
Abstract
The regulated expression of therapeutic genes may become crucial in gene therapy when their constitutive expression interferes with cell fate in vivo. The efficient regulation of transgene expression requires tightly controlled inducible promoters, as shown for the tetracycline regulatory system (tet-system). However, its application requires the introduction of two components into the target cell genome: the tet-responsive transactivator and the regulated expression cassette. In order to facilitate the usage of the tet-system for approaches in gene therapy, both components have to be transferred by a single vector, thus eliminating the preselection of transactivator positive cells. Published “all-in-one” vectors for regulated transgene expression display a relatively low signal-to-noise ratio, resulting in regulatory windows of around 500-fold even in selected clones. In this study, we show that a modified vector architecture combined with the introduction of new tet-responsive promoters, Ptet, improved the dynamic range of such all-in-one vectors to levels up to 14,000-fold for viral and 25,000-fold for nonviral transfer vectors in nonclonal human cell lines, and up to 2,800-fold in murine hematopoietic cell lines. This improved regulation was the result of a strong reduction of background expression in the off-state, even if cells were transduced at high multiplicity of infection, while induction remained at high levels. In addition, the results indicated that successful regulation of gene expression in different target cells depended on vector architecture as well as the choice of the Ptet-promoter.
Introduction
In the past, diverse modifications were introduced to improve the tet-regulated transgene expression system. These modifications have included improvements of the tet-responsive promoter, where a reduction of the tet-operator sequences (tetO) toward 36-nucleotide (nt) spacing from core to core improved the regulatory behavior (Agha-Mohammadi et al., 2004). The transactivator has also been subject to various refinements (Baron et al., 1997; Urlinger et al., 2000; Das et al., 2004). The original transactivator binds only in the absence of tetracycline (TetOff), whereas modifications have led to the reverse function (Gossen et al., 1995), i.e., the reverse transactivator binds only in the presence of doxycycline, a tetracycline derivative. This so-called TetOn system has been preferentially used since its invention, particularly in vivo, as it does not require permanent administration of doxycycline to switch off transgene expression. Furthermore, the transactivator has been optimized by replacing the original VP16 transactivation domain by a so-called minimal domain. Additionally, the codon usage has been optimized for higher expression levels and stability in eukaryotes. One of the most promising transactivator is the TetOn variant rtTA2s-M2 (Urlinger et al., 2000), which is used in this study.
Effective and stable transfer of the tet-system components into a wide range of different cell types can be achieved via viral delivery systems (Kenny et al., 2002; Vigna et al., 2002; Chtarto et al., 2003; Barde et al., 2006; Loew et al., 2006). The components of the tet-system can be delivered either on separate vectors (the so-called two-vector system) or in the more advanced approach by a single vector (the all-in-one vector system). The major hindrances for the successful use of the tet-system are reliable introduction of both components into the same cell and a high regulatory window, i.e., high induction of the transgene and tight regulation in the absence of induction. Both approaches, therefore, face some problems. The two-vector system often relies on preselection for the integration of the transactivator-expressing unit (Gopalkrishnan et al., 1999; Qu et al., 2004). In the next step, the second vector containing the inducible cassette is introduced, followed by a second selection to identify clones displaying tightly regulated transgene expression. The all-in-one vector system circumvents the need for preselection, because a single transduction is sufficient for introducing all components needed for tet-regulated transgene expression (Paulus et al., 1996). The major drawback of this system is a typically much higher background activity compared with the two-vector system. The all-in-one vector approach, therefore, showed regulatory windows of maximum 500-fold, even after clonal selection (Kafri et al., 2000; Haack et al., 2004; Barde et al., 2006). Recently, transposon based systems have attracted interest for the stable transfer of expression cassettes into target cells (Ivics et al., 1997; Wilson et al., 2007; Mátés et al., 2009). So far, however, like for viral one-vector systems, the tet-regulation suffered from high background activity, even after clonal selection, after transposon-mediated transfer into target cells (Saridey et al., 2009).
In this study, we describe a novel all-in-one vector system, allowing tight control of transgene expression without loss of inducibility. The central refinement of our vectors consists of modifications of the regulatory cassettes, employing new Ptet-promoters. Combined with small alterations of vector architecture, the modifications improved transgene regulation in murine and human bulk cell cultures after retroviral and nonviral gene transfer, which greatly simplifies the application of tet-regulated expression in basic biology and somatic gene therapy.
Materials and Methods
Plasmid cloning
All components needed for tet-regulated transgene expression were subsequently introduced into plasmids containing either the basic ES.1-γ-retroviral backbone (Loew et al., 2010a) or the terminal repeats for Sleeping Beauty transposase, kindly provided by Z. Ivics (Mátés et al., 2009). The Ptet-promoter variants Ptet-T2, -T6, and -T11 were generated by PCR with overlapping oligos by standard procedures as recommended by the suppliers (Phire-Taq, Biozym, Oldendorf, Germany), respectively. Structure and sequence details are given in Supplementary Figs. S1 and S2 (Supplementary Data are available online at
Cultivation of cells
HT1080 cells were cultured in Dulbecco's modified Eagle's medium (Biochrom, Berlin, Germany) supplemented with 10% fetal calf serum (FCS) (PAA, Pasching, Austria) and 0.1 mg/ml sodium pyruvate (PAA). BaF3 and 32D cells were cultivated in RPMI 1640 medium (PAA) supplemented with 10% FCS, 0.1 mg/ml sodium pyruvate and 5 ng of mIL-3/ml (Peprotech, London, UK). All cells were incubated at 37°C and 5% CO2. For induction of tet-regulated transgene expression, cells were cultivated in the presence of doxycycline (Sigma) at 1 μg/ml for 4 days. Medium containing doxycyline was refreshed every 2 days. Transduced cells were sorted according to eGFP fluorescence in the presence of doxycyline on a FACSAria (BD Bioscience, Heidelberg, Germany). Cells were subsequently cultivated for 12 days in the absence of doxycyline before the background activity was measured.
