
Abstract
Bone marrow mesenchymal stem cells (BMSCs) represent an important source of cells for tissue repair. The tropism of these cells to the sites of injury and tumors has been well established. Their tumor-homing properties make BMSCs good candidates as antitumor agent delivery vehicles. In this study, we showed that BMSCs have the ability to migrate toward various cancer cells, including prostate cancer cells in vitro and in vivo and incorporating into the tumor mass. When infected with herpes simplex virus thymidine kinase (TK) gene by lentiviral transduction, TK-BMSCs maintained their tumor tropism capabilities and significantly inhibited the growth of subcutaneous PC3 prostate cancer xenografts in nude mice, in the presence of prodrug ganciclovir (GCV). Xenogenic TK-BMSCs also survived and exerted a significant antitumor effect in an animal model bearing metastastic RIF-1 (fibrosarcoma) tumor in the presence of prodrug GCV. The present study demonstrated that overexpression of TK in BMSCs did not affect their multidifferentiation potentials and their specific homing capacities toward the tumor mass, and the TK-BMSCs alone did not cause any harmful side effects in vivo. The use of TK-BMSCs as tumor-specific delivery vehicles together with controlled prodrug treatment may be a safe and novel anticancer therapy approach.
Introduction
A cell-based delivery strategy that exploits the tumor-homing property of bone marrow-derived mesenchymal stem cells (BMSCs) has the potential to solve inherent gene therapy delivery problems. Intravenous/systematic delivery of BMSCs resulted in their specific migration to sites of injury and improved recovery in animal models of skin wounds (Sasaki et al., 2008), stroke, and myocardial infarction (Kawada et al., 2004). Tumor/cancer is considered as wounds that never heal (Dvorak, 1986); tumor microenvironments have many similarities to the tissue repair processes that attract specific homing of mesenchymal stem cells (MSCs) (Dwyer et al., 2007; Menon et al., 2007). Stem/progenitor cells of human or murine origin have been demonstrated to migrate to multiple tumor types, including glioblastoma, melanoma, pancreatic and breast carcinoma, and neuroblastoma in immunocompromised mouse models, regardless of the location of the tumors (Marini et al., 2005; Dwyer et al., 2007; Hall et al., 2007; Menon et al., 2007; Lu et al., 2008; Mishra et al., 2008). Thus, tumor-specific delivery of antitumor agents may be achieved by using genetically modified BMSCs.
In addition to their tumor-targeting properties, BMSCs have potential inhibitory effects on tumor cell growth in vitro and in vivo without host immunosuppression through induction of apoptotic cell death and G0/G1 phase arrest of cancer cells (Khakoo et al., 2006; Lu et al., 2008). MSCs were further shown to home to the tumor microenvironment (Marini et al., 2005; Hall et al., 2007; Mishra et al., 2008) and have been used as carriers for in vivo delivery of various clinically relevant anticancer agents, including cytokines (Elzaouk et al., 2006), interferon (Studeny et al., 2002, 2004; Nakamizo et al., 2005), prodrugs (Kucerova et al., 2007; Miletic et al., 2007), or adenovirus (Chan et al., 2005; Hakkarainen et al., 2007; Stoff-Khalili et al., 2007; Sonabend et al., 2008). Effective and tumor-selective therapy has also been achieved with the use of a genetically modified progenitor cell line in a tumor metastasis model, for which no other effective treatments are currently available (Aboody et al., 2006). BMSCs are easy to obtain, proliferate readily, and are immune privileged cells. Therapeutic transgenes can be transduced and expressed in the BMSCs. All of these characteristics make BMSCs a valuable resource for cell-based therapy and account for their widespread use in clinical applications (Bianco et al., 2005).
In this study, we focus on the ability of BMSCs to migrate toward various tumor cells in vitro and their distribution within a PC3 prostate xenogenic tumor model. We have also evaluated the antitumor effect of TK gene-modified BMSCs in combination with a prodrug in vitro and in vivo. We show that TK-BMSCs retain their BMSC characteristics and tumor tropism potentials. TK-BMSCs have significant cytotoxic effects on tumor cell lines in vitro in the presence of the prodrug GCV. When administrated systematically, TK-BMSCs tracked to the tumor sites and exerted significant inhibitory effects on tumor growth in tumor-bearing animal models.
Materials and Methods
Tumor cell lines
The cell lines used were human breast cancer (MCF-7) and prostate cancer (PC3 and DU145) from the American Tissue Culture Collection (ATCC, Manassas, VA. The murine fibrosarcoma tumor cell line RIF-1 was from Cancer Research UK (London, UK). All tumor cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) with standard antibiotics at 37°C.
