
Abstract
ProSavin is an equine infectious anemia virus vector-based gene therapy for Parkinson's disease for which inducible HEK293T-based producer cell lines (PCLs) have been developed. These cell lines demonstrate stringent tetracycline-regulated expression of the packaging components and yield titers comparable to the established transient production system. A prerequisite for the use of PCL-derived lentiviral vectors (LVs) in clinical applications is the thorough characterization of both the LV and respective PCL with regard to identity and genetic stability. We describe the detailed characterization of two ProSavin PCLs (PS5.8 and PS46.2) and resultant ProSavin vector. The two cell lines demonstrate stable production of vector over a time period sufficient to allow generation of master and working cell banks, and subsequent large-scale vector production. ProSavin generated from the PCLs performs comparably in vivo to that produced by the standard transient transfection process with respect to transduction efficiency and immunogenicity. The development of ProSavin PCLs, and the detailed characterization described here, will aid the advancement of ProSavin for clinical application.
Introduction
A prerequisite for the use of LVs in clinical applications is to ensure that the product is stable and well-characterized. This includes not only detailed characterization of the LV itself but also of the respective PCL, with regard to identity and genetic stability. A study reporting the generation of HIV-1 vector producer cell lines revealed a decrease in vector production over time, in a number of the PCLs. Further investigation found this to be due to low-level expression of vesicular stomatitis virus G glycoprotein (VSV-G), in the uninduced state, of the regulated parental VSV-G cell line. This expression caused a selective pressure against the stably integrated VSV-G DNA over long-term culture, and caused limited stability of the cell line (Farson et al., 2001). This study highlights the importance of performing stability studies to ensure that the clones that are selected are genetically stable. Further to this study, another group also found that during extended culture of an HIV-1-packaging cell line vector production declined. However, the copy number of the integrated vector components remained stable for the duration of the study (Ni et al., 2005). This indicated that the decrease in vector production was not due to genetic instability, but was due to gene silencing. Gene silencing is a phenomenon known to have evolved in eukaryotic cells as a defense mechanism against foreign DNA, particularly repeat arrays (Bestor, 2000; McInerney et al., 2000). It is known that plasmid DNA incorporated in stably transfected cell lines is often concatemeric, and thus it is essential to assess vector production after prolonged culture of producer cell lines that have been generated by stable transfection of plasmid DNAs. These stability studies need to be conducted for a period of time that would be sufficient to enable generation of master and working cell banks, and a subsequent large-scale production process.
It is also possible that the genotype of producer cells could be modified over time as a result of autotransduction, which would be signified by an increase in genome copy number, a phenomenon that has previously been demonstrated with unregulated VSV-G-pseudotyped retroviral vectors (Vogt et al., 2001; Segall et al., 2003). In our system expression of the vector genome is constitutive; however, expression of both packaging components (VSV-G and EIAV gag/pol) is controlled by the tetracycline (Tet) repressor (TetR) regulatory protein. This has a number of advantages: first, it is likely that the cells can be grown for extended periods before vector production with no adverse effects on cell growth/viability due to VSV-G expression. Second, in the absence of vector shedding autotransduction of producer cells should be minimal or prevented entirely. Our previous analyses indicate that in the absence of an inducing agent the level of repression conferred by the TetR protein derived from the codon-optimized TetR (coTetR) gene is such that expression of either of the packaging components is undetectable (Stewart et al., 2009). Whether levels are sufficiently low to prevent production of any functional vector particles has been examined further in this study by monitoring the genome copy number of noninduced producer cells over time.
Here we describe the detailed characterization of two ProSavin PCLs (PS5.8 and PS46.2) and the resultant ProSavin vector products. The cell lines demonstrate stable production of vector during prolonged culture. We also assessed the in vivo performance of ProSavin from the PCLs, to evaluate comparability with ProSavin generated from the standard transient transfection process. This comprised a comparison of the in vivo transduction efficiency of the vector preparations and also their relative immunogenicity. This was of particular importance as the high-level VSV-G protein expression observed in the parental packaging cell line (Stewart et al., 2009) could result in higher levels of VSV-G associating with vector particles relative to transiently made vector. As VSV-G is known to be immunogenic (Burns et al., 1993; Yee et al., 1994) this could potentially enhance the immunogenicity of the PCL-derived vector preparation. This is the first report describing the detailed characterization and in vivo performance of a therapeutic lentiviral vector derived from stable PCLs.
Materials and Methods
Cell lines
HEK293T cells used for transient transfection were obtained from M. Calos (Stanford University, Stanford, CA). HEK293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM, cat. no. D5671; Sigma-Aldrich, Poole, UK) containing 10% (v/v) fetal calf serum (FCS) obtained from Moregate BioTech (Munich, Germany) and supplemented with 2 mM
ProSavin PCLs PS5.8 and PS46.2 and the PC48.2 packaging cell line were generated as previously described (Stewart et al., 2009). These cell lines were maintained in DMEM containing γ-irradiated tetracycline-negative FCS of Australian origin (cat. no. A15-527; PAA Laboratories, Yeovil, UK) supplemented with 2 mM
Determination of copy number by quantitative polymerase chain reaction
To determine copy numbers of the various integrated components the PCLs (PS5.8 and PS46.2) and negative control HEK293T cells were cultured in the presence and absence of appropriate antibiotics. For the autotransduction analysis PCLs were cultured in the presence of antibiotics only. For all cell lines 1.5 × 106 cells were harvested and DNA was extracted with a QIAmp DNA blood mini kit (cat. no. 51106; Qiagen, Crawley, UK). The DNA concentration was ascertained by spectrophotometry and adjusted to 30 ng/μl. Samples (n > 8) were analyzed with 150 ng of DNA per 25-μl reaction, and the quantitative polymerase chain reaction (qPCR) was performed as described previously (Rohll et al., 2002), except that TaqMan assays were performed with a 7900HT fast real-time PCR system (Applied Biosystems, Foster City, CA). For determination of the number of copies of vector genome an EIAV-packaging signal primer–probe set was used, details of which have been described previously (Radcliffe et al., 2008). For determination of EIAV gag/pol, 50 nM EIAV gag/pol forward (5′-GAGGCCCCAGAAACAAACTTT) and 300 nM reverse (5′-ATTCCTTTTTTATTTCGCTCAGATCT) primers and 125 nM EIAV gag/pol probe (5′-FAM-CCGATACAACAGAAGAGTCAGCACAACAAATCT-TAMRA) were used in the PCR mix. For determination of VSV-G, 300 nM VSV-G forward (5′-GGAAAGCATTGAACAAACGAAAC) and reverse (5′-CGGCATCCGTCACAGTTG) primers and 125 nM VSV-G probe (5′-FAM-AGGAACTTGGCTGAATCCAGGCTTCC-TAMRA) were used in the PCR mix. To prepare standard curves the following plasmid DNAs containing the appropriate amplicon were used to prepare standards of known copy number: pONY8.0Z (Mazarakis et al., 2001) for detection of packaging signal, pVSVG (Stewart et al., 2009) for VSV-G, and pGagPol (Stewart et al., 2009) for EIAV gag/pol. DNA prepared from a clonal HEK293T cell line containing a single EIAV (pONY8.4ZCG) (McGrew et al., 2004) vector copy (single integrant, SI), verified by Southern blotting, was used as an internal reference standard and to calculate copy numbers of the vector genome. A clonal HEK293T cell line containing a single copy of an EIAV vector (pONYK.hygro-ires-GFP.MAD) was generated. The vector genome contained within it multiple amplicon DNA (MAD) regions, including amplicons of VSV-G and EIAV gag/pol. DNA was prepared and the integration of a single copy of the vector genome was verified by comparison with the SI DNA. The copy numbers of VSV-G and EIAV gag/pol within the PCLs were calculated by normalization to this MAD internal reference standard.