Preparation of viral supernatant, transduction, and transfection
For preparation of viral supernatant, 293T cells were seeded onto 10-cm dishes and cotransfected (calcium phosphate) with plasmids coding for the viral vector (10 μg/dish), gag/pol (10 μg), and VSV-G (0.5 μg) envelope protein in the presence of 20 mM HEPES (PAA) and 25 μM chloroquine (Sigma). Every 12 h, the medium of the transfected producer cells was replaced by 8 ml of fresh culture medium including 20 mM HEPES. The supernatant was harvested 36 h after transfection, sterile-filtrated, and stored in aliquots at −80°C. Titration of viral supernatants was performed on SC-1 cells in the presence of protamine sulfate (Sigma; 4 μg/ml) and doxycycline (Sigma; 1 μg/ml). Cells (1 × 105) were seeded on six-well plates 24 h prior to infection. The transduction was performed using serial dilutions in 1 ml final volume, and fresh medium including doxycycline was added 24 h after transduction. The cells were FACS-analyzed 6 days after transduction. The titers were compared on the basis of transducing units per milliliter of supernatant (TU/ml). For analyzing the tet-regulated transgene expression, 1 × 105 HT1080 cells were seeded on 24-well plates 24 h prior to transduction. Supernatants of known titer were added at a multiplicity of infection (MOI) of 0.1 or 3, as described in Results, in the presence of protamine sulfate and doxycyline. Populations obtained with MOI 0.1 (1–3% positive cells) were enriched by one round of sorting, whereas MOI 3 populations were not enriched. For comparison, the luciferase values were normalized to 100% eGFP-positive cells.
Cotransfection of HT1080 cells with plasmids for transient expression of Sleeping Beauty transposase (2 μg of pCMV-SB100x; Mátés et al., 2009) and the transposable element (2 μg) was performed using polyethylenimine (PEI; 25 kDa linear; Polyscience, Niles, IL). Cells were seeded on six-well plates. One hundred microliters of PEI (0.1 g/L in 150 mM NaCl; pH 5.5) reagent and 100 μl of DNA solution (5 μg of DNA in 150 mM NaCl; pH 5.5) were added to 1.8 ml of medium. Transfection reagent was replaced with normal medium including doxycyline after ∼12 h.
Reporter gene analysis
eGFP fluorescence was measured with a FACSCalibur (BD Bioscience) and analyzed using FlowJo software (Tree Star Inc., Ashland, OR). Dead cells were excluded for analysis through exclusion of propidium iodide (Sigma; 1 μg/ml)-positive cells. Luciferase activity was measured as previously described (Loew et al., 2010a). In brief, cells were centrifuged and resuspended in lysis buffer. Typically 1–10 μl of the cell lysate was measured at room temperature on a tube luminometer (LB 9507; Berthold Technologies, Bad Wildbad, Germany). Values were normalized against the total protein content of the cell lysate. Protein determination was performed using the Coomassie Plus (Bradford) Assay kit (Pierce Biotechnology, Rockford, IL). One microliter of cell lysate was treated according to the manufacturer's instruction in triplicate. Analysis was performed on a microplate reader and analyzed with SoftMax Pro 4.0 software (Molecular Devices, Sunnyvale, CA). For a standard curve, different amounts of bovine serum albumin (0.5–8 μg) were used. Statistical analysis was performed using Student's t test.
Quantitative PCR
Mean copy number was determined via quantitative PCR (qPCR) using primers detecting the wPRE (woodchuck posttranscriptional element; forward, 5′-GAGGAGTTGTGGCCCGTTGT-3′; reverse, 5′-TGACAGGTGGTGGCAATGCC-3′) (Modlich et al., 2006) and PTBP2 (polypyrimidine tract binding protein 2; tm_PTBP2_optimized
Results
Retroviral “all-in-one” vector: design and experimental outline
We constructed bidirectional tetracycline-inducible all-in-one vectors, integrating all components required for tet-regulated transgene expression into the ES.1-γ-retroviral SIN vector backbone (Loew et al., 2010a). The reverse (TetOn) transactivator rtTA2S-M2 (Urlinger et al., 2000) was expressed under the control of the human phosphoglycerate kinase promoter (hPGK), which is constitutively active in a wide range of mammalian cells. The inducible unit was inserted as an antisense expression cassette (3′–5′relative to the viral genome), which consisted of an optimized tet-responsive promoter (Ptet-T2) containing a tet-operator heptamer fused to a minimal promoter followed by the cDNA of choice, a constitutive transport element from Mason Pfizer Monkey Virus (CTE) (Schambach et al., 2000) and polyA signals from either the SV40 (SV40pA) or the human growth hormone (hghpA). These initial MoMLV-based bidirectional vectors were termed MOV-scT2 and MOV-hcT2 (Fig. 1A). Based on the Ptet-1 (Gossen and Bujard, 1992), the minimal promoter Ptet-T2 (Supplementary Fig. S1) was developed, which contains consensus sequences for the TATA box (cTATA) and the binding site of the transcription factor IIB (cTFIIB) (Lagrange et al., 1998). In the context of the monocistronic vectors, these modifications increased the regulatory window by ∼10-fold, due to increased inducible activity and a reduced background activity in the off-state of the system (Loew et al., 2010b).

