
Abstract
Liver ischemia–reperfusion (I/R) injury is a multifactorial process that affects graft function after liver transplantation. Inflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β, and IL-18, have been shown to play key roles in the pathophysiology of liver I/R injury. Studies have indicated that NALP3 (NACHT domain, leucine-rich repeat [LRR] domain, and pyrin domain [PYD]-containing protein-3) inflammasome is pivotal in the processing and releasing of IL-1β and IL-18. The aim of this study was to test whether NALP3 silencing has a protective effect in murine liver I/R injury. Using a partial lobar liver warm ischemia model, mice were hydrodynamically injected with pNALP3shRNA, pshRNANC, or saline 48 hr before ischemia. Those mice pretreated with pNALP3shRNA showed decreased serum alanine aminotransferase levels; inhibited production of proinflammatory cytokines such as IL-1β, IL-18, TNF-α, and IL-6 by downregulation of caspase-1 activation and NF-κB activity; as well as decreased release of HMGB1 (high-mobility group box-1) and inflammatory cell infiltration, leading to the prevention of liver I/R injury, when compared with controls. Histology revealed that pretreatment with pNALP3shRNA significantly ameliorated hepatocellular damage after I/R. Thus, by using a small hairpin RNA approach, our study confirms that NALP3 signaling is involved in liver I/R and that silencing of NALP3 can protect the liver from I/R injury by reducing IL-1β, IL-18, TNF-α, IL-6, and HMGB1 release through downregulation of caspase-1 activation and NF-κB activity.
Introduction
The process of liver I/R injury is a cascade of inflammatory events involving multiple interconnected factors, including hepatic sinusoidal endothelial cell injury and disturbances of microvascular circulation, activation of Kupffer cells, production and release of reactive oxygen species (ROS) and inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β, IL-18 and HMGB1 (high-mobility group box-1) (Lentsch et al., 2000; Jaeschke, 2003; Takeuchi et al., 2004; Tsung et al., 2005b).
Inflammasomes are multiprotein cytoplasmic complexes that mediate the activation of inflammatory caspases: caspase-1 can be activated by both the NALP1 and NALP3 (NACHT domain, leucine-rich repeat [LRR] domain, and pyrin domain [PYD]-containing protein-3) (Agostini et al., 2004; Mariathasan et al., 2006; Martinon et al., 2006, 2009; Sutterwala et al., 2006). Caspase-1 cleaves pro-IL-1β to IL-1β and also activates IL-18 and IL-33 (Dinarello, 1998; Schmitz et al., 2005). NALP3 is involved in the recognition of numerous exogenous and host ligands, including bacterial RNA, ATP, and uric acid crystals (Kanneganti et al., 2006; Mariathasan et al., 2006; Martinon et al., 2006; Sutterwala et al., 2006), and is also triggered by low concentrations of intracellular potassium (K+ efflux) and increased levels of reactive oxygen species (Pétrilli et al., 2007a,b; Martinon, 2010).
A growing number of systemic inflammatory diseases, characterized by fever, anemia, and elevated levels of acute-phase proteins, have been linked to excessive production and bioactivity of IL-1β (Martinon and Tschopp, 2004; Church et al., 2008). Inflammasomes have been suggested to be involved in liver I/R injury, as the requisites for their activation, such as ROS, K+ efflux, and various damage-associated molecular pattern molecules (DAMPs), including HMGB1, DNA, and RNA, are released during hepatic ischemia (Meissner et al., 2008; Imaeda et al., 2009; Bamboat et al., 2010b). Furthermore, IL-1 family cytokines, including IL-1β and IL-18, which are secreted after inflammasome activation, have been implicated as promoters of liver I/R injury (Kato et al., 2002; Takeuchi et al., 2004) and some therapeutic strategies targeting IL-1β or IL-18 have proved to be effective in the protection of hepatocytes from I/R injury (Shito et al., 1997; Harada et al., 2002; Takeuchi et al., 2004). In addition, there is evidence of a key role for IL-1β in the pathogenesis of I/R injury, as IL-1β knockout mice exhibit significant reduction of ischemic and reperfusion injury (Furuichi et al., 2006) and, conversely, IL-1 receptor antagonist knockout mice exhibit increased I/R injury (Pinteaux et al., 2006). However, whether blocking the NALP3 inflammasome signaling pathway could protect the liver from I/R injury is unknown.