BMSC preparation
Green fluorescent protein (GFP) transgenic rats (kindly provided by Professor M. Okabe, Osaka University, Japan) or Sprague-Dawley rats were used. Bone marrow was harvested from the long bones and layered onto Lymphoprep (1.077 g/ml; Nycomed Norway, Asker, Norway) and centrifuged at 850 g for 25 min. The isolated mononuclear cells were cultured in Dulbecco's modified Eagle medium containing 10% FBS and standard antibiotics at a density of 1–3 × 105 cells/cm2 and incubated at 37°C. BMSCs were defined by chondrogenic, osteogenic, and adipogenic differentiation in vitro according to standard conditions reported previously (Chen et al., 2006a,b, 2007).
Vector design and plasmid constructs
For construction of the Lenti-Luc-GFP-Topo plasmid, luciferase and eGFP cassettes were cloned by PCR methods from the plasmid containing luciferase-eGFP (a kind gift from Yao-Cheng Li, Ph.D., The Salk Institute, La Jolla, CA). The primers used were as follows: sense primer, ACCATGGAAGACGCCAAAAACAT; and antisense primer, TTACTTGTACAGCTCGTCCA. The PCR product of 2,395 bp was purified and ligated with the restriction enzyme digestions and confirmed by sequencing analysis.
Virus production and gene transduction
Lentiviral particles were produced in 293T cells by transient cotransfection involving a four-plasmid expression system. In brief, 293T cells were plated into 10-cm2 plates (2 × 106 cells/well); 24 hr later, transfer vector plasmid (either Lenti-luc-GFP or pLOX-GFP-IRES-TK, kindly provided by Dr. Didier Trono, Switzerland) DNA (8 μg), helper plasmid plp-1 DNA (5.28 μg), plp-2 DNA (4 μg), and envelope plasmid plp-VSVG DNA (2.8 μg) were added. Transfection by calcium phosphate in the presence of 25 μM chloroquine was carried out for 12 hr. The virus particles in the medium were harvested 72 hr later and concentrated by centrifugation at 30,000 g at 4°C for 2 hr; then virus was titered according to protocols from Invitrogen (Carlsbad, CA). Gene transductions were carried out at the appropriate multiplicities of infection (MOIs; 50 for BMSCs) in the presence of 8 μg/ml Polybrene. The transfected cells were selected with 10 μg/ml blasticidin. The transfection was confirmed by either luciferase assay or RT-PCR examination for TK expression. The primer sequences for TK were as follows: sense primer, TCCGAGACAATCGCGAACA, and antisense primer, TGTTATCTGGGCGCT-TGTCA, all with a product of 102 bp.
Two-color flow cytometry analysis of luciferase-transduced BMSCs
All antibodies used were purchased from Dako Cytomation Ltd. (Cambridgeshire, UK) unless otherwise specified. The cells were first washed in PBS buffer and then incubated with antibodies for 30 min at 4°C in the dark. The antibodies were polyclonal goat anti-rat CD31, CD44, CD45, and CD90, followed by secondary phycoerythrin-conjugated rabbit anti-goat antibodies, and cells without primary antibodies served as negative controls. The flow cytometry analysis was performed using a Partec Cytoflow Space flow cytometer (Partec GmbH, Münster, Germany) according to the manufacturer's instructions.
Detection of luciferase expression using IVIS 200 in vivo imaging system
For in vitro imaging using IVIS 200 (Xenogen, Caliper Life Sciences, Hopkinton, MA), cells were serially diluted at a ratio of 1:2 from 6.4 × 104 to 500 cells in black polystyrene 96-well plates. D-Luciferin (Biosynth International Inc., Naperville, IL) at 150 μg/ml was added to each well and incubated at 37°C for 10 min prior to imaging examination for 1 min. Bioluminescent images were acquired, and the bioluminescent intensity was quantified as photons per second using Living Image 2.5 software (Xenogen) accordingly.
Cell migration and chemotaxis assays
To assess cell migration, 1 × 105 BMSCs were dispersed onto the inserts of Transwell dishes (Falcon, Becton Dickinson Labware, Lincoln Park, NJ) with 8.0-μm pore size and allowed to adhere for 1 hr at 37°C. The Transwell inserts were then transferred to the bottom chamber, which contained 2 × 105 tumor cells in 600 μl of medium and cultured for 12 hr. Then cells in the upper surfaces of the filter were removed with cotton swabs, and cells that had migrated to the lower surfaces were fixed with 95% alcohol, stained with Giemsa buffer, and counted in five random fields on each filter. The chemotaxis assay was carried out using the Dunn chemotaxis analysis system for PC3 and DU145 cells as described previously (Wells and Ridley, 2005). In brief, 106 tumor cells were put into the outer ring of the chamber as the chemoattractant, and the migration of BMSCs on the overslip was observed and recorded by a time-lapse microscope once every 10 min for 11 hr. Migration was analyzed by AQM 2001 software (Kinetic Imaging Ltd., Manchester, UK) and Mathematic 3.0 (Wolfram Research, Champaign, IL).
