with receptor-modified T cells use T cells that are exposed to a range of different conditions both in vitro and in vivo, use T cells modified with a large number of different receptor formats, and those T cells target a wide variety of different antigens. Thus, each individual trial can be seen as providing one single data point in an—at least—three-dimensional space. As most clinical trials will differ from one another in many of these dimensions, a systematic analysis of the factors that are critical to success is precluded.
The solution to this second and much more significant challenge is clearly more drastic: it requires researchers to perform “one-parameter trials.” Specifically, for all three parameters discussed previously, one or at most two standard formats would have to be defined. This approach could be facilitated by European Union or international consortia focused on developing this field. Clinical teams interested in examining the merits of a novel target antigen would then commit to using the agreed-on conditions for cell growth/engraftment and a standard receptor format. Likewise, teams interested in developing improved conditions for cell growth and engraftment would use a standard receptor format and target antigen. Finally, teams interested in developing improved receptor formats would keep both growth and engraftment conditions and target antigen constant. In this manner, and arguably only in this manner, the effects of defined changes in clinical trial protocols can unambiguously be identified.
Why is implementation of these recommendations likely to be difficult, and why has there in fact been a lack of a similar type of standardization in many fields that would have benefited from it? A key problem in the proposed standardization is that although this may be of great value for the community as a whole, the gain for any individual clinical team that must adhere to these standards might in fact be negative. Thus, the lack of standardization may be seen as a variant of the tragedy of the commons (Hardin, 1968), in which the direct benefits for an individual who complies are small and the tendency toward noncompliance is therefore large. We speculate that as long as the incentive for individual scientific teams remains modest or even negative, standardization in this field will be difficult to achieve, and this is where we believe that involved funding agencies can play a critical role.
Funding bodies that support the clinical translation of adoptive therapy with gene-modified T cells or other cells should be able to support standardization by providing a defined financial incentive. As argued previously, at an aggregate level, the information emerging from such concerted clinical efforts is likely to be superior. Furthermore, as a consequence of standardization, clinical trials performed by such consortia can also be performed at greatly reduced cost. Finally, this model for incentive-driven generation of a “clinical trials common” should evidently also apply to other areas of academia-driven clinical development.
Conclusions
In conclusion, adoptive cellular therapy for cancer is promising and certainly the use of tumor-infiltrating lymphocytes for melanoma seems ready for development as a routine therapy. Beyond this, engineered T cells hold broad promise but must be carefully evaluated clinically. The use of first-generation TCR- and antibody-based receptors seem safe with early evidence of efficacy, and this can be built on. Many ingenious ways to enhance the efficacy of T cells have been developed, but early trials suggest toxicity due to normal tissue expression in some cases. This paradoxically reveals the enormous potential power of the approach but also highlights the need to target T cells appropriately and to introduce controls when necessary. Overall, enormous progress has been made and the power of adoptive T cell therapy and engineered T cells in particular is clear. Continued efficient development of this complex but promising type of therapy both clinically and preclinically is perhaps best achieved through coordinated testing by funded cooperative groups, such as the ATTACK project, as the size and scope of the research effort needed are beyond most single groups.
Footnotes
Acknowledgments
Authors would like to acknowledge the European Commission, Sixth Framework Programme, for funding the ATTACK project (LSHC-CT-2005-018914) which supported research in this area.
Author Disclosure Statement
The authors have no conflicts of interest.