In the present study, we use a small hairpin RNA (shRNA) plasmid targeting NALP3 to examine the inhibitory effect of shRNA on the expression of NALP3 in vitro and in vivo, and explore the function and potential mechanisms of NALP3 in a murine model of liver I/R. Our studies demonstrate that gene silencing of NALP3 results in protection from inflammation and hepatocyte injury after liver I/R. In addition, we show that the protective effect of NALP3 silencing in liver I/R injury is caspase-1 dependent and is associated with reductions of inflammatory mediators IL-1β, IL-18, TNF-α, IL-6, and HMGB1, and infiltration of macrophages and neutrophils.
Materials and Methods
Animals
Male C57BL/6J mice (8–10 weeks old) were used (Experimental Animal Center, Chinese Academy of Sciences, Shanghai, China). Animals were housed in the animal facility under specific pathogen-free (SPF) conditions with a 12-hr light-to-dark cycle and free access to food and water. Animals received humane care in compliance with the University of Huazhong Science and Technology Animal Care and Use Committee.
Small interfering RNA design/preparation
Small interfering RNAs (siRNAs) against NALP3 were designed with an siRNA selection program (Ambion, Austin, TX). The sense and antisense strands of murine NALP3 siRNA were as follows: siRNA1 sequence: 5′-GGAGUAUUUCUUUAAGUAUTT-3′ (sense) and 5′-AUACUUAAAGAAAUACUCCTT-3′ (antisense); siRNA2 sequence: 5′-GGAUGAACGUGUUCCAGAATT-3′ (sense) and 5′-UUCUGGAACACGUUCAUCCTC-3′ (antisense); siRNA3 sequence: 5′-CCACAAUUCUGACCCACAATT-3′ (sense) and 5′-UUGUGGGUCAGAAUUGUGGAG-3′ (antisense); in addition, a nonspecific scrambled siRNA sequence: 5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′ (antisense). All siRNAs were synthesized by Qiagen (Chatsworth, CA).
Construction of NALP3 shRNA plasmid
The shRNA sequence of the target gene was the same with the siRNA3 sequence. The shRNA expression cassette (containing the siRNA3 interference sequence) under the control of the U6 promoter was cloned into HuSH shRNA plasmids (Origene, Rockville, MD) to form shRNA-interfering plasmid pNALP3shRNA. In the same vector, pshRNANC plasmids containing the same lengths of irrelevant sequence (scrambled siRNA sequence) were constructed as controls. Sequences for both shRNA plasmids were verified as correct. All plasmids were purified with EndoFree plasmid mega kits (Qiagen, Hilden, Germany).
Transient transfection
Chinese hamster ovary (CHO) cells were cultured in 6-well plates to about 80–90% confluence. siRNA1, siRNA2, siRNA3, or scrambled RNA in serum-free F12–Dulbecco's modified Eagle's medium (DMEM) was mixed with Lipofectamine 2000 (Invitrogen, Rockville, MD), according to the manufacturer's protocol. Mock controls were transfected with Lipofectamine 2000 (Invitrogen) alone. After incubation at room temperature for 20 min, this mixture was distributed into duplicate wells of CHO cells, and transfection was performed at 37°C in 5% CO2. The medium was replaced with fresh complete medium 4–6 hr after transfection. Kupffer cells were transfected with pNALP3shRNA or pshRNANC, using a mouse macrophage Nucleofector kit (Amaxa/Lonza, Cologne, Germany) according to the manufacturer's instructions. Mock controls were transfected with mouse macrophage Nucleofector solution (Amaxa/Lonza) alone. Forty-eight hours after transfection, cells were subsequently harvested for real-time polymerase chain reaction (PCR) and Western blotting.