Experimental outline and basic all-in-one vectors. (
The basic experimental design is shown in Fig. 1B. The regulatory characteristics of the vectors were investigated by the expression of the lmg* dual reporter gene, consisting of luciferase and eGFP, that allowed the simultaneous measurement of both gene activities (Loew et al., 2010b). Human HT1080 cells were transduced to an efficiency of a maximum 20% eGFP-positive cells after induction, and highly purified populations of transduced cells (>90%) were obtained by FACS. The vector copy number was determined via qPCR (Table 1), revealing a similar average of vector integrates for all tested populations. After cultivation of these sorted populations in either the presence (i.e., “on-state”) or absence (“off-state”) of doxycycline, the cells were analyzed for eGFP fluorescence and luciferase activity.
Exploring effects of vector elements on the regulatory properties of the MOV vector
We determined the tet-inducible transgene expression of the vectors MOV-scT2 and MOV-hcT2, in HT1080 cells. The dynamic range of gene regulation based on the induced eGFP expression and the luciferase activity was similar (∼220-fold) for both constructs (Fig. 1C). Thus, MOV-hcT2 was chosen as an initial reference for further experiments. Next, we tested whether the removal of the splice acceptor from MOV-hcT2 would lead to higher vector titers. For this purpose, we removed the pol/env border fragment containing the native splice acceptor (SA) site, resulting in the vector MOV.1-hcT2 (Fig. 1D). As shown in Fig. 2, no significant differences in the retroviral titers were observed, with titers in the range of 1–3 × 106 TU/ml. However, comparison of the luciferase activities determined in the enriched cell populations (Fig. 1D, right panel) revealed a fivefold reduction of background expression in the off-state in populations transduced by MOV.1-hcT2, whereas the level of induction remained unchanged. Thus, the dynamic range of gene regulation was increased three- to fourfold (Fig. 1D, left panel) by this modification of the vector backbone.
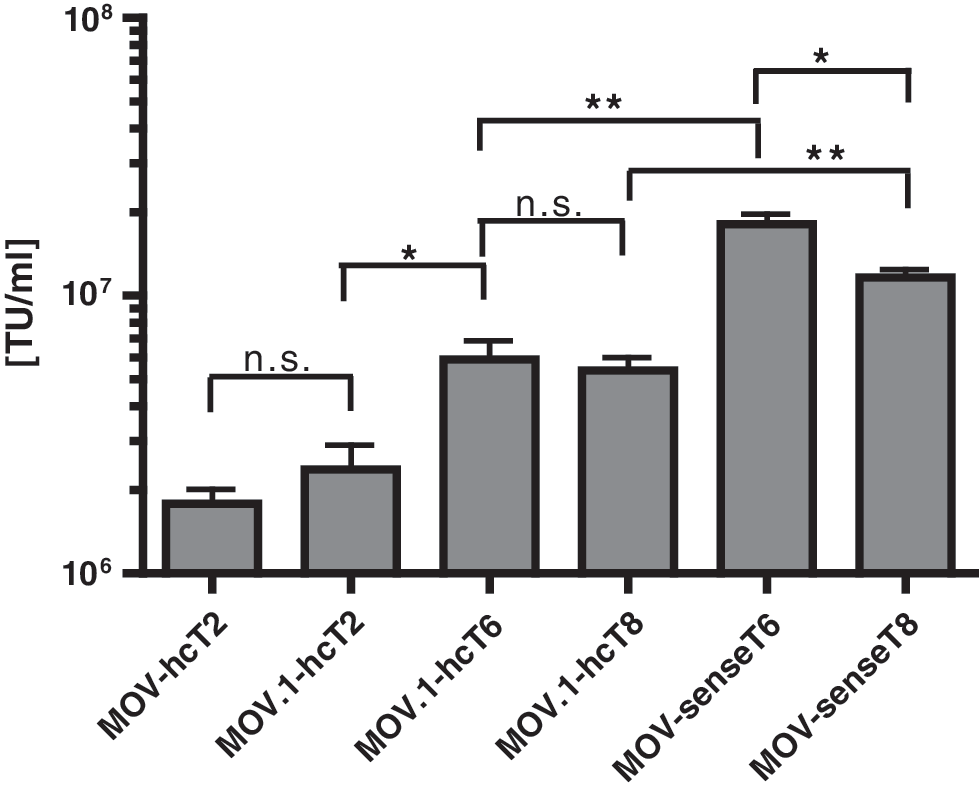
Comparison of titers. Titers were determined for the vectors used in this study. Titration was based on eGFP fluorescence and was performed on SC-1 cells. Values are given as transducing units per milliliter (TU/ml). Data are shown as means ± SD (n = 3). n.s., data not significantly different (P > 0.05); *P < 0.05; **P < 0.01.