ProSavin production
Stability study and vector production from ProSavin PCLs
PCLs were passaged regularly in the presence or absence of antibiotics. At regular time points (days 0, 13, 27, 55, 83, and 111) the cells were tested for their ability to produce vector. Four ten-cm dishes were seeded at a density of 8 × 106 cells per dish (note that this seeding density is slightly lower than previously used [Stewart et al., 2009] and is not optimal for maximal vector yields: this is considered of no consequence for this study as stable vector production over time is under scrutiny and not absolute vector titers). The next morning doxycycline (cat. no. D9891; Sigma-Aldrich), at a concentration of 1 μg/ml, was added to three of these, so as to test vector production on induction in triplicate, and the remaining dish was used as a noninduced control. Sodium butyrate (10 mM) (cat. no. B5887; Sigma-Aldrich) was added to all dishes and 6–8 hr later the medium was replaced with fresh medium (to which dox [1 μg/ml] was added in the case of induced cells). Twenty-two hours later vector was harvested and filtered (0.45 μm).
A time course experiment, ranging from 7 to 55 hr postinduction, was performed to establish at which time point maximal vector production was achieved from each of the PCLs. This was found to be 22 hr postinduction for both PCLs, which is comparable to the optimal harvest time point used for harvesting vector for the standard transient transfection system (data not shown).
The titers of ProSavin vector were estimated by integration (DNA) titer assay, as described previously (Stewart et al., 2009).
Large-scale vector production from ProSavin PCLs
Large-scale ProSavin preparations from the PCLs were produced by the previously described procedure in 80–90 ten-centimeter dishes. Cell culture supernatants were harvested and the vector was concentrated 2000-fold by centrifugation. This comprised an initial low-speed centrifugation at 6000 × g at 4°C for a minimum of 18 hr, followed by ultracentrifugation at 50,000 × g at 4°C for 90 min. The titers of concentrated ProSavin were estimated by integration (DNA) titer assay; the titer of ProSavin produced from PS5.8 was 1.3 × 108 transducing units (TU)/ml, and that from PS46.2 was 8.2 × 107 TU/ml.
Production of research-grade ProSavin by transient transfection
Research-grade ProSavin was produced by the transient vector production method, with three-plasmid cotransfection of HEK293T cells using Lipofectamine 2000 CD (cat. no. 12566-014; Invitrogen) as previously described (Stewart et al., 2009), except that the transfections were carried out in a 10-layer Nunc Nunclon Δ Cell Factory (Thermo Fisher Scientific, Waltham, MA). The following quantities of plasmid were used in the Cell Factory: 400 μg of ProSavin genome plasmid (pONYK1-ORT) (Farley et al., 2007), 200 μg of EIAV synthetic gag/pol (pESGPK) (Stewart et al., 2009), and 8 μg of VSV-G envelope (pHG) (Farley et al., 2007). Vector was concentrated by centrifugation as described previously. The titer of research-grade ProSavin was ascertained by integration (DNA) titer assay as 2.4 × 108 TU/ml.
Production of ProSavin “in the spirit of GMP” by transient transfection
ProSavin produced in a manner analogous to clinical-grade vector was generated as described previously but was purified and concentrated by ion-exchange chromatography and ultrafiltration. Briefly, the vector-containing supernatant is filter-clarified (Sartoclean CA 1.2 μm/0.45 μm filter capsule, cat. no. 5621306F7-00-B; Sartorius, Goettingen, Germany). To digest DNA, Benzonase (cat. no. 1.01695.0002; Merck) is added to a final concentration of 5 U/ml. After incubation overnight at 2–8°C, the supernatant is applied to an anion-exchange membrane chromatography unit (Sartobind Q MultiSep, cat. no. 91-Q-02K-15-06; Sartorius). A stepwise gradient of sodium chloride is passed through the membrane and the vector-containing fraction of the eluate is collected and then concentrated and purified by hollow fiber tangential flow ultrafiltration/diafiltration. During this process the vector medium is exchanged into formulation buffer. The vector material is then filter-sterilized (mini Kleenpak filters, 0.2 μm, cat. no. KA02EKVP2S; Pall, Port Washington, NY) and further concentrated by hollow fiber ultrafiltration. This ProSavin preparation was labeled IH21 and the titer was ascertained by integration (DNA) titer assay as 2.3 × 108 TU/ml.
Northern blotting
HEK293T cells, packaging cell line PC48.2, and PCLs PS5.8 and PS46.2 (±doxycycline induction) were harvested into 5.0 × 106 cell aliquots, in duplicate. The cells were subsequently homogenized by passage through QIAshredder columns (cat. no. 79654; Qiagen) according to the manufacturer's instructions. Total RNA was extracted with an RNeasy mini kit (cat. no. 74104; Qiagen) according to the manufacturer's instructions. From the total extracted RNA the mRNA was isolated with an Oligotex mRNA mini kit (cat. no. 70022; Qiagen) according to the manufacturer's instructions. Total and mRNA was also extracted from HEK293T cell pellets that had been transduced with unconcentrated ProSavin generated from PS5.8 or PS46.2, or control ProSavin that had been generated by the standard transient transfection process as a control. Cell culture supernatants from PS5.8 and PS46.2 were concentrated 71-fold and vector RNA was extracted with a QIAmp viral RNA mini kit (cat. no. 52904; Qiagen) combined with an RNase-free DNase set (cat. no. 79254; Qiagen). RNA (1–10 μg) and 2 μg of RNA Millennium Marker (cat. no. AM7150; Ambion, Warrington, UK) were electrophoresed on a 1% agarose–formaldehyde gel and transferred to a nylon membrane (Nytran SuPerCharge, cat. no. 732-4197; VWR, Lutterworth, UK). The membrane was hybridized with 32P-labeled probes prepared with a DNA-labeling kit (Amersham Rediprime II, cat. no. RPN1633; GE Healthcare UK, Little Chalfont, UK), at 65°C. The probe for the ProSavin genome was derived from a 595-bp ClaI DNA fragment that represents the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) region of pONY1.Blast. The probe for EIAV gag/pol was derived from a 970-bp XbaI and SspI DNA fragment of pGagPol, for VSV-G the probe was derived from a 900-bp StuI DNA fragment of pVSVG, and for coTetR the probe was derived from a 700-bp NotI and NheI DNA fragment from pPuro.coTetR. The hybridized membrane was washed for 20 min at room temperature in buffer containing 0.1% sodium dodecyl sulfate (SDS)–2 × saline sodium citrate (SSC), followed by two washes, the first for 15 min and the second for 10 min, at 65°C in 0.1% SDS–0.1 × SSC. The membrane was then exposed to Kodak BioMax MS film (Sigma-Aldrich).