Stimulation of Kupffer cells in vitro
Stimulation of Kupffer cells in vitro was done as previously described (Sutterwala et al., 2006). Kupffer cells were transfected with pNALP3shRNA or pshRNANC, using a mouse macrophage Nucleofector kit (Amaxa/Lonza) according to the manufacturer's instructions. Mock controls were transfected with mouse macrophage Nucleofector solution (Amaxa/Lonza) alone. Forty-eight hours later, the cells were stimulated with lipopolysaccharide (LPS, 50 ng/ml) from Escherichia coli serotype O111:B4 (Sigma-Aldrich, St. Louis, MO) for 12 hr and cultured for an additional 20 min in the presence of 5 mM ATP (Sigma-Aldrich), washed, and then incubated for an additional 3 hr in fresh culture medium. Culture supernatants were then assayed for IL-1β.
Model of liver I/R injury and treatment
We employed a segmental (70%) warm liver I/R injury model in mice (Tsung et al., 2005a). Briefly, mice were anesthetized with sodium pentobarbital (20 mg/kg, intraperitoneal). Under an operating microscope, the liver hilum was dissected free from surrounding tissue. All structures in the portal triad (hepatic artery, portal vein, and bile duct) to the left and median liver lobes were interrupting with a traumatic clamp (Fine Science Tools, Foster City, CA). After 1 hr of ischemia, reperfusion was initiated by removal of the clamp. Throughout the ischemic interval, evidence of ischemia was verified by visualizing the pale blanching of the ischemic lobes. The clamp was then removed and gross evidence of reperfusion, based on immediate color change, was confirmed before closing the abdomen. Sham controls underwent the same procedure, but without vascular occlusion.
In the treatment groups, each mouse was injected via the tail vein with 100 μg of pNALP3shRNA, pshRNANC (dissolved in 1.8 ml of normal saline), or 1.8 ml of normal saline alone by hydrodynamic injection over 5 to 6 sec, 48 hr before the onset of warm ischemia (Wang et al., 2006; Zhu et al., 2006; Gao et al., 2010).
Detection of plasmid distribution by hydrodynamic injection via the tail vein
Mice were injected via the tail vein with 100 μg of pEGFP-N1 plasmid (Clontech, Palo Alto, CA) or pcDNA3.1 plasmid (Invitrogen) (dissolved in 1.8 ml of normal saline) by hydrodynamic injection over 5 to 6 sec. Livers were harvested 48 hr after injection and sectioned for fluorescence microscopy examination.
Assessment of liver damage
At 6 hr of reperfusion, sALT (serum alanine aminotransferase) and sAST (serum aspartate aminotransferase) levels, indicators of hepatocellular injury, were measured with a Beckman LX20 autoanalyzer (Beckman Coulter, Fullerton, CA). Ischemic lobes were fixed with 10% formalin. Samples were embedded in paraffin and cut into 4-μm-thick sections for hematoxylin–eosin (H&E) and terminal deoxyribonucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) staining.
Isolation of hepatic nonparenchymal cells
Liver nonparenchymal cells (NPCs) were harvested and isolated as previously described (Llacuna et al., 2009). In brief, after in situ 0.05% collagenase digestion, the cell suspension was centrifuged at 50 × g for 2 min to remove most of the parenchymal cells. The nonparenchymal cell supernatant was centrifuged (300 × g, 10 min, 4°C) to collect the cells, followed by centrifugation (900 × g, 20 min, 4°C) through a two-step Percoll gradient (25 and 50%), and the interface layer enriched in the Kupffer cells (KCs) and endothelial cells was collected for Western blotting and real-time PCR. Separation of pure KCs from endothelial cells was done by selective adherence of KCs to plastic flasks after 60 min of incubation. Cells with 90 to 95% viability as determined by trypan blue exclusion showed typical macrophage morphological features and stained positively for F4/80.