Effect of TK-BMSCs and GCV on cell proliferation
TK-BMSCs and four tumor cell lines were cocultured in 96-well plates at 5,000 cells/well for 24 hr, and then 100 μg/ml GCV was added to the culture. Cell proliferation was assessed every 8 hr for a 56-hr period using the CellTiter 96 assay kit (Promega UK, Southampton, Hampshire, UK) according to the manufacturer's instructions. To examine the cytotoxic effects of TK-BMSCs with various concentrations of GCV, Luc-PC3 and Luc-RIF-1 cells (3,000 cells/well) were cultured with TK-BMSCs at ratio of 1:1 and 1:10 with 0–100 μg/ml GCV. After 5 days, cells were examined using the IVIS 200 system, and the results were expressed as a percentage of luciferase intensity of the control group. To measure the cytotoxic effect of varying ratios of TK-BMSCs on tumor cells, 5,000 Luc-PC3 or Luc-RIF-1 cells were cultured with TK-BMSCs at the tumor cells/TK-BMSCs ratios of 1:100, 1:50, 1:20, 1:14; 1:10; 1:5, and 1:2 in the presence of 100 μg/ml GCV or the TK-BMSCs/GCV conditioned medium, and then cell proliferation was measured at day 6 by luciferase expression using the IVIS 200 system. For the clonogenic assay, tumor cells were mixed with TK-BMSCs at the ratios of 1:1 and 1:10 and treated with 100 μg/ml GCV or conditioned medium for 48 hr, and after 14 days the resulting colonies were quantified.
In vivo distribution of BMSCs in subcutaneous and lung metastasis tumor models
All the animal experiments were carried out under approval of the animal experimental ethics committee from our institutions. Tumor implants were established by subcutaneous injection of 2 × 107 PC3 or DU145 cells at the dorsal site of nude mice (three mice for each cell type). A lung tumor metastasis model was established in nude mice (n = 3) by intravenous injection of 5 × 106 PC3 cells in 200 μl of PBS through the tail vein. Luc-GFP-BMSCs (1 × 106) in 200 μl of PBS were injected intravenously at day 14 in the subcutaneous tumor model or at day 7 in the lung tumor metastasis model following administration of tumor cells. To examine the distribution of Luc-BMSCs using the IVIS 200 system, D-luciferin (150 mg/kg) was administrated