Detection of catecholaminergic enzymes by Western blot analysis
HEK293T cells were transduced, in the presence of Polybrene (8 μg/ml) (cat. no. H9268; Sigma-Aldrich), at a multiplicity of infection (MOI) of 1 with ProSavin generated from a standard HEK293T transient cotransfection process (research-grade ProSavin) or from PCLs PS5.8 or PS 46.2. Three days posttransduction cells were harvested and lysed in fractionation buffer (0.1 M Tris-HCl [pH 7.3], 0.2% [v/v] Nonidet P-40 [cat. no. EM-B56009-70; EMD Chemicals/VWR]). Twenty micrograms of total protein was loaded onto 4–20% polyacrylamide denaturing gels. Western blotting was performed with one of the following: anti-CH1 antibody (obtained from E. Werner, Innsbruck Medical University, Innsbruck, Austria [Golderer et al., 2001]), anti-TH antibody (cat. no. AB152; Chemicon, Livingstone, UK), or anti-AADC antibody (cat. no. AB136; Chemicon). Primary antibody incubation was followed by incubation with a peroxidase-conjugated anti-rabbit secondary antibody (cat. no. P0448; Dako, Ely, UK). Visualization was performed with an enhanced chemiluminescence (ECL) Advance Western blotting detection kit (cat. no. RPN2135; GE Healthcare UK).
Animals
Twenty-six adult male Wistar rats weighing 225–250 g were obtained from B&K Universal (Hull, UK) and housed in a standard 12-hr light/dark cycle with ad libitum access to food and water. All surgical procedures were approved by the local veterinarian and ethics committee and were carried out according to U.K. Home Office regulations. General anesthesia in animals was induced and maintained during the procedure via an intraperitoneal injection of xylazine–ketamine mix.
Intrastriatal administration of ProSavin
Once animals were fully anesthetized they were placed into a stereotaxic frame (Stoelting, Wood Dale, IL). The test article was drawn up in a 10-μl Hamilton syringe attached to a sterile 33-gauge stainless steel needle. Bilateral intrastriatal injections were performed, and the stereotaxic coordinates used for injections were taken from the bregma. The coordinates used were as follows: anteroposterior (AP), 0.0 mm; mediolateral (ML), ± 3.5 mm; dorsoventral (DV), −4.7 mm; with the incisor bar set at 0.0 mm. A small burr-hole was drilled in the skull at the position of injection and the needle lowered to the dural surface, at which point the DV coordinates were calculated. The needle was lowered to the desired DV coordinate and an injection volume of 4 μl of test article (ProSavin or TSSM formulation [tromethamine, NaCl, sucrose, and mannitol]) was slowly infused at a speed of 0.2 μl/min, using an infusion pump (World Precision Instruments, Stevenage, UK). On completion of the infusion the needle was left in place for 3 min, after which it was withdrawn slowly from the brain. The skin was sutured to close the wound.
The 26 rats were divided into 5 groups as follows: group 1, six animals received bilateral injections of 4 μl of ProSavin generated from PS5.8 (dose, 5.2 × 105 TU per hemisphere); group 2, six animals received bilateral injections of 4 μl of ProSavin generated from PS46.2 (dose, 3.3 × 105 TU per hemisphere); group 3, six animals received bilateral injections of 4 μl of research-grade ProSavin (produced by the transient process) (dose, 9.6 × 105 TU per hemisphere); group 4, six animals received bilateral injections of 4 μl of ProSavin IH21 (dose, 9.2 × 105 TU per hemisphere); group 5 (negative control), two animals received bilateral injections of 4 μl of TSSM formulation buffer.
Four weeks after test article administration the rats were killed and then transcardially perfused with heparinized phosphate-buffered saline (PBS) and processed as described subsequently.
Blood collection for immunology
Blood (at least 0.5 ml) was collected from individual rats before test article administration, 14 days after vector administration, and before termination (terminal bleed, day 28). Blood was allowed to clot at room temperature for a minimum of 30 min before centrifugation at 2500 rpm for 15 min, at 4°C. On completion the serum fraction was collected and frozen at −80°C for immunological analysis.
Histology
Half the animals from each group (n = 13) were used for histology. After administration of the final dose of anesthetic the animals were perfused transcardially with 4% paraformaldehyde Brains were dissected, postfixed overnight in the same solution, and then transferred to 30% sucrose. Brains were sectioned serially at a thickness of 40 μm, using a Leica 3050 cryostat (Leica Microsystems, Wetzlar, Germany), and placed in multiwell plates containing PBS and 0.01% sodium azide.
For immunochemistry, 1 in 10 sections through each brain were blocked for 2 hr at room temperature in PBS containing 10% normal goat serum. Sections were then incubated with primary antibodies specific to the human CH1 or AADC protein, neither of which cross-reacts with endogenous rat proteins: anti-AADC (cat. no. AB136; Chemicon) for four nights at 4°C; anti-GTP-CH1 (obtained from E. Werner) overnight at 4°C. After incubation with primary antibody, sections were washed and incubated with secondary antibody from a VECTASTAIN Elite ABC kit (anti-rabbit IgG) (cat. no. PK-6101; Vector Laboratories, Peterborough, UK). For 3,3′-diaminobenzidine (DAB) visualization sections were washed and then incubated with ABC tertiary complex from the VECTASTAIN Elite kit followed by treatment with a DAB peroxidase substrate kit (cat. no. SK-4100; Vector Laboratories). Sections were mounted on glass slides and coverslipped, using DPX permanent medium (cat. no. 360292F; VWR).
Counts of human CH1- and human AADC-positive cells in each of the sections were assessed by an observer blinded to the experimental conditions. Images were captured with AxioVision software (AxioVision Systems/Carl Zeiss, Welwyn Garden City, UK).
EIAV qPCR end points
The remaining animals from each group (n = 13) were used for EIAV qPCR. After terminal anesthesia and subsequent perfusion, brains were removed and the left and right striata were carefully dissected and placed immediately in RNAlater (cat. no. 76104; Qiagen). Samples were incubated overnight at 5 ± 3°C, the liquid was removed, and striatum samples were stored at −80°C until additional processing.
DNA was isolated from each of the striatal sections and qPCR was performed to detect the number of copies of EIAV packaging signal in each of the samples as previously described (Lamikanra et al., 2005), except that 150 ng of DNA per 25-μl reaction and a 7900HT fast real-time PCR system combined with TaqMan genomic assays were used.
Immunological analyses of humoral responses in serum samples by Western analysis
Untransfected HEK293T cells (negative control) and transfected HEK293T cells transiently producing ProSavin (positive control) were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.2], 0.15 M NaCl, 0.1% SDS, 1% sodium deoxycholate, 1% Triton X-100/Igepal CA-630). The proteins from the negative and positive control cell pellets were separated by SDS–PAGE on 10% polyacrylamide gels and blotted onto polyvinylidene difluoride (PVDF, cat. no. RPN303F; Amersham). The membranes were blocked in PBS with 5% nonfat dried milk powder to prevent nonspecific binding before incubation with test serum samples that had been diluted 1:50 in 5% nonfat dried mild powder in wash buffer (0.05% Tween 20 in PBS). To enable ProSavin antigens in the rat sera to be identified, membranes were incubated with positive control ProSavin-specific monoclonal/polyclonal antibodies (anti-Gag/Pol [cat. no. EIAP6A1 VMRD, Pullman, WA], anti-neomycin phosphotransferase [cat. no. 06-747; Millipore, Livingston, UK], anti-TH, anti-CH1, anti-AADC) or rat VSV-G-positive or -negative sera. After washing, the membranes were incubated with an appropriate horseradish peroxidase (HRP)-conjugated secondary antibody. To enable detection of antibodies in the rat sera an HRP-conjugated anti-rat immunoglobulin polyclonal antibody (cat. no. P0450; Dako) that reacts with rat immunoglobulins of all antibody isotypes was used. Binding of antibodies to ProSavin-associated antigens on the membrane was visualized with an ECL Advance Western blotting detection kit and detected by chemiluminescence, and recorded by autoradiography.