Enzyme-linked immunosorbent assay
IL-1β, IL-18, TNF-α, and IL-6 were measured with a sandwich enzyme-linked immunosorbent assay (ELISA) kit (eBioscience, San Diego, CA), according to the manufacturer's protocol. Briefly, samples were added to the appropriate wells of precoated ELISA plates and incubated at room temperature for 2 hr. Plates were rinsed and detection antibodies were added, followed by avidin–horseradish peroxidase (HRP). After washing, substrate solution was added. The plates were read at 450 nm in an ELISA reader.
Real-time PCRs and semiquantitative PCR
Total RNA was extracted from mouse liver samples with TRIzol reagent (Invitrogen) and reverse-transcribed into cDNA (Fermentas, Burlington, ON, Canada) according to the manufacturer's instructions. Equal quantities of cDNA from each animal were used for real-time PCR (RT-PCR). Analysis of expression levels of genes of interest was done with an ABI PRISM 7000 sequence detection system (Applied Biosystems/Invitrogen, Foster City, CA) and a SYBR green quantitative PCR (qPCR) kit (Invitrogen). cDNA was amplified by PCR with specific primers as follows: NALP3: 5′-TGTGAGAAGCAGGTTCTACTCT-3′ (sense), 5′-GGATGCTCCTTGACCAGTTGG-3′ (antisense); glyceraldehyde-3-phosphate dehydrogenase (GAPDH): 5′-TTCACCACCATGGAGAAGGC-3′ (sense), 5′-GGCATGGACTGTGGTCATGA-3′ (antisense). The relative expression levels for each target gene were normalized to GAPDH and are presented as fold increases versus liver tissue from sham mice. Quantification was performed according to the comparative C t method (ΔΔC T). Results are expressed relative to GAPDH values. mRNAs for NALP3, NALP1b, and NLRC4 (NLR [nucleotide-binding domain leucine-rich repeat] family CARD [caspase activation and recruitment domain]-containing protein-4) in murine Kupffer cells were detected by semiquantitative PCR with 5′-AGATGACGAGTGTCCGTTGC-3′ as the forward primer and 5′-GCGTTCCTGTCCTTGATAGAGT-3′ as the reverse primer for NALP3, 5′-TGGGATGGTTCTAGAAACGCC-3′ as the forward primer and 5′-AGGGTCCACTGATGTCACTCG-3′ as the reverse primer for NALP1b, and 5′-TGGGATGGTTCTAGAAACGCC-3′ as the forward primer and 5′-CGTTCAATGCAAAGAGGTCA-3′ as the reverse primer for NLRC4. PCR products were separated on 3% agarose gels and digital photographs were taken on a transilluminator.
Electrophoretic mobility shift assay
Nuclear extracts of NPCs were prepared according to the nuclear extraction kit manual (Panomics/Affymetrix, Santa Clara, CA). Protein concentrations were determined by bicinchoninic acid assay with trichloroacetic acid precipitation, using bovine serum albumin as a reference standard (Pierce Biotechnology, Rockford, CA). Nuclear factor (NF)-κB DNA-binding activity was determined by electrophoretic mobility shift assay, using a LightShift chemiluminescence electrophoretic mobility shift assay (EMSA) kit (Pierce Biotechnology). Double-stranded oligodeoxynucleotides containing an NF-κB-binding motif (sequence, 5′-AGTTGAGGGGACTTTCCCAGGC-3′) were labeled with biotin and used as a probe.
Western blot analysis
Protein was extracted from isolated NPCs, transfected CHO cells, and Kupffer cells with lysis buffer containing 50 mM Tris, 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% sodium deoxycholate, 1% Triton X-100, and protease inhibitors, pH 7.2. Proteins were subjected to electrophoresis on SDS–polyacrylamide (8–12%) gels. The separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (HYBOND, Escondido, CA). Membranes were blocked with 5% nonfat milk in Tris-buffered saline–Tween 20 (TBST) for 2 hr at room temperature with shaking, and then incubated at 4°C overnight with primary antibodies for caspase-1 (sc-514; Santa Cruz Biotechnology, Santa Cruz, CA), NALP3 (sc-66846; Santa Cruz Biotechnology), and HMGB1 (Zhou et al., 2009) (kindly provided by J. Tang, Institute of Biophysics, Chinese Academy of Science, Beijing, China). Blots were visualized with SuperSignal West Pico chemiluminescent substrate (Pierce Biotechnology). Relative quantities of proteins were determined with a densitometer.