intraperitoneally 5 min before the imaging examination. The animals were anesthetized using 3% isoflurane and placed inside the camera box. The light emitted from the bioluminescent tumor cells was digitized and electronically displayed as a pseudocolor overlay onto the grayscale animal image. Regions of interest were drawn around the tumor and quantified as photons per second using the software provided. The RIF-1 tumor cell lung metastasis model was also established by intravenous injection of 1 × 105 RIF-1 cells into C3H mice (n = 5) and assessed in a similar fashion.
Therapeutic effects of TK-BMSCs on tumor growth in vivo
The subcutaneous PC3 tumor model and RIF-1 lung metastasis model were established as described above. The experimental details are given in Fig. 1. In brief, the groups are as follows: group 1, TK-BMSCs intravenous injection plus GCV intraperitoneal injection; group 2, TK-BMSCs intravenous injection only; group 3, GCV intraperitoneal injection only; and group 4, PBS intraperitoneal injection control. The number of TK-BMSCs injected was 106; GCV was diluted in PBS, and 30 mg/kg was given per injection intramuscularly. The injections were repeated twice as shown in Fig. 1. To determine subcutaneous tumor growth, tumors were measured by calipers, and tumor volume was calculated as length × width2/2. For the lung metastases, the tumor size was measured by photon emission of the luciferase bioluminescence using the IVIS 200 system.
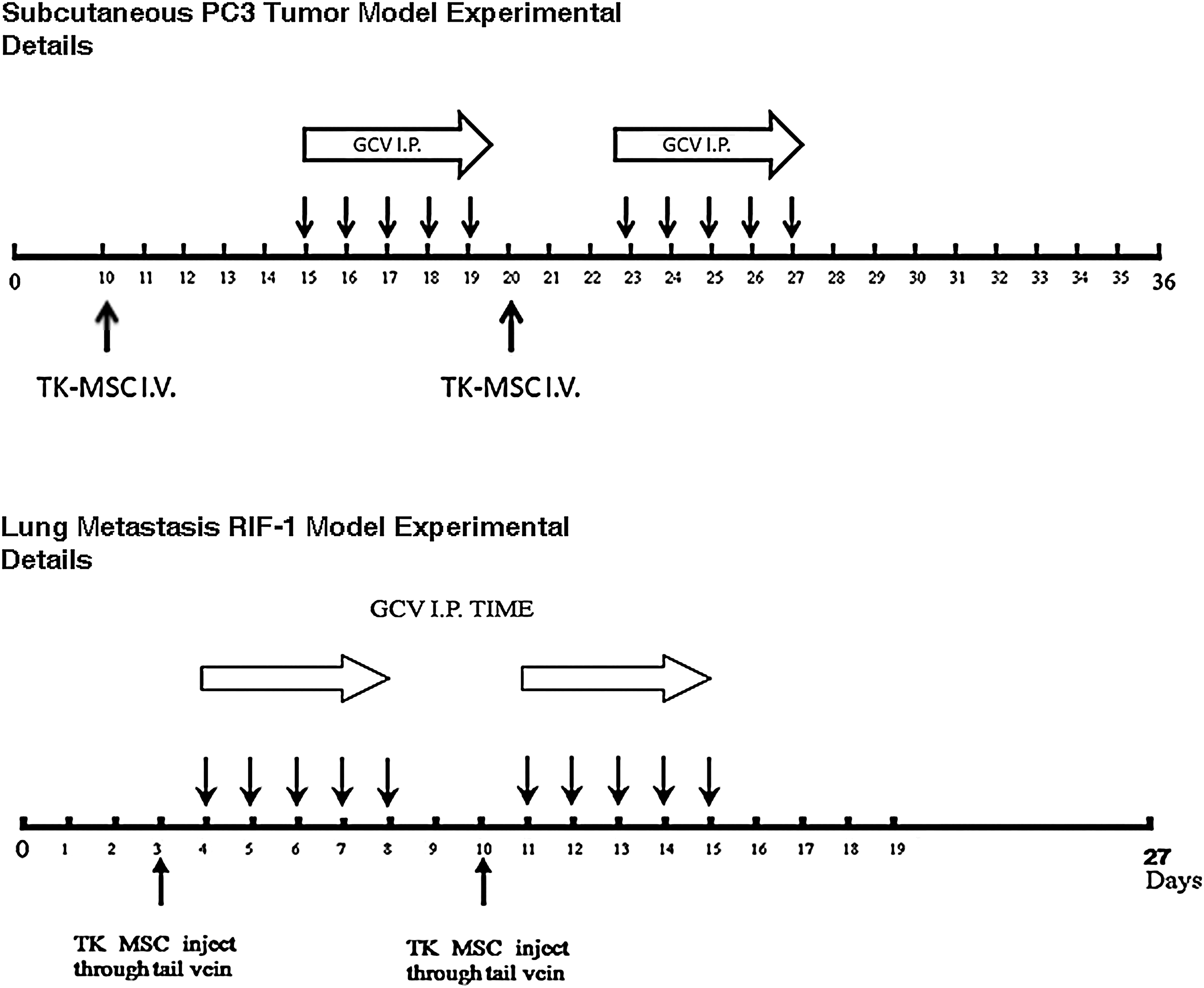
Experimental details for testing the therapeutic effects of TK-BMSCs with or without GCV on tumor growth. For the PC3 subcutaneous model, 106 TK-BMSCs were injected at day 10 and day 20 following PC3 tumor cell subcutaneous implantation, and 30 mg/kg GCV in PBS was injected intramuscularly at day 15 and day 23, each time for 5 consecutive days. For the RIF-1 lung metastasis model, 106 TK-BMSCs were injected at day 3 and day 10 following RIF-1 tumor cells systemic injection, and GCV (30 mg/kg in PBS) was injected intramuscularly at day 4 and day 11, each time for 5 consecutive days.
Statistics