Introduction of improved Ptet-promoters further reduces background expression
To further increase the regulatory window, new Ptet-promoters, Ptet-T6, Ptet-T8, and Ptet-T11 (Supplementary Figs. S1 and S2) were integrated into the MOV.1-hcT2 backbone, resulting in the vectors MOV.1-hcT6, MOV.1-hcT8, and MOV.1-hcT11, respectively (Fig. 1D). The introduction of the new Ptet-promoters resulted in an improved signal-to-noise ratio in transduced HT1080 cell populations, i.e., the background expression in the off-state was reduced while high induction levels were maintained, clearly demonstrating the advanced regulatory properties of these novel Ptet-promoters (Fig. 3A, left panel). Based on the luciferase measurements, the introduction of Ptet-T6 led to an approximately threefold reduction of background activity relative to the Ptet-T2 promoter, whereas induction was only moderately decreased (∼15% decline). The introduction of Ptet-T8 and Ptet-T11 promoters (for details, see Materials and Methods) further reduced background expression. Measurement of luciferase revealed an ∼13- to 35-fold decrease of background activity in the off-state, whereas the activity upon induction was reduced only ∼1.4- to 1.9-fold when compared with Ptet-T2. Moreover, the introduction of all modified minimal promoters into the MOV.1 backbone resulted in approximately twofold higher vector titers compared with the Ptet-T2 variant (Fig. 2). Taken together, the introduction of the novel Ptet-promoters into the bidirectional viral vector context increased the dynamic range of gene regulation in transduced HT1080 cell populations from ∼700-fold for Ptet-T2 up to ∼2,000-, 7,000-, and 14,000-fold for the Ptet-T6, Ptet-T8, and Ptet-T11 promoter, respectively (Fig. 3A, right panel), combined with the additional advantage of increased vector titers.
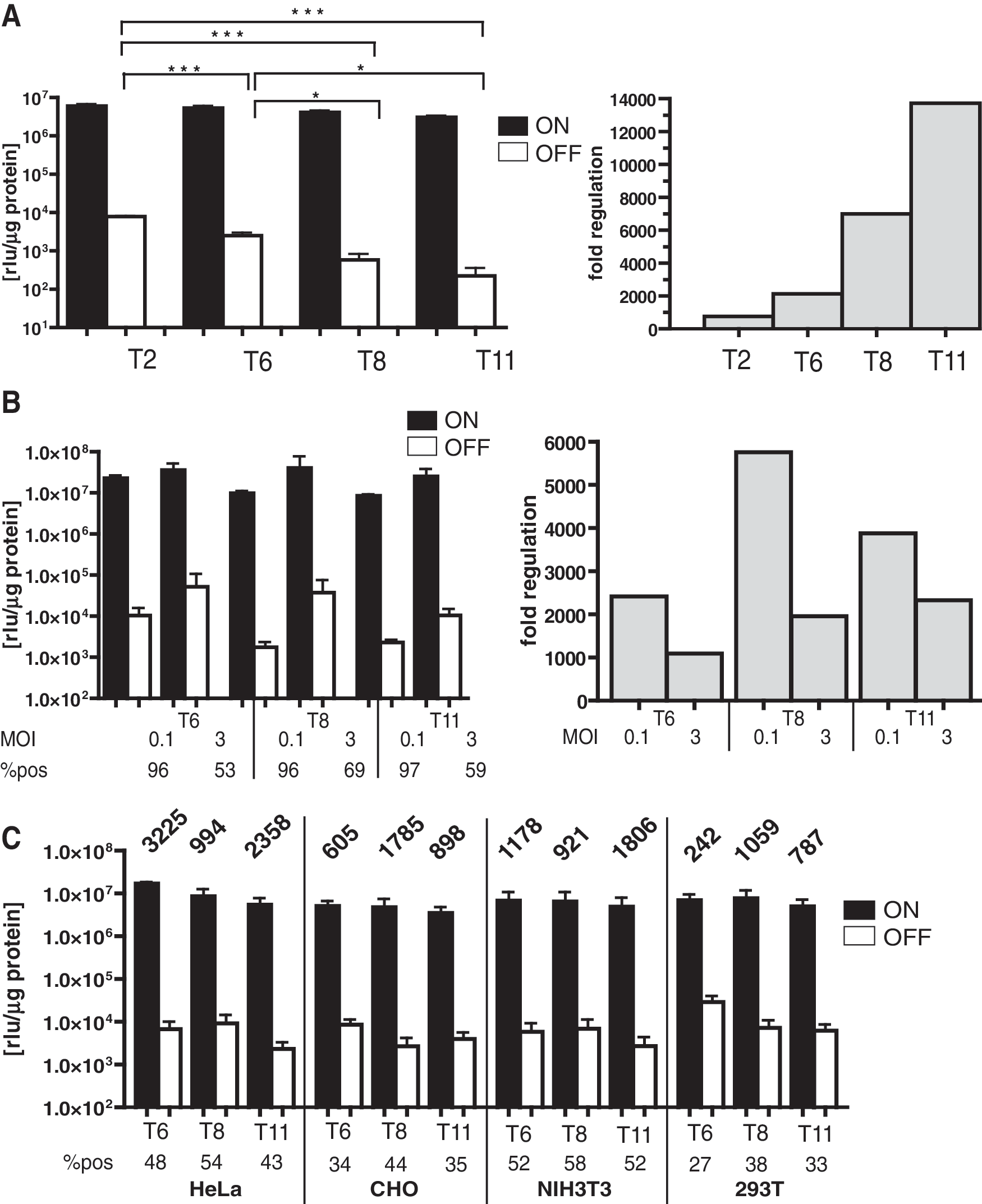
Novel Ptet-promoters improved the dynamic range of gene regulation after viral transfer. (
Impact of high gene transfer rates on the tet-induced gene regulation
In many gene therapy trials, multiple transduction cycles were performed to ensure sufficient gene transfer (30–60%), potentially resulting in two to four vector copies per cell in >30% of the transduced cells (Kustikova et al., 2003; Fehse and Roeder, 2008). Thus, we further explored whether the improved regulatory performance of our novel all-in-one vectors is maintained in cells containing more than one vector insertion. We compared the luciferase activity in the on- and off-state between HT1080 cell pools that were transduced at an MOI of either 0.1 or 3. Transduction of HT1080 cells at an MOI of 3 resulted in gene transfer rates ranging from 47% to 69% for the different MOV.1 vectors, whereas at an MOI of 0.1 the gene transfer efficiencies were below 3%. For all three MOV.1 vectors, the range of transgene regulation observed in HT1080 cells transduced at an MOI of 3 was only slightly lower (1.6- to 2.9-fold) than that in cell pools transduced at an MOI of 0.1; thus, the dynamic window of gene regulation was maintained at levels above 1,000-fold (Fig. 3B). Furthermore, as summarized in Fig. 3C, the improved regulatory window of our novel all-in-one vectors at high MOIs was also retained in four other established lines derived from a variety of tissues.