Determination of reactive oxygen species
Reactive oxygen species was determined with 2′,7′-dichlorofluorescein diacetate (DCFDA; Molecular Probes/Invitrogen, Eugene, OR) in an LSR II flow cytometer (BD Biosciences, San Jose, CA) according to the manufacturer's instructions.
Immunohistochemistry
Liver slices (4 μm) were blocked with 0.3% H2O2 followed by goat serum, and then stained with primary antibody to NALP3 (Santa Cruz Biotechnology), F4/80 (CI:A3-1; Abcam, Cambridge, UK), or Gr-1 (RB6-8C5; eBioscience) at 4°C overnight. Isotype- and species-matched irrelevant monoclonal antibodies (mAbs) were used as controls. The next day, the slices were incubated with immunoperoxidase-conjugated goat IgG fraction to rabbit IgG Fc (Dako, Glostrup, Denmark) at room temperature for 60 min, followed by three washes in phosphate-buffered saline (PBS) containing 0.05% Tween 20. Slides were counterstained with Harris hematoxylin and mounted with Crystal Mount (Biomeda, Foster City, CA), after which they were air dried and photographed with a microscope.
Terminal deoxyribonucleotidyltransferase-mediated dUTP nick-end labeling staining
TUNEL staining (Roche, Mannheim, Germany) was applied to paraffin-embedded, 4-μm-thick liver sections to detect DNA fragmentation as a measure of the number of apoptotic cells, according to the manufacturer's instructions. Results were scored semiquantitatively by averaging the number of apoptotic cells per microscopic field at × 400 magnification (high-power field) in a blinded fashion. Six fields were evaluated per tissue sample.
Flow cytometry
Flow cytometry was performed with an LSR II flow cytometer (BD Biosciences). Fc receptors were blocked with anti-mouse CD16/32 antibody (clone 93; BioLegend, San Diego, CA). Cells were stained with fluorescein isothiocyanate (FITC)-labeled anti-mouse CD146 (ME-9F1; BioLegend) and phycoerythrin (PE)-labeled anti-mouse F4/80 (clone BM8; eBioscience). Intracellular staining was assessed with rabbit anti-mouse NALP3 antibody (Santa Cruz Biotechnology) and secondary Alexa 647-conjugated goat anti-rabbit IgG (Invitrogen). Control rabbit IgG (Millipore, Billerica, MA) was used as control. Appropriate isotype controls were used when necessary. Data were analyzed with CellQuest software (BD Biosciences) as instructed.
Statistical analysis
Data are presented as means ± SEM. The statistical significance of differences was determined by Student t test or one-way analysis of variance (ANOVA) with SPSS 12 statistical software (SPSS, Chicago, IL). Values of p less than 0.05 were considered significant.
Results
Construction of NALP3 shRNA plasmids
To construct an effective shRNA plasmid for NALP3, we first sought to determine an effective sequence for interfering NALP3. We designed and selected three NALP3 siRNA sequences: siRNA1 (S1), siRNA2 (S2), and siRNA3 (S3), as well as a scrambled siRNA (SS), followed by transfection into CHO cells, which constitutively express NALP3 protein. siRNA3 (S3) was shown to inhibit NALP3 significantly, whereas the scrambled siRNA (SS) had no measurable effect on NALP3 expression (Fig. 1A). We determined that the siRNA3 sequence was the most effective sequence specifically targeting NALP3 protein.