Results
Producer cell lines are stable in the absence of selective pressure
To establish whether the ProSavin PCLs were capable of producing high-titer vector after extensive passaging a stability study was carried out by culturing each of the two PCLs (83 days for PS5.8 and 111 days for PS46.2) with and without antibiotic selection (hygromycin, blasticidin, Zeocin, and puromycin). At regular intervals the cell lines were tested for their ability to produce vector under induced (+dox) or uninduced (−dox) conditions.
TetR-mediated regulation was maintained in both cell lines with and without antibiotic selection, as vector was not detected in the absence of dox (data not shown). Figure 1 shows the data for vector production for PS5.8 and PS46.2 under induced conditions. Each of the PCLs yielded comparable vector titers, with and without antibiotic selection, for the duration of the study. These data indicate that both PCLs are stable, in terms of vector production both in the presence and absence of antibiotic selective pressure.

Long-term stability of vector production. The ability of the producer cell lines (PCLs) PS5.8 and PS46.2 to produce ProSavin vector on induction was assessed at regular time points after culture with/without antibiotic selection pressure. The stability study was carried out for 111 days for PS46.2 and for 83 days for PS5.8. Vector titers were established by DNA integration assay. Data shown represent mean values ± SD.
Copy number analysis of vector components in the producer cell lines
At three time points (day 0, mid- and end time points; see Fig. 2) during the stability study, producer cells from both culture conditions (±antibiotic selection) were harvested. Genomic DNA was prepared and analyzed by qPCR to assess the copy number per cell for each of the integrated vector components: vector genome, EIAV gag/pol, and VSV-G (Fig. 2). This enabled screening for the possible loss of any vector component over the duration of the study. For each cell line and culture condition combination the copy number per cell was compared with the value obtained on day 0. The data show that there is 1 vector genome copy per cell, for both PS46.2 and PS5.8. The average number of EIAV gag/pol copies per cell was 3 for PS46.2, and 9 for PS5.8. The copy numbers per cell for EIAV gag/pol and vector genome were stable for the duration of the study (108 days for PS46.2 ± antibiotic selection and 95 days for PS5.8 ±antibiotic selection), indicating that for these components gene loss did not occur. Conversely, qPCR analysis showed that VSV-G gene copies were lost during culture for both PCLs, and this loss was observed to have stabilized by the mid-time point (day 57) for both PCLs with and without antibiotic selection pressure. In both cases the reduction in VSV-G copies was approximately 2-fold: for PS5.8, 32 copies were lost (average on day 0 was 54 copies per cell); and for PS46.2, 9 copies were lost (average on day 0 was 22 copies per cell). The remaining VSV-G gene copies were stable for the remainder of the stability study. The reduction in the number of VSV-G expression cassettes did not appear to affect vector production, as titers remained comparable over the duration of the study (Fig. 1).

Analysis of the genetic stability of the vector components in the PCLs. The copy number of the three components was determined by TaqMan qPCR. Cells from PS46.2 and PS5.8 cultured with/without antibiotic selection in the absence of induction were harvested at various time points during the stability study and copy numbers of vector genome, EIAV gag/pol, and VSV-G were determined. Data shown represent mean values.
Producer cells are autotransduced after induction of vector production
The vector genome copy number data demonstrated that in the absence of induction the vector genome was stable over time, with neither loss nor gain in the number of integrated copies per cell (Fig. 2). An increase in copy number over time would have indicated autotransduction resulting from leaky expression of the packaging components. To demonstrate autotransduction and confirm that superinfection interference (Coffin, 1997) was not occurring, the PCLs were cultured in the induced state for 10 days, and at regular time points cells were harvested. Genomic DNA was prepared and analyzed by qPCR to assess the vector genome copy number per cell (Fig. 3). For both PS5.8 and PS46.2 the genome copy number increased rapidly from the time of induction to day 7 and by day 10 the cells had died, and therefore copy numbers could not be determined for this time point. The cell death was most likely due to cytotoxicity of the VSV-G protein but could also be due to genotoxicity as a result of exposure of the cells to high vector MOIs over the course of the experiment. On day 7, cell lines PS5.8 and PS46.2 had reached genome copy numbers of 60 and 76, respectively.
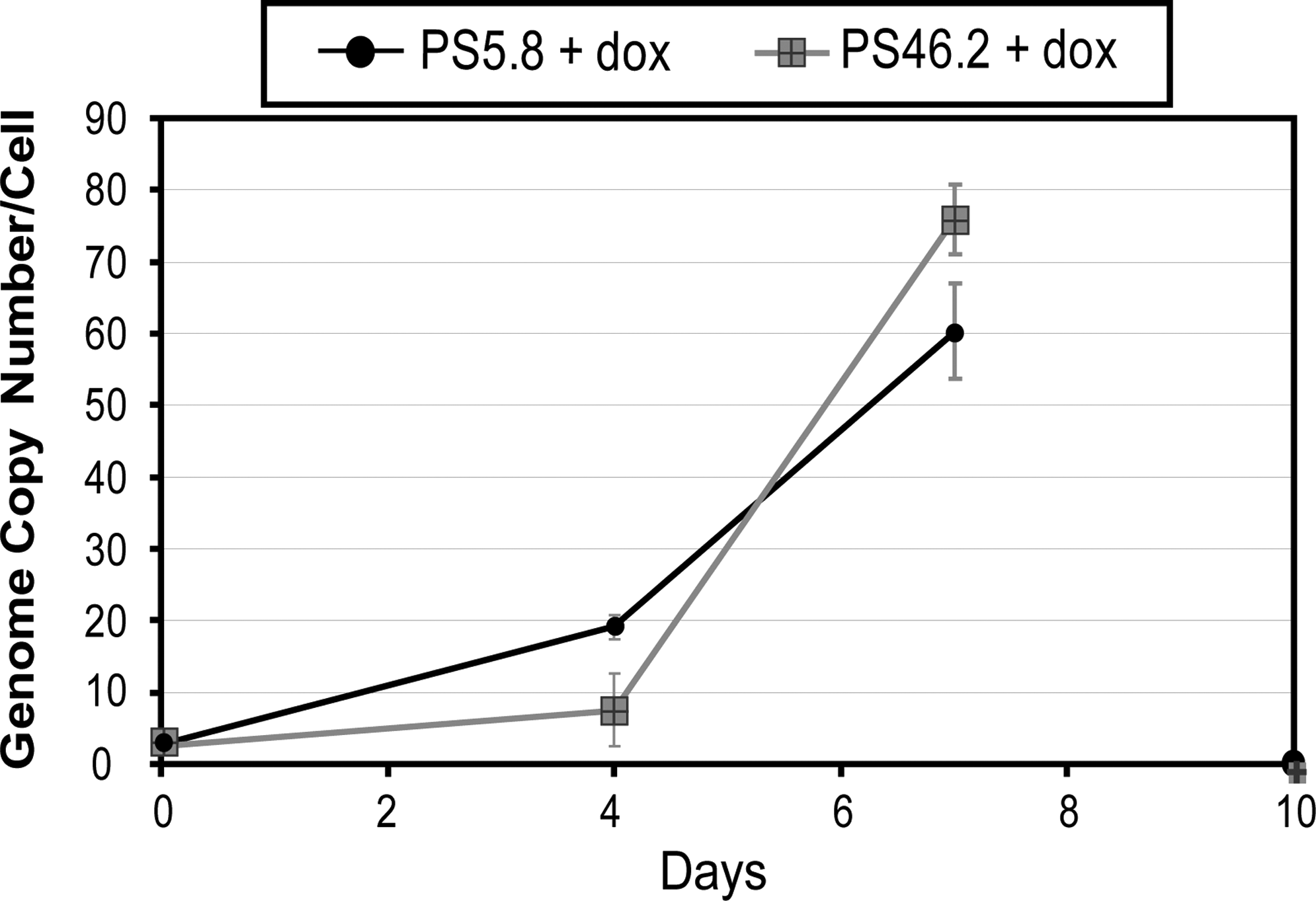
Analysis of the genetic stability of vector components in the PCLs. To assess autotransduction both of the PCLs were cultured in the on-state and cells were harvested at various time points postinduction. Vector genome copy number was assessed by qPCR on the DNA extracted from these cells. Data shown represent mean values ± SD.