Values are presented as means ± SD or mean percentage of the control. Student's t test was used for comparison of mean values between different groups using SPSS statistical software, and p < 0.05 was considered significant.
Results
BMSC migration toward tumor cells in vitro
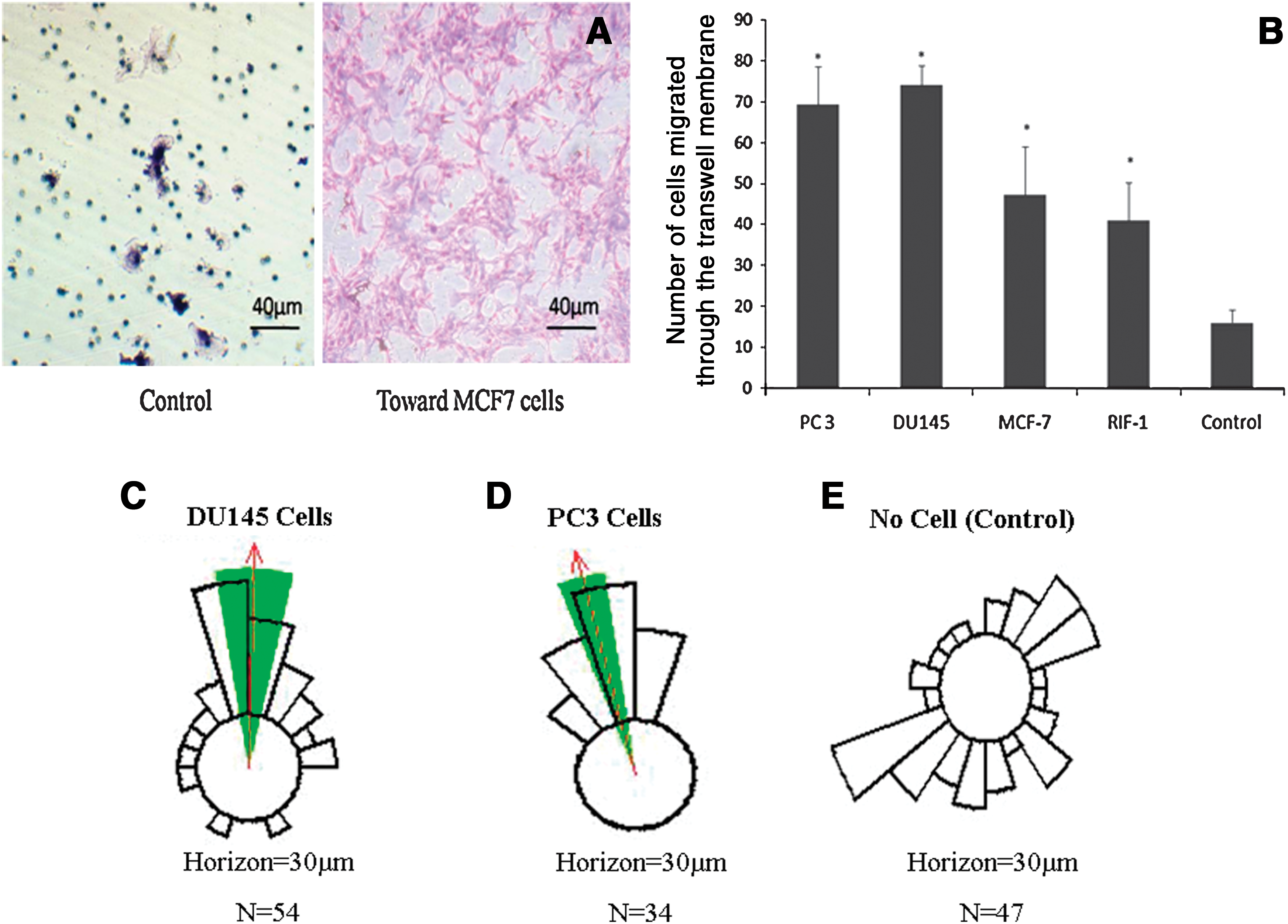
Figure 2A shows the results of a typical BMSC Transwell cell migration assay toward the tumor cells. The numbers of BMSCs migrating toward the tumor cell lines were significantly higher (p < 0.05) than those in the control group (Fig. 2B). Dunn chamber chemotaxis analysis showed that, of the total 54 and 34 BMSCs tracked, there was a significant clustering of cell trajectories toward the direction of DU145 and PC3 cells, respectively (p < 0.01; Fig. 2C and D). In contrast, only randomly dispersed trajectories were recorded for the cells in the group with control medium, suggesting the absence of a chemotactic response (Fig. 2E).
(
Transgene expression in TK-BMSCs and tumor cell lines
The luciferase gene was stably transduced into BMSCs and tumor cell lines (DU145, MCF-7, RIF-1, and PC3), as shown by the in vivo imaging luciferase assay. Prior to in vivo use, Luc-BMSCs were confirmed as having luciferase expression after eight passages, and the bioluminescence intensity correlated with the cell numbers very well (Fig. 3A; R = 0.998). Similar correlations were found between other cell lines: R = 0.999 for DU145, R = 0.996 for MCF-7, R = 0.988 for RIF-1, and R = 0.996 for PC3 cells. In all cells, expression of the luciferase gene was found after at least 10 passages, and cryopreservation recovery cycles did not affect luciferase expression. No cytotoxic effects were observed in normal BMSCs or any of the cell lines (PC3, DU145, MCF-7, and RIF-1) with only GCV treatment (Fig. 3B). TK gene expression in TK-BMSCs was demonstrated by RT-PCR (Fig. 3C). TK-BMSCs had the ability to convert GCV into triphosphate GCV, which exerts its cytotoxic effects by inhibiting cellular DNA polymerases (Ilsley et al., 1995) and competing with deoxyguanosine triphosphate for incorporation into nascent DNA molecules during cell division. The cytotoxic effects were confirmed 24 hr after GCV was added to TK-BMSC culture.