Specific knockdown of the NALP3 gene by pNALP3shRNA in vitro. (
Next, we successfully constructed the pNALP3shRNA and pshRNANC plasmids, using the siRNA3 interference sequence and scrambled siRNA sequence, respectively, and performed verification by restriction enzyme mapping and sequence analysis (data not shown).
pNALP3shRNA silences NALP3 expression in vitro
To test the efficiency of pNALP3shRNA for inhibition of NALP3 expression, Kupffer cells were transfected with pNALP3shRNA or pshRNANC. As shown in Fig. 1B, the expression of NALP3 in Kupffer cells was significantly suppressed by pNALP3shRNA transfection compared with the mock treatment or pshRNANC transfection (p < 0.05). In accordance with protein levels, pNALP3shRNA transfection reduced NALP3 mRNA levels compared with the pshRNANC and mock groups (Fig. 1C; p < 0.05). To further check the function of NALP3 silencing, we analyzed IL-1β release by Kupffer cells transfected with pNALP3shRNA or pshRNANC, as described in Materials and Methods. The release of IL-1β was significantly decreased in pNALP3shRNA-treated cells compared with mock- and pshRNANC-treated cells (Fig. 1D; p < 0.05). In addition, specific silencing was verified by measuring the expression of irrelevant target genes after pNALP3shRNA treatment (Fig. 1E). These data indicated that pNALP3shRNA transfection could silence NALP3 and specifically inhibit the function of NALP3 in vitro.
NALP3 is involved in liver I/R injury
To explore whether NALP3 was involved in liver I/R injury, Western blot analysis was performed on NPCs from sham mice and mice subjected to liver I/R. NALP3 protein expression was significantly upregulated after 6 hr of reperfusion and lasted until 12 hr (Fig. 2A).
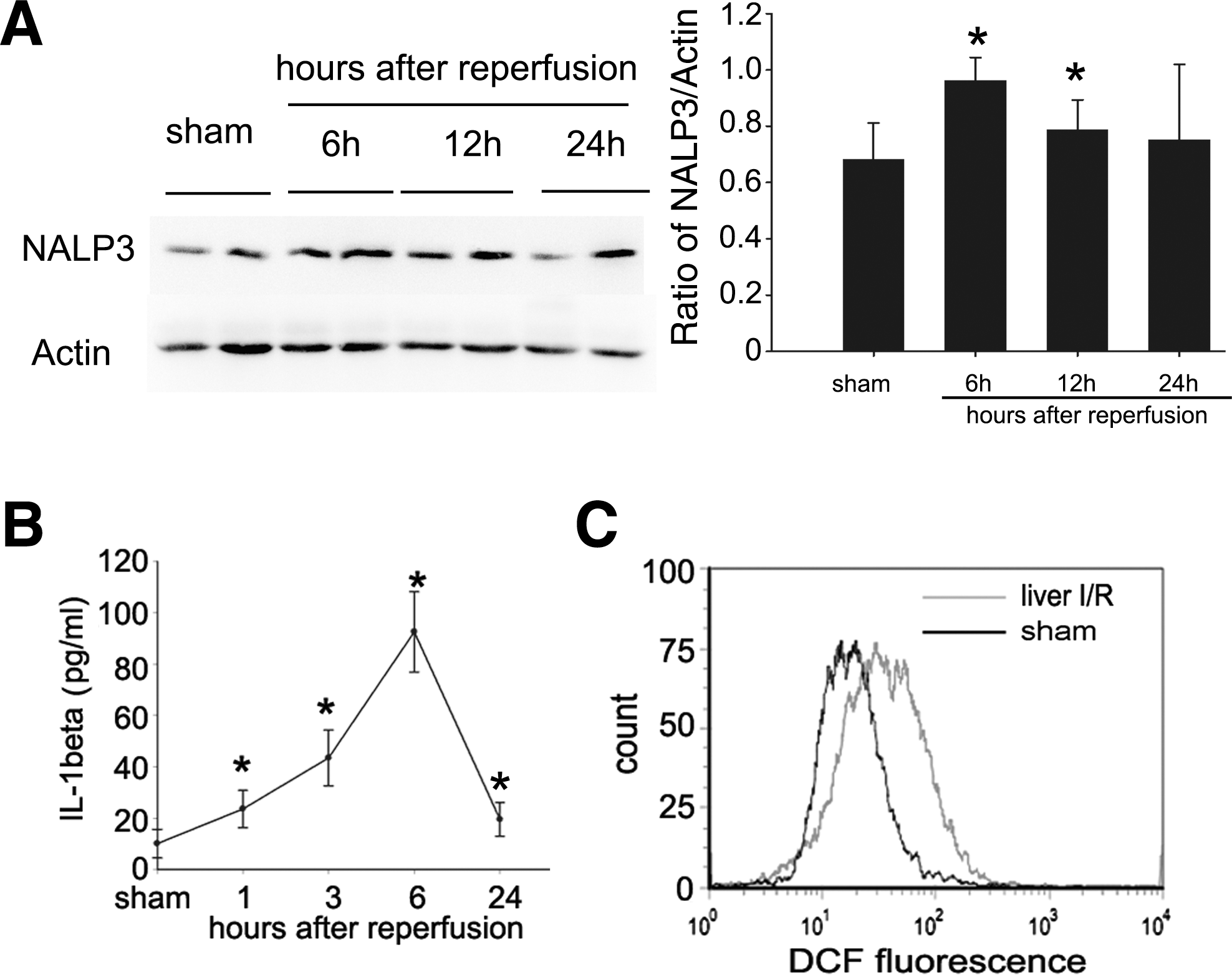
Levels of NALP3 and reactive oxygen species (ROS) in nonparenchymal cells (NPCs) and serum IL-1β are increased during liver ischemia–reperfusion (I/R). (