Transcripts of the expected size are present in producer cell lines, vector particles, and transduced cells
The integrity of vector-related mRNA transcripts in the PCLs (vector genome RNA, gag/pol, VSV-G, and TetR) was examined by Northern blot analysis. This was performed to ensure that the transcripts are of the predicted sizes, that the integrated expression cassettes had not undergone any deletions or rearrangements, and that polyadenylation is occurring as expected with no detectable readthrough.
To examine whether the correct ProSavin transcripts were being generated in the PCLs, mRNA was extracted and Northern blot analysis was carried out with a probe designed against the WPRE region. The two expected transcripts of 8.0 and 5.8 kb, generated from the full-length genomic and internal tricistronic mRNAs, respectively, were clearly present (Fig. 4A, lanes 1 and 2). Smaller transcripts (less than 5.8 kb) were also observed for both PCLs, and these transcripts were of the same sizes in both cell lines (Fig. 4A, lanes 1 and 2). It is unlikely that these smaller transcripts are due to mutation(s) in the ProSavin genome cassette, as identical mutations would have to have occurred in both PCLs. Because the probe is targeted to the WPRE region at the 3′ end of the genome transcript the smaller transcripts on the Northern blot must represent fragments of the transcript from the 3′ end. Therefore, the most likely explanation for the presence of these small transcripts is partial degradation of the larger mRNA transcripts. These same smaller transcripts have been detected by Northern analysis of mRNA from transiently transfected producer cells and resulting vector RNA (data not shown), indicating that the small transcripts detected in this Northern blot are not unique to the PCLs. Correctly sized internal ProSavin transcripts (kb) were detected in HEK293T cells that had been transduced with vector generated from the PCLs, albeit at lower levels (Fig. 4A, lanes 4 and 5). This RNA transcript length is the same size as the transcript detected in HEK293T cells that had been transduced with vector generated by the standard three-plasmid cotransfection method (Fig. 4A, lane 6). Probing of vector RNA demonstrated the presence of a predominant band at 8 kb, the correct size for full-length genomic ProSavin RNA (Fig. 4A, lanes 7 and 8). Smaller weaker bands, of comparable size, were detected in the vector RNA samples (Fig. 4A, lanes 7 and 8), again the most likely reason being due to vector RNA degradation. For all samples no transcripts were detected that were greater in length than the expected ProSavin transcripts, illustrating that polyadenylation is working well with no detectable readthrough.
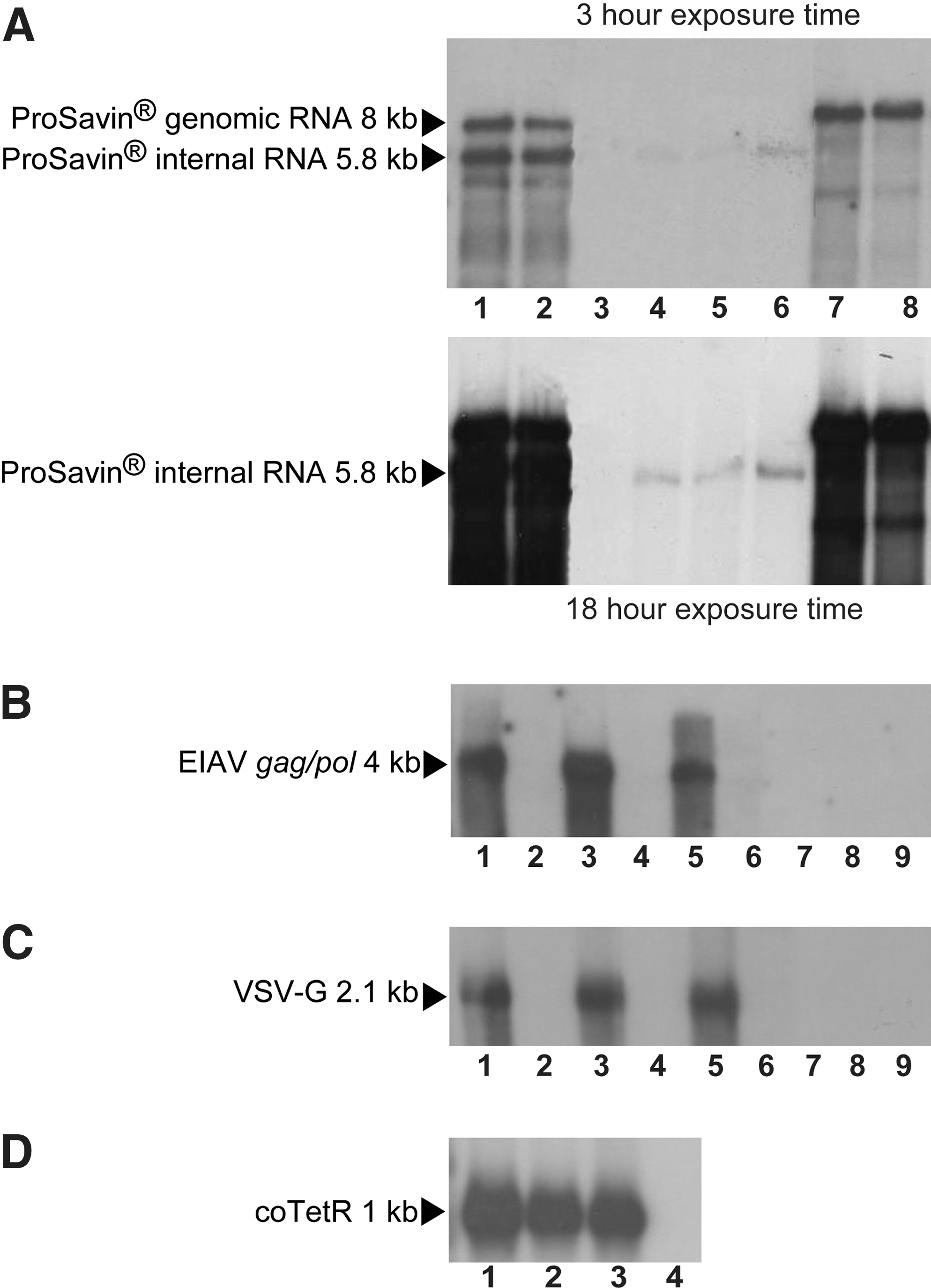
Northern blot analyses to examine PCL component transcripts: Northern analysis to examine transcript length of the ProSavin genome (
Northern blot analyses to examine EIAV gag/pol (Fig. 4B), VSV-G (Fig. 4C), and coTetR (Fig. 4D) transcripts were performed on mRNA from PS5.8 and PS46.2 alongside mRNA from the parental packaging cell line (PC48.2). This was to confirm that these transcripts remain unchanged within individual PCLs as compared with the parental packaging cell line. For VSV-G and EIAV gag/pol Northern blot analyses, mRNA was examined from the packaging cell line and PCLs that had been cultured with and without dox; this allowed the TetR-mediated regulation of these packaging components to be assessed. Figure 4B–D shows that transcripts of the correct lengths were detected for EIAV gag/pol, VSV-G, and coTetR mRNAs. As expected, transcripts for EIAV gag/pol, VSV-G, and coTetR were not detected in mRNA isolated from HEK293T cells (Fig. 4B and C, lane 8; Fig. 4C, lane 4). No EIAV gag/pol and VSV-G transcripts were detected in cells that had been cultured in the absence of dox (Fig. 4B and C, lanes 2, 4, and 6). The transcript sizes for EIAV gag/pol, VSV-G, and coTetR in the induced PCLs are the same as those seen in the parental packaging cell line (PC48.2) (Fig. 4B and C, lanes 1, 3, and 4). This confirms that no changes affecting EIAV gag/pol, VSV-G, or coTetR expression have occurred in the PCLs because of the introduction of the ProSavin genome cassette. As expected, EIAV gag/pol and VSV-G RNA transcripts were not detected in the vector RNA samples (Fig. 4B and C, lanes 8 and 9).