(
Stem cell properties of TK-BMSCs are not affected by transgene expression
The phenotypic profile of the Luc-GFP-BMSCs was examined by flow cytometry and confirmed that the transduced MSCs had a similar phenotype to the nontreated BMSCs. Luc-GFP-BMSCs were positive for CD44 and CD90 and negative for CD45 and CD31 (Supplementary Fig. 1A; supplementary data are available online at
TK-BMSCs are cytotoxic to tumor cells in the presence of GCV through bystander effects
GCV alone did not demonstrate any cytotoxic effect on BMSCs, PC3, DU145, MCF-7, and RIF-1 cells at all concentrations tested. However, in the presence of 0.1 μg/ml GCV, cell proliferation of TK-BMSCs was reduced to 43% of the control level (Fig. 4A). Cytotoxicity of TK-BMSCs in the presence of GCV was demonstrated at a GCV concentration of 1 μg/ml in PC3 (Supplementary Fig. 3A) and RIF-1 cells (Fig. 4C). There was a dose-dependent cytotoxic effect of the GCV and the ratios of TK-BMSCs to tumor cells. TK-BMSCs exerted cytotoxic effects over all tumor cells, even at the ratio of 1 TK-BMSC to 100 tumor cells (p < 0.05; Fig. 4B), but different tumor cells displayed varied sensitivities. TK-BMSCs did not kill RIF-1 cells efficiently at a low TK-BMSC to RIF-1 ratio, whereas significant effects were achieved at higher ratios of 1:5 (TK-BMSCs/RIF-1 cells) or above (Supplementary Fig. 3B). The numbers of colonies in the TK-BMSCs + GCV group were significantly reduced compared with that of the control groups (p < 0.01; Fig. 4D). Significant cytotoxicity was also seen in the three tumor cell lines tested with the TK-BMSC conditioned medium treatment (Supplementary Fig. 3C; p < 0.01) with differences in sensitivity.

(
Homing of BMSCs to tumor sites in vivo
In nude mice when Luc-BMSCs were injected through the tail vein, the majority were found in the lungs at 1–6 days following injection (Supplementary Fig. 4). In contrast, Luc-BMSCs were engrafted mainly at the transplanted tumor site (RIF-1 tumor cells) as early as 3 days after intravenous injection, and the engrafted BMSCs survived and expressed the luciferase in the tumors for up to 12 days (Fig. 5A). Some of the BMSCs also migrated into the lung and spleen at the early time points. The redistribution of BMSCs was observed in the DU145 tumor model, with more BMSCs migrating to the lung at 12 days following injection compared with 9 days (Fig. 5B). The various organs of the mice were also examined separately at the time of sacrifice, as shown in Supplementary Fig. 5A. BMSCs were identified, by immunostaining with anti-eGFP antibody, in the tumor stroma and parenchyma in both PC3 and DU145 tumor lung metastasis models (Supplementary Fig. 5B–D). In the PC3 cells lung metastasis model, after intravenous infusion of a single dose of Luc-BMSCs, mice were followed for 30 days using the in vivo imaging system. Changes in the distribution of BMSCs over time are shown in Fig. 5B, and the percentage of bioluminescent signal in the lung compared with that in the whole body was quantified and is shown in Fig. 5C. On day 1 after infusion, 95.02 ± 4.8% of the BMSCs were seen in the lung; 39.39 ± 14.86% of the BMSCs were still in the lung on day 3; after that, the BMSC numbers in the lung remained relatively stable: 26.63 ± 5.05% in the lung at day 14 and 36 ± 1.37% at day 30 (Fig. 5D). In the RIF − 1 cells lung metastasis model, Luc-BMSCs accumulated in the RIF-1 cells lung metastasis sites and survived for at least for 1 day (Fig. 5D).

(
TK-BMSCs-mediated tumor growth inhibition in vivo
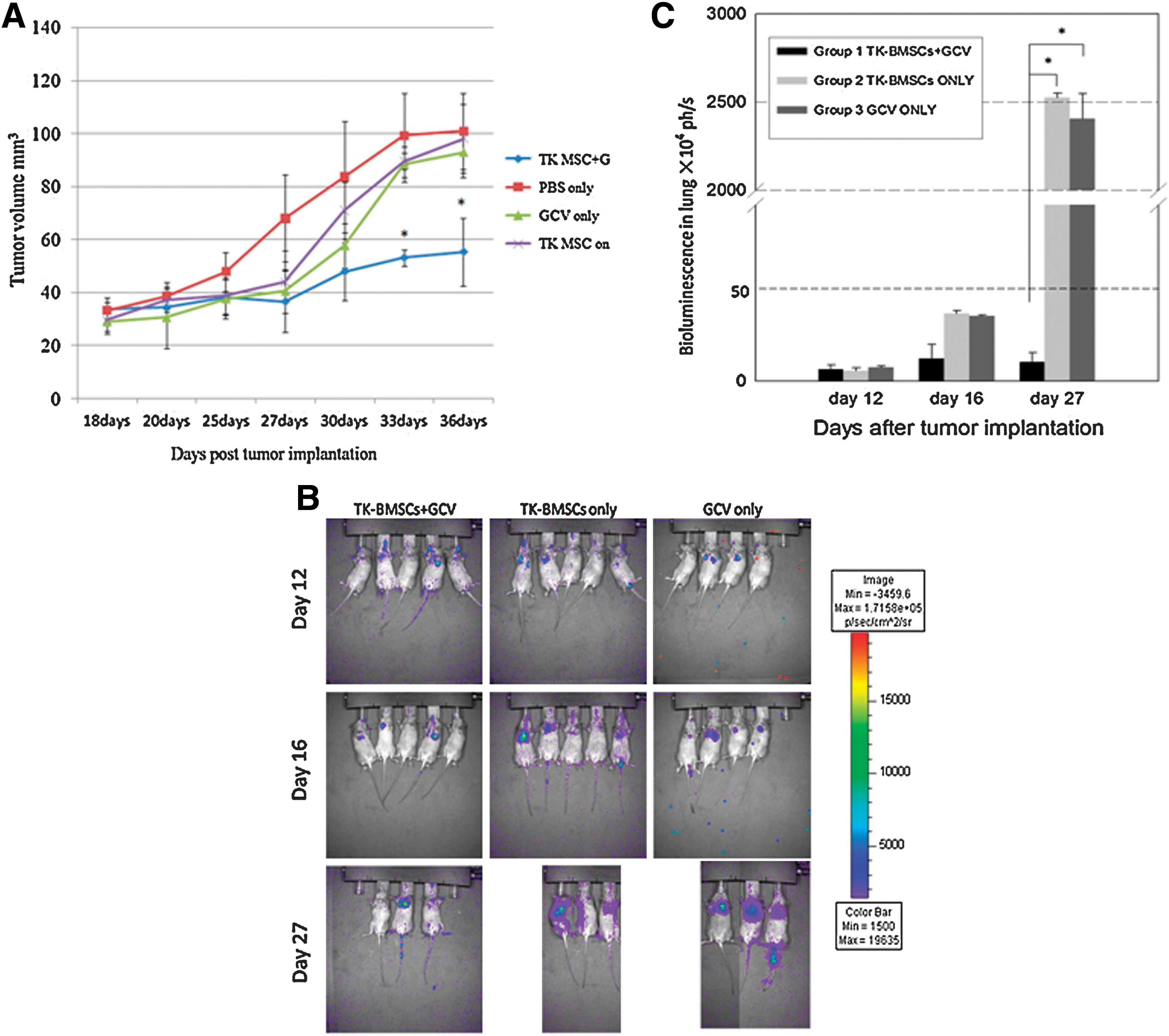
Tumor growth in animals injected with TK-BMSCs + GCV was significantly inhibited by 46.5% and 45.1% on day 33 and day 36, respectively (significant difference between group 1 and other groups on day 33 and day 36, p < 0.05). None of the animals exhibited any obvious signs of toxic side effects during the experiment. No inhibitory effect on tumor growth was found in the groups without GCV administration or when only GCV was used. TK-BMSC administration alone did not show any effect on tumor growth (Fig. 6A). In the Luc-RIF-1 cell lung metastasis model in C3H mice, lung tumor growth was significantly inhibited (up to 99.5%) in the TK-BMSCs + GCV group compared with the other groups at day 27 (p < 0.01; Fig. 6B and C).