Comparison of bidirectional vs. unidirectional vector architecture
For gene therapeutic approaches, the vector titers that can be achieved for a certain construct are important. Titers generated by transient transfection of packaging cells with the bidirectional vectors MOV.1-hcT6 and MOV.1-hcT8 were about 5–6 × 106 TU/ml. The increased titers after Ptet-T2 was replaced by Ptet-T6, Ptet-T8, or Ptet-T11 within the bidirectional MOV.1 vectors (Fig. 2) suggested that the new Ptet-promoters generated less antisense transcripts during vector production. Thus, we assumed that the inversion of the regulatory unit, preventing the generation of antisense transcripts in packaging cells, might further improve vector titers. Indeed, the inversion of the regulatory unit, resulting in the unidirectional vectors MOV.1-senseT6, MOV.1-senseT8, and MOV.1-senseT11 as illustrated in Fig. 4A, led to two- to threefold higher viral titer (∼1–2 × 107 TU/ml; Fig. 2). However, determination of luciferase activity in transduced HT1080 cells (Fig. 4B), revealed a 1.3- to 6-fold reduced dynamic range of transgene regulation for these unidirectional vectors when compared with their bidirectional counterparts (Fig. 4C). Nevertheless, the unidirectional vectors still mediated a 1,000- to 10,000-fold gene regulation.
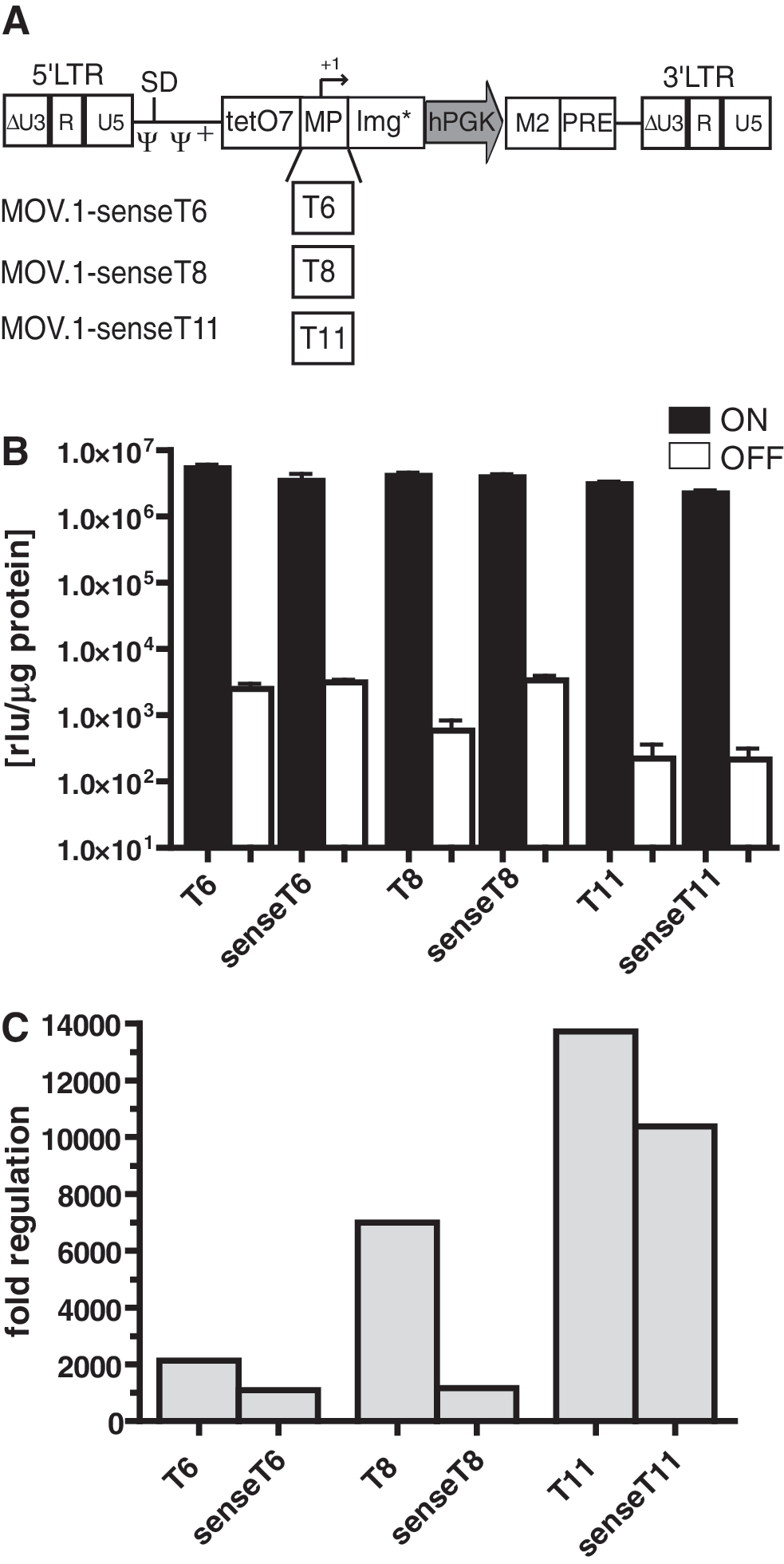
Comparison of bi- and unidirectional vectors in HT1080 cells.