Producer cell lines express functional catecholaminergic enzymes
To confirm that ProSavin generated from the PCLs was capable of transduction and subsequent transgene expression, HEK293T cells that had been transduced with ProSavin generated from the PCLs were examined by Western blot analysis, using antibodies to TH, CH1, and AADC. As a positive control cell extracts from HEK293T cells that had been transduced with ProSavin generated from an HEK293T standard transient transfection process were examined and untransduced HEK293T cell lysates were used as a negative control. All three catecholaminergic enzymes (56-kDa AADC, 42-kDa TH, and 26-kDa CH1) were detected from all ProSavin-transduced cells (Fig. 5, lanes 1, 2, and 3). None of these proteins were detected in the untransduced HEK293T negative control samples (Fig. 5, lane 4).
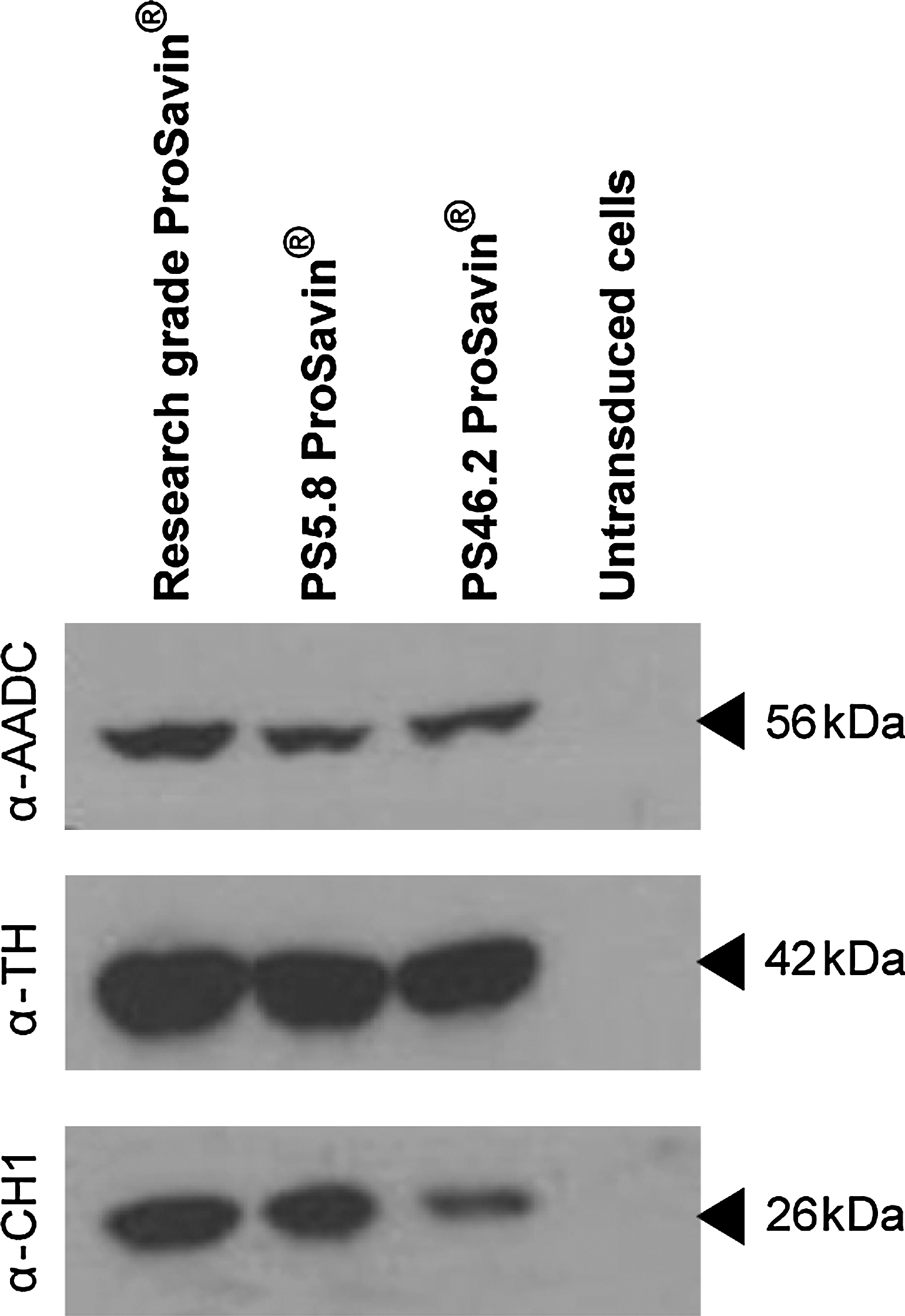
Western blots of catecholaminergic proteins expressed from cells transduced with ProSavin generated from the PCLs. Shown are Western blots of total cell extracts prepared from HEK293T cells transduced at an MOI of 1 with ProSavin generated from an HEK293T standard transient transfection process (research-grade ProSavin) and from PS5.8 and PS46.2 PCLs. As a negative control, cell extracts from untransduced HEK293T cells were included. The blots were probed with antibodies against the catecholamine proteins (AADC, aromatic
To assess enzyme function HEK293T cells (a noncatecholaminergic cell line) were transduced with ProSavin vector preparations produced from PS5.8 and PS46.2 and by the standard transient transfection process. Catecholamines were separated by high-performance liquid chromatography and were detected electrochemically in cell extracts. Dopamine was detected in cells transduced with each of the ProSavin preparations, indicating that the expressed proteins form catalytically active enzymes. The levels of dopamine detected from cells that had been transduced with each of the ProSavin preparations ranged from 27 to 70 ng per 106 integrated genome copies, with no difference observed among the various ProSavin preparations (data not shown).