(
Discussion
Our results indicate that lentiviral vectors can efficiently transduce BMSCs and remain capable of long-term transgene expression for at least eight to 10 passages. The luciferase and TK gene did not show any cytotoxic effects on the transduced BMSCs. The luciferase and TK gene were expressed in BMSCs and other cell lines and showed good correlations between the bioluminescence and the cell numbers when examined using quantitative imaging techniques. This technique allows the homing of Luc-BMSCs to various tissues/organs in vivo to be followed and quantified in a timely fashion. The luciferase-labeled tumor cells can, therefore, be used to evaluate tumor growth in deep organs in vivo longitudinally without the need of sacrificing the animals.
The lentiviral transduction of the reporter gene or TK gene did not affect the self-renewal capability of the BMSCs or their multipotent differentiation ability and tumor tropism potential. Our results show that lentiviral vectors containing an internal cytomegalovirus promoter induced high levels of target gene expression in long-term MSC cultures, and that cryopreservation recovery cycles did not affect the BMSC phenotype.
In the present study, we tested the migration of rat BMSCs toward four tumor cell lines: two human prostate cancer cell lines (PC3, DU145), one human breast cancer cell line (MCF-7), and a mouse fibrosarcoma cell line (RIF-1). Of these, PC3, DU145, and MCF-7 are all metastatic human tumor cell lines, and RIF-1 is a radiation-induced mouse fibrosarcoma cell line. The migration of BMSCs toward all four different tumor cell lines was demonstrated in vitro, consistent with previous reports (Menon et al., 2007). In vivo experiments further confirmed the homing of BMSCs to PC3 and DU145 tumor sites in both subcutaneous and lung metastasis tumor models. The homing of BMSCs to tumor sites occurred rapidly following intravenous infusion: 3 days for the subcutaneous tumor and 1 day for the lung metastasis tumors. Although some of the BMSCs were found in the organs without tumors, the numbers was significantly low compared with those in the tumor sites. Furthermore, BMSCs survived and expressed functional luciferase or TK gene in the tumor microenvironments, suggesting that these cells are ideal vehicles for delivery of antitumor agents. As shown by us and other groups (Studeny et al., 2002), the BMSCs were found in both the parenchyma and stroma of tumors, so that the biological substances secreted by the BMSCs could reach most of the cells inside tumors. We demonstrated that infused BMSCs could migrate to and engraft in tumor tissues. However, the mechanisms by which BMSCs home and engraft to tumors are not yet fully understood. It is likely that tumor tissues express specific ligands to facilitate trafficking, adhesion, and infiltration of BMSCs. The specific migration of BMSCs toward tumors is a multistep process. The tumor cells and their microenvironments secreted chemokines or cytokines, as demonstrated in the conditioned medium study, which could unregulate the expression of chemokine and cytokine receptors on the BMSCs (Croitoru-Lamoury et al., 2007; Shi et al., 2007). Shortly after exposure to tumor stimuli, some chemokine receptors, such as CXCR4, are up-regulated in BMSCs (Son et al., 2006), and the SDF-1 secreted by BMSCs could also lead to an autocrine effect on their migration (Menon et al., 2007). Many cytokines and chemokine receptors are expressed on BMSCs (Von Luttichau et al., 2005; Honczarenko et al., 2006; Croitoru-Lamoury et al., 2007; Ringe et al., 2007), and differential gene regulation may occur in BMSCs when exposed to different microenvironments. These factors may influence the chemotactic properties of BMSCs that are mobilized into the circulation (O'Donoghue et al., 2003; Chen et al., 2008) or in response to injury situations (Barbash et al., 2003; Satake et al., 2004).