Properties of the unidirectional retroviral vectors in murine hematopoietic cell lines
To test whether the unidirectional all-in-one vectors ensure also a robust high-level transgene regulation in other tissue culture models, we examined their regulatory properties in murine hematopoietic cell lines. BaF3 (a pro B cell line) and 32D cells (a bone marrow-derived myeloid cell line), both widely used for signaling studies, were transduced and enriched by FACS as described for HT1080 cells. In contrast to the induction levels achieved in HT1080 cells, transgene expression levels in both hematopoietic cell lines were highly reduced (four- to 30-fold) in the on-state for both the bidirectional and the unidirectional vectors (Fig. 5A). However, unidirectional vectors showed up to sixfold and 16-fold reduced background expression in BaF3 and 32D cells, in a side-by-side comparison with the bidirectional vectors (Fig. 5A). Overall, the unidirectional vector MOV.1-senseT11 conferred the highest dynamic range of gene regulation (∼1,000-fold) in BaF3 cells, whereas in 32D cells the best regulatory window (2,800-fold) was achieved with the unidirectional vector MOV.1-senseT6 (Fig. 5B).

Regulated gene expression in hematopoietic cell lines. (
A nonviral approach results in highly reduced activity in the uninduced state
DNA transposon-mediated gene delivery systems have recently been developed as an alternative tool for retroviral gene transfer because they offer important advantages over viral vectors (VandenDriessche et al., 2009). Therefore, we tested the unidirectional all-in-one vector concept in a transposon-based, nonviral vector system. The two components of the tet-system were integrated as distinct expression units into the Sleeping Beauty transposable element resulting in TOV-T6, TOV-T8, and TOV-T11 vectors (Fig. 6A). In contrast to the viral vectors, the terminal repeats of the transposon do not contain a polyA signal; thus, both units received a polyA signal (SV40pA for the tet-inducible and HGHpA for the constitutive expressing cassette). HT1080 cells were cotransfected with TOV-T6, TOV-T8, and TOV-T11 and an expression plasmid encoding the hyperactive Sleeping Beauty transposase (SB100x) (Mátés et al., 2009). After stable integration, eGFP-positive cells were enriched by FACS and cultivated as described for the viral approach. Determination of luciferase activity of TOV-T6, TOV-T8, and TOV-T11 transduced cells revealed similar levels of transgene induction, as was observed in HT1080 populations transduced by the viral vector counterparts (Fig. 6B). In comparison with the viral approach, nonviral gene delivery conferred an even stronger reduction of luciferase activity in the off-state (Fig. 6B, left panel). Compared with the viral vectors, the background activity was reduced four- and sixfold for Ptet-T6 and Ptet-T8 controlled gene expression and, as a consequence, the dynamic range of gene regulation increased to 18,000- and 25,000-fold. In contrast to the findings obtained with the viral vectors in transduced HT1080 cells, the introduction of Ptet-T11 showed no further improvement compared with Ptet-T8.