Producer cell line-derived ProSavin displays efficient in vivo transduction and minimal immunogenicity
A study was performed to assess the in vivo transduction efficiency and immunogenicity of ProSavin generated from PCLs PS5.8 and PS46.2 compared with “research-grade” ProSavin produced by the transient transfection process after stereotaxic injection into the striata of adult rats. As a secondary objective the transduction profile of ProSavin vector manufactured in the spirit of GMP (IH21) was also compared with the research-grade and PCL ProSavin material. The research-grade batch was produced in parallel with vector generated from the two PCLs (see Materials and Methods). In this process vector was generated from the PCLs, or from transiently transfected HEK293T cells, and concentrated by centrifugation. The IH21 ProSavin material was harvested from transfected HEK293T cells in a similar manner but was purified and concentrated by ion-exchange chromatography and ultrafiltration as per clinical-grade vector. Each vector was injected bilaterally into the striata of six adult male rats and a further two rats were injected with buffer as negative controls. Animals were killed 1 month after vector administration. Half the animals from each group were used for histological analyses and the other half for qPCR vector copy number analysis. Sera from all animals were taken before vector administration, on day 14 after vector administration, and at termination (day 28). Immunological tests were performed on the blood to assess antibody generation to the ProSavin vector components and producer cells.
Histological analyses evaluated human CH1 and human AADC expression in rat striata, enabling comparison of expression patterns from the various ProSavin vectors. CH1- and AADC-positive cells were detected in the brains from all groups of rats injected with the various ProSavin vector preparations. Figure 6A shows images from one section from one animal of each group, for both CH1 and AADC immunostaining. In Fig. 6B the average positive cell counts per hemisphere, for each group of animals, is shown. It is clear that both transgenes (encoding CH1 and AADC) were expressed in brains from all animals for all ProSavin-injected groups. A lower number of cells expressing AADC was observed compared with those expressing CH1; this is a reproducible phenomenon thought to be due either to differences in antibody avidity or to the fact that AADC is not as stable as CH1 in vivo (Azzouz et al., 2002). ProSavin produced by the transient transfection processes (research-grade and IH21 material) produced higher levels of CH1 transgene expression in vivo than was seen from ProSavin produced by either of the PCLs. However, the differences in CH1 expression were less than 3-fold and can be explained by differences in the doses that were administered, because the fold difference in positive cells (Fig. 6B) closely resembled the fold differences in administered dose (see Materials and Methods). Striatal sections from three animals per group were taken for DNA extraction, and this DNA was then used to measure total vector DNA copy number by qPCR. It is important to consider that the vector DNA may be either integrated or episomal (in quiescent cells, any unintegrated vector DNA will not be diluted out by cell division). Figure 7 shows the average total vector copies normalized to transducing units (TU) of vector administered, for each group of animals. Overall, it appears that each of the ProSavin preparations was successfully delivered to the striata of the rats and that high levels of vector DNA were present for at least 28 days, whether integrated or episomal. The in vivo transduction efficiency of all the ProSavin preparations was similar, with the highest level being observed from the group of animals that had received ProSavin produced from PCL PS46.2.

Transduction efficiency in the brains of rats that received bilateral intrastriatal injections of the various ProSavin preparations. ProSavin produced from PCLs (PS5.8 or PS46.2) or by the standard transient transfection process (research grade or IH21) was injected bilaterally into the striata of three rats; one rat was also injected with TSSM (tromethamine, NaCl, sucrose, and mannitol) formulation buffer. One in every 10 sections throughout each brain hemisphere was immunostained with an antibody to either human CH1 or human AADC and positive cells were counted. Human CH1 and AADC expression was observed in the brains of rats that had received each of the ProSavin preparations. Representative images (original magnification, × 100) from one section from one animal of each group are shown (

Transduction efficiency in the brains of rats that had received bilateral intrastriatal injections of the various ProSavin preparations. DNA from left and right striatal sections, from three animals of each group, was analyzed for the quantity of EIAV vector sequences, by qPCR. This measured the number of vector copies per striatal section. Data shown represent geometric mean total vector copies normalized to transducing units (TU) of vector administered, for each group, ± SD.
Western analysis was performed on serum samples generated from blood samples collected on days 0, 14, and 28 from each animal to assess whether antibody responses were generated against various vector components. No antibody responses against any ProSavin component were detected in any animal that received TSSM formulation buffer (Fig. 8). Antibody responses directed against neither the ProSavin enzymes (CH1, AADC, and TH) nor neomycin phosphotransferase (Neo-PT gene is encoded by the ProSavin genome) were detected in the sera of any animal. However, antibodies against VSV-G were detected in at least one serum sample from at least one animal per group after administration of the various ProSavin vectors. Antibodies against the p26 EIAV capsid were detected in three of the four groups of animals that received ProSavin vector, and in one of the four groups of animals an HEK293T-associated antigen response was observed. The highest frequency of antibody responses was detected in animals that had received ProSavin derived from PS5.8, where 50% of animals demonstrated ProSavin-associated humoral responses on both days 14 and 28. Antibody responses to VSV-G and the p26 EIAV capsid were each detected in two of six animals from the PS5.8 group. In the same group, antibodies against an HEK293T-associated antigen were detected in sera from three animals, in both day 14 and day 28 samples. Antibodies were not detected against this HEK293T-associated antigen in any of the other groups of animals studied. Animals injected with research-grade and IH21 ProSavin demonstrated slightly lower frequencies of ProSavin-associated antibody responses, namely VSV-G and p26, than was observed for animals that had received ProSavin derived from PS5.8. ProSavin derived from PCL PS46.2 evoked only a single antibody response (anti-VSV-G) at one time point in one animal. This group thus demonstrated the lowest frequency of ProSavin-associated antibody responses as compared with the other three groups.

Humoral responses in sera from rats that had received bilateral intrastriatal injections of the various ProSavin preparations. Serum samples prepared from blood samples collected from individual rats on day 0 (d0), day 14 (d14), and day 28 (d28) postinjection of ProSavin or TSSM formulation buffer. Humoral responses to ProSavin were assessed by Western blot analysis. Antibodies were detected against VSV-G (VSV-G), p26 EIAV capsid protein (p26), and an HEK293T-associated antigen (HEK293T) in a limited number of animals after administration of the various ProSavin vectors.
Discussion
We previously reported the development of a stable HEK293T-based lentiviral packaging cell line demonstrating tetracycline-regulated expression of the packaging components (EIAV gag/pol and VSV-G) and the use of this cell line to generate two inducible PCLs for the production of ProSavin, a gene therapeutic for the treatment of Parkinson's disease (Stewart et al., 2009). Here we provide a detailed characterization of these PCLs and show that (1) vector production by both PCLs is stable after prolonged culture, even in the absence of selective pressure; (2) transcripts of the expected sizes are present for each vector component in producer (and transduced) cells; (3) cells transduced with PCL-derived ProSavin vector express catalytically active enzymes; and (4) PCL-derived and transiently prepared ProSavin vectors are comparable in terms of transduction efficiency and humoral responses after in vivo administration.