BMSCs can be readily modified by lentiviral systems and stably express the transgene. The transfection rate was close to 90% with the MOI 50 in our system, and the stable cell lines can be established when drug selection is employed. A possible risk associated with lentiviral transduction is insertional oncogenesis following multiple vector integrations into the host genome. The lentiviral systems used in this study are self-inactivating (SIN) vectors, which are the safest lentiviral vectors currently available. SIN vectors were modified to abolish their self-replicating ability and cannot recombine with wild-type viruses. No transformation of the modified BMSCs was found in our studies. Cell-based therapy with systemic delivery of BMSCs has been used in both animal experiments and clinical trials, and no adverse effects have been reported so far (Studeny et al., 2004; Stoff-Khalili et al., 2007). Malignant transformation of BMSCs is very rare and may happen only in extreme situations, such as long-term culture with many passages, and in some animal species. Although lentiviral modification of MSCs may increase the risk, the TK gene product made by the TK-BMSCs with the aid of prodrug GCV will also kill proliferating MSCs and eliminate the risk of tumorigenesis.
The so-called bystander effect was the main mechanism involved in the TK + GCV antitumor strategy, as demonstrated in the present study. The TK-BMSCs + GCV system can achieve higher concentrations of TK enzyme production inside the tumor, while the side effects caused by TK and GCV to the normal tissues are minor and well tolerated, as demonstrated by the in vivo animal experiments in the present study. Furthermore, the use of TK-BMSCs to target tumors may be the best choice for treating metastatic tumors, taking advantage of their tumor-specific homing capacity. Cells labeled with the TK gene can also be monitored in vivo by positron emission tomography with fluorine-18–labeled FEAU (Love et al., 2007; Ray et al., 2007); only when the TK-BMSCs arrive and accumulate at the tumor mass will the prodrug GCV be added, allowing more controlled and tumor-targeted therapy and increasing the safety.
There is much evidence to show that BMSCs are immunoprivileged cells and have unique immunosuppressive properties (Di Nicola et al., 2002; Potian et al., 2003; Tse et al., 2003; Chen et al., 2006a,b). We have shown that rat BMSCs can survive in the immunocompetent C3H mice, at least for a short time, and exert a significant antitumor effect in the RIF-1 cells lung metastasis model. Other studies carried out in our laboratory have also confirmed that rat BMSCs can survive in normal mice for at least 8 weeks (Wang et al., 2007). This suggests that allogenic or xenogenic BMSCs may be used as vehicles for delivery of antitumor agents, which can be readily modified, tested, and expanded beforehand for routine clinical use.
Although we have demonstrated in the present study the potent cytotoxic effects of TK-BMSCs + GCV on tumor cells in vitro and in vivo, TK-BMSCs + GCV did not eradicate the tumors in the tumor-bearing animals. Because the TK + GCV works by disrupting DNA synthesis in rapidly proliferating cells, they can only inhibit tumor growth by killing the proliferating cells, and are ineffective in killing the nondividing cells inside the tumor. New strategies have been developed to tackle these problems, such as using conditional replication-competent adenoviral and suicide-gene chemotherapy systems according to the specific tumor type (Stoff-Khalili et al., 2007; Sonabend et al., 2008).
In conclusion, the current study demonstrated the great therapeutic potential of TK-BMSCs with GCV in cancer treatment. They may be used in combination with traditional chemotherapy and radiotherapy to improve the survival rate of cancer patients, especially for those with metastatic or rapidly growing cancers or tumors.
Footnotes
Acknowledgments
We gratefully acknowledge financial support from China Natural Science Foundation (grant no. 30872635/C160705) and Hong Kong Innovation and Technology Commission (grant no. ITS/305/09) to Gang Li for supporting the research work.
Author Disclosure Statement
No competing financial interest exists.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.