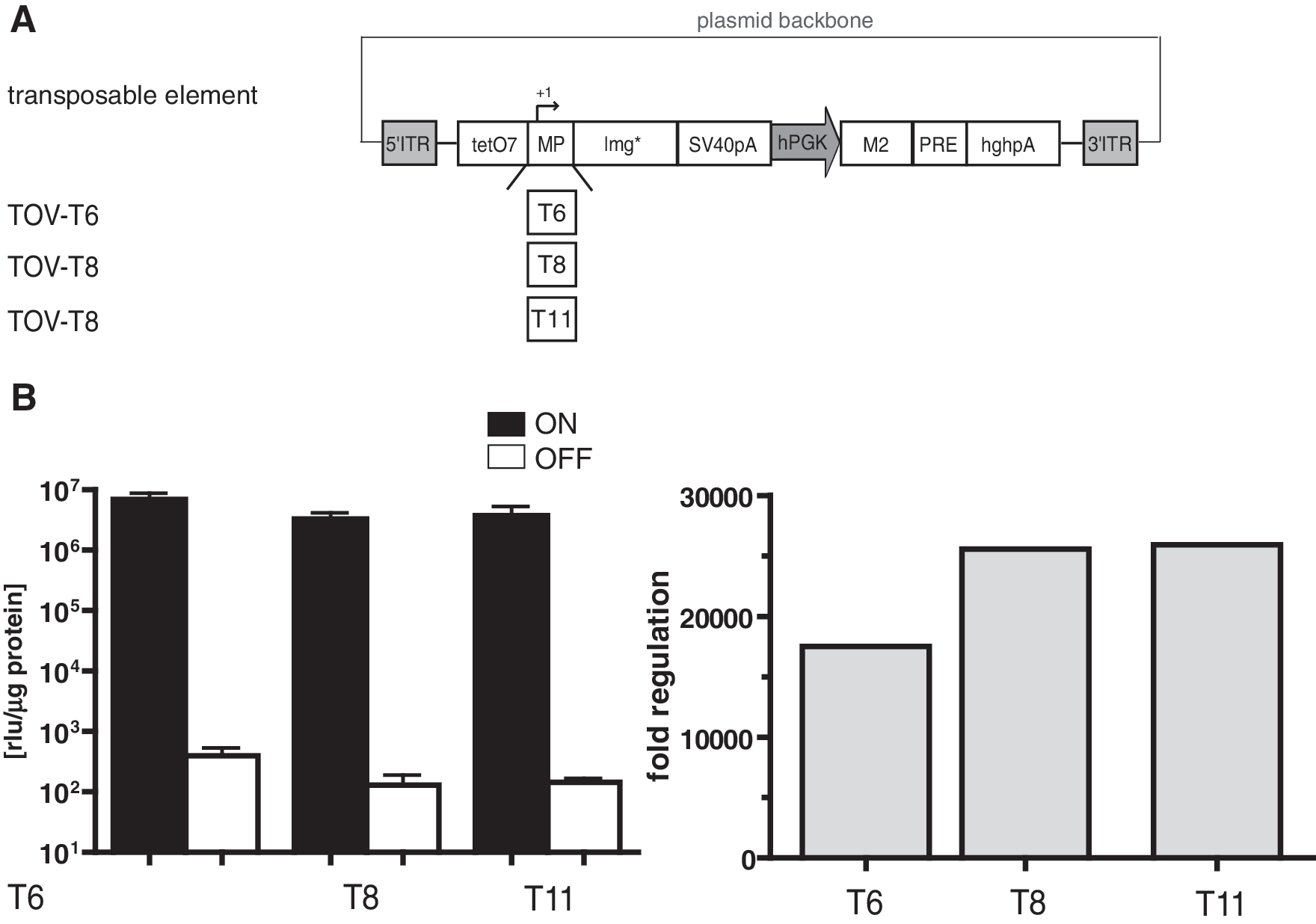
Tet-inducible Sleeping Beauty transposase system. (
In summary, we showed that improved tet-regulated all-in-one vectors, based on the incorporation of novel minimal promoters and modification of the vector architecture, resulted in highly improved regulatory windows in hematopoietic and nonhematopoietic cell lines. In particular, our novel all-in-one vectors display very low background expression in the off-state, which is of major interest for many approaches in gene therapy and cell biology.
Discussion
The development of vectors, viral or nonviral, able to transfer both components of the tet-system simultaneously into a target cell, has been the subject of many studies. Interestingly, almost all research on this topic was done by colleagues engaged in the development of gene therapy, which is a consequence of the work with primary cells that do not allow extended ex vivo handling, e.g., preselection. However, the tight regulation of gene expression in cell populations transduced by such all-in-one vectors was hampered by the interference of the Ptet-promoters (mostly Ptet-1) and the promoters (e.g., CMVie-promoter) used to express the tet-transactivators. Therefore, in most all-in-one vectors described so far, the induction of gene expression did not exceed 100- to 500-fold and displayed relatively low signal-to-noise ratios, even in single-cell clones (Gopalkrishnan et al., 1999; Johansen et al., 2002; Vigna et al., 2002).
In this study, we described tet-inducible all-in-one vector systems optimized for tightly regulated transgene expression in nonclonal cell populations, as required for gene therapy approaches. Optimization was achieved by alterations of the retroviral architecture and incorporation of novel tet-responsive promoters, together leading to improved regulatory windows greater than 10,000-fold in nonclonal cell populations. Remarkably, we were able to further improve the regulatory performance two- to eightfold by incorporation of distinct expression cassettes for the tet-dependent transactivator and the regulatory unit into a Sleeping Beauty transposable element, resulting in up to 25,000-fold induction.