Stability studies to examine vector yield from the PCLs over time were required, as it is possible that expression of the vector components could be lost. Potential causes are genetic instability and/or gene silencing, phenomena that have been observed during extended culture of HIV-1-packaging and producer clones (Farson et al., 2001; Ni et al., 2005). During the stability study vector was not detected in the absence of induction at any time point and under all conditions examined, confirming that the regulation conferred by the presence of the coTetR gene is stably maintained. On induction each PCL demonstrated comparable vector production at all time points throughout the study. This indicates that vector production by both PCLs is stable during prolonged culture, even in the absence of selective pressure. Furthermore, the duration of the stability study was calculated to encompass a number of cell doublings in excess of those required to generate master and working cell banks and a large-scale production process. An additional benefit was the ability to culture the producer cells in the absence of antibiotics.
Examination of the copy numbers of each vector component at the beginning and end of the stability study indicated that the PCLs are genetically stable with regard to the number of copies per cell of vector genome and EIAV gag/pol, even after long-term culture. However, both PCLs were genetically unstable in terms of the number of VSV-G copies, with loss of approximately half the copies when analyzed at a time point mid-way through the study (day 57), after which the number of copies of VSV-G remained stable. Most importantly, the titers of vector produced from the PCLs are stable for the duration of the study. This indicates that the decrease in VSV-G copies does not adversely affect vector production, most likely because sufficient copies of VSV-G remain to confer adequate expression for efficient pseudotyping of the vector particles.
Data from the stability study indicate that, in the off state, expression of the packaging components in the PCLs is tightly repressed. In agreement with this, results from Northern analyses show that no VSV-G or EIAV gag/pol transcripts are detected in the absence of induction. However, there is a chance that low-level leakage of VSV-G and EIAV gag/pol expression may occur. This could result in the production of small quantities of vector, which would not be detectable by the DNA integration assay. This vector would be capable of transducing the vector producer cells, a phenomenon known as autotransduction, which is undesirable as it could potentially compromise the genetic stability of the PCLs. As discussed previously, the number of vector genome copies for both PCLs, in the absence of induction, remained stable for the duration of the study, indicating that autotransduction was not detectable during the course of this study. To investigate this further and to confirm that the undetectable level of autotransduction was due to the absence of vector production and not some other factor (such as superinfection interference; Coffin, 1997) the PCLs were cultured for several days under induced conditions. The ProSavin PCLs demonstrated rapid autotransduction after vector production, which confirms the conclusion that stringent control of the packaging components is conferred by the TetR system. These results support previous findings that superinfection interference does not occur for VSV-G-pseudotyped retroviral vectors (Vogt et al., 2001; Segall et al., 2003).
Transcripts of the expected size for the internal ProSavin tricistronic mRNA, and for the three ProSavin-encoded catecholaminergic proteins, were detected in cells that had been transduced with PCL-derived ProSavin vector. In addition, dopamine was detected in the culture medium of the transduced cells, indicating that the ProSavin transgene proteins are catalytically active.
To assess the in vivo transduction profile of the various ProSavin preparations, rats received bilateral striatal injections with ProSavin produced from either PS5.8 or PS46.2 or from transiently transfected HEK293T cells. Results show that injection of the various ProSavin preparations led to transduction and transgene expression. Vector was detected by qPCR in striatal sections from all animals that had received ProSavin. This indicated successful delivery of each of the ProSavin preparations to the striata of rats and persistence of the vector for at least 1 month. Differences in transduction efficiency of the different vectors were minimal, although ProSavin generated from PCL PS46.2 did demonstrate higher in vivo copy numbers per transducing unit administered compared with the other preparations. Immunological analysis demonstrated that ProSavin produced by the various methods elicited only minimal immune responses to ProSavin, namely to VSV-G, the p26 capsid, and an HEK293T-associated antigen. Overall it appears that ProSavin produced by cell line PS5.8 elicited a higher frequency of antibody responses against the broadest range of antigens compared with those animals that had received ProSavin generated from PS46.2 or produced by transient transfection. However, a large proportion of the observed antibody response detected in this PS5.8 group (6 antibody responses out of a total of 13) was raised against an HEK293T-associated antigen, which was not observed in any of the other groups of animals. The reason for this is unknown but may be due to slight differences in the production of ProSavin. It could be that HEK293T-associated antigens were copurified from PS5.8 cell culture supernatant alongside ProSavin vector, whereas no or fewer HEK293T-associated antigens were copurified in the other ProSavin preparations. This HEK293T-associated antigen response is not specific to PCL PS5.8 as it has been observed in other in vivo studies in which ProSavin or EIAV-LacZ (produced by the standard transient transfection process) was administered (data not shown). Any HEK293T-associated antigen should not pose a problem in patients receiving ProSavin because they are human and therefore “self.” However, if the HEK293T-associated antigen response is excluded from the analysis the frequency of antibody responses from animals treated with ProSavin from PS5.8 is still higher than from the other ProSavin-treated groups in spite of the transduction efficiency in the brains of rats being lowest with ProSavin produced from PS5.8. One explanation is that perhaps a greater number of nonfunctional vector was generated from PS5.8 compared with the other ProSavin preparations. The nonfunctional vector would not be capable of transducing cells and therefore a higher number of immunogenic vector particles would be available to which an immune response could be elicited. In this study, ProSavin produced from PS46.2 cells appears to be the least immunogenic vector in vivo.
The presence of multiple copies of the VSV-G gene in PCLs PS5.8 and PS46.2 was of concern as it was hypothesized that the resultant ProSavin vector particles could potentially be pseudotyped with large amounts of VSV-G protein, making PCL-derived ProSavin more immunogenic than transiently produced vector. Overall, the results from the immunological analysis indicates that the high copy number of VSV-G in these cell lines has not increased the immunogenicity of the ProSavin vector when compared with ProSavin produced by the standard transient transfection process.
An important consideration regarding any lentiviral vector PCL intended for the generation of material for clinical application is to confirm the absence of any replication-competent lentivirus (RCL). The development and validation of an RCL assay for EIAV-based vectors have been previously described (Miskin et al., 2006). These assays are laborious and must be performed on material made at large scale (i.e., on a scale that produces enough material for a clinical trial) for the results to be meaningful. To date, no RCLs have been detected in four clinical/proof-of-concept batches of ProSavin generated by the current transient transfection-based manufacturing process (J. Miskin, personal communication). A large-scale manufacturing process utilizing the PCLs is currently under development and needs to be defined before production of material for relevant RCL testing can take place. However, this is an important parameter that must be addressed before any lentiviral vector generated from a PCL can be used in a clinical setting.
In summary, we have conducted a comprehensive characterization of HEK293T-based inducible ProSavin PCLs with respect to their identity and stability, and the in vivo transduction efficiency and immunogenicity of vector product. The data suggest that both PCLs and resultant vector behaved similarly to ProSavin produced by the transient transfection process, both in vivo and in vitro. PS46.2 is potentially a better candidate PCL for further development than PS5.8 because of the observed relatively low immunogenicity and slightly higher transduction efficiency in vivo of the resultant ProSavin product. It is acknowledged that the large-scale production of ProSavin would benefit from the ability to grow PCLs in serum-free (SF) suspension culture. Thus, adaptation of PS46.2 to SF suspension culture is being examined. In addition, the large-scale production of ProSavin from PCLs growing in scalable adherent systems (Cell Factories and microcarriers) is under investigation.
Footnotes
Acknowledgments
The authors are grateful to the manufacturing team at Oxford BioMedica for the production of ProSavin IH21 used as a control in the assessment of in vivo vector transduction efficiency.
Author Disclosure Statement
The authors declare no competing financial interests.