The tet-responsive promoters Ptet-T6, Ptet-T8, and Ptet-T11 (Supplementary Figs. S1 and S2), introduced into the all-in-one vector systems, displayed reduced background expression in the off-state of transduced cell populations when compared with Ptet-T2, a tet-responsive promoter closely related to Ptet-1 (Gossen and Bujard, 1992). The structure and functional properties of Ptet-T2 and Ptet-T6 promoters have been described in detail in a recent study (Loew et al., 2010b). The background expression as well as the induction level of Ptet-T8 was reduced compared with those of Ptet-T6, thus confirming earlier results with this minimal promoter (Loew et al., 2006). Interestingly, the MMTV-core promoter, as designed by evolution, contains several cis-elements that were assumed to be involved in its native regulation of hormone-responsive activity. In addition to one GRE (glucocorticoid-receptor element), NF-1 and Oct-1 (Fox-AI) binding sites especially were of importance. Their coordinate binding is proposed to be responsible for nucleosome repositioning during activation of transcription by the steroid hormone (Belikov et al., 2004; Holmqvist et al., 2005). Thus, the tightly controlled expression of Ptet-T8 may be related to a particular promoter structure during the off-state of tet-controlled gene expression. This finding prompted us to modify the Ptet-T6 promoter accordingly. All residual CMVie-promoter sequences (−50/–36 and −23/+16) including the initiator element (Inr) were replaced by MMTV sequences. The resulting promoter, Ptet-T11, indeed showed superior background control in HT1080 cell populations (Fig. 3). However, further experiments to investigate whether removal of the Inr element and/or the introduction of the Oct-1 (Fox-AI) binding site contributed to the enhanced control of background expression in the off-state, when transferred by otherwise identical viral vectors, were beyond the scope of this study. Importantly, the regulatory properties of the vectors were fairly maintained even at high MOI, a test done to mimic the situation in gene therapy trials (Kustikova et al., 2003; Fehse and Roeder, 2008). Similar investigations for the performance of one-vector systems so far have been rarely included in publications (Barde et al., 2006).
The transiently produced vector titers differed significantly depending on the Ptet-promoter and vector architecture. Increased titers were observed for bidirectional vectors, transferring Ptet-promoters displaying tightly controlled background expression in the off-state. The decreased background activity may generate less antisense transcripts, able to interfere with homodimer formation of the viral genome during viral particle formation and/or eliciting a cellular response against double-stranded RNA (Maetzig et al., 2009). The same mechanism would also explain the approximately threefold increase of titers obtained with the unidirectional vectors.
Interestingly, although in HT1080 cell populations the bidirectional design of the viral vectors resulted in a higher dynamic range compared with the unidirectional expression system, the latter was superior in hematopoietic cell lines. This cell type-dependent shift of the dynamic range of tet-regulation toward lower activity might be explained by different compositions of the basal transcription machinery in these cells, as demonstrated for myoblast and terminally differentiated myotubes (Deato and Tjian, 2007). The individual composition of the transcription machineries may also explain preferences for regulatory units observed in standard cell lines, e.g., NIH3T3, CHO, HeLa, or 293T, after introduction of Ptet-promoters by viral vectors (Fig. 3C). Other studies have also found different regulatory windows in different cell types, affecting both inducibility and background activity (Haack et al., 2004; Markusic et al., 2005). Overall, we concluded that a strong interdependence between the vector architecture, the Ptet-promoters, and the cell type exists, underlining the need to optimize the setting for specific applications.
Recently, transposon-based vector systems were used for the delivery of expression cassettes (Ivics et al., 1997; Ding et al., 2005; Mátés et al., 2009). Using the tet-system in the context of a transposon based DNA vector, only low regulatory windows have been reported (Saridey et al., 2009). We assumed that the novel Ptet-promoters should also mediate tightly controlled gene expression in a transposon-based approach. Indeed, nonclonal HT1080 cultures stably transformed by Sleeping Beauty displayed very low background activity in the off-state, resulting in an outstanding high regulation of gene expression (>25,000-fold) in bulk cultures. The presence or absence of viral components, e.g., the pol/env region (Fig. 1D), or the altered integration preferences offer possible explanations. Preferential integrations of the γ-retroviral vectors in the vicinity of transcriptional start sites or CpG islands might lead to stronger interactions of the minimal promoter with nearby cellular enhancers (Lewinski et al., 2006; Beard et al., 2007). Accordingly, the more random integration of Sleeping Beauty transposon throughout the genome (Yant et al., 2005) should reduce interference with cellular enhancers, resulting in lower background activity of the minimal promoter in the off-state. Studies using clonal analysis clearly showed that different clones behave differently in terms of their regulation, indicating an influence of the integration site on regulatory behavior (Saridey et al., 2009). However, the examination of basic mechanisms involved in chromosomal interference with the Ptet-promoters was beyond the scope of this study, which aimed to provide suitable transfer vectors for regulated gene expression in bulk cultures.
Together, the new vectors described here will likely improve the utility of the TetOn system in gene therapy approaches. The regulatory properties of the Ptet-promoters were target cell-specific, affected by the vector architecture and greatly improved when a transposon-based transfer system was used. Because all data were obtained on cell populations, misleading results that might arise from influences of the chromosomal context in clonal analysis were avoided.
Footnotes
Acknowledgments
This work was supported by grants of the German ministry for Research and Education (CB-Hermes, 01GN0930), the Deutsche Forschungsgemeinschaft (KL 1311/4-1 and Cluster of Excellence REBIRTH Exc 62/1), and the European Union (PERSIST, HEALTH-F5-2009-222878). The Sleeping Beauty Transposase System was kindly provided by Zoltan Ivics, Zsuzsanna Izsvák, and Lajos Mátés (MDC Berlin). We would like to acknowledge the assistance of the Cell Sorting Core Facility of the Hannover Medical School supported in part by Braukmann-Wittenberg-Herz-Stiftung and Deutsche Forschungsgemeinschaft. The authors thank Tamaryin Godinho for critical reading of the manuscript.
Author Disclosure Statement
A competing financial interest exists: A patent application for the novel Ptet promoters has been filed by Rainer Loew and Herrmann Bujard.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.