We also assessed over time the serum IL-1β level of sham mice and mice subjected to liver I/R. Serum IL-1β protein expression increased after 1 hr of reperfusion, continued to increase, and was maximal at 6 hr of reperfusion. However, the IL-1β protein level after 24 hr of reperfusion were similar to that detected at 1 hr of reperfusion, and remained significantly greater than in sham controls (Fig. 2B). As the level of IL-1β peaked 6 hr after reperfusion, subsequent experiments were performed at this time.
Because ROS are required for NALP3 inflammasome activation, which contributes to release IL-1β (Martinon, 2010), we examined the level of ROS in NPCs. As shown in Fig. 2C, ROS of NPCs in liver I/R mice increased markedly compared with sham mice, indicating that NALP3 was activated during liver I/R. Thus, NALP3 signaling is required for the development of liver I/R injury.
Distribution of pEGFP-N1 by hydrodynamic injection via the tail vein
To visualize the distribution of plasmids, mice were rapidly injected via the tail vein with plasmid carrying a gene encoding green fluorescent protein (pEGFP-N1) or with non-GFP plasmid (pcDNA3.1), as described in Materials and Methods. GFP was strongly expressed in liver pretreated with pEGFP-N1 (Supplementary Fig. S1; supplementary data are available online at
pNALP3shRNA inhibits NALP3 expression in vivo
To explore the possibility of using pNALP3shRNA to downregulate NALP3 in vivo, we first determined the regulatory pharmacokinetics of pNALP3shRNA in terms of downregulating the expression of NALP3 target gene by hydrodynamic injection via the tail vein. mRNA levels for NALP3 24, 48, and 96 hr after injection of pNALP3shRNA were analyzed. As shown in Fig. 3A, it was found that mRNA for NALP3 was decreased at 24 hr, hit a trough by 48 hr, and then rose by 96 hr after injection (p < 0.05). We therefore chose 48 hr after injection to perform ischemia and reperfusion in mice. We also determined the optimal dose of pNALP3shRNA by evaluating ALT levels in mice pretreated with various concentrations of pNALP3shRNA. As show in Fig. 3B, mice pretreated with 100 μg of pNALP3shRNA were protected to the greatest extent from live I/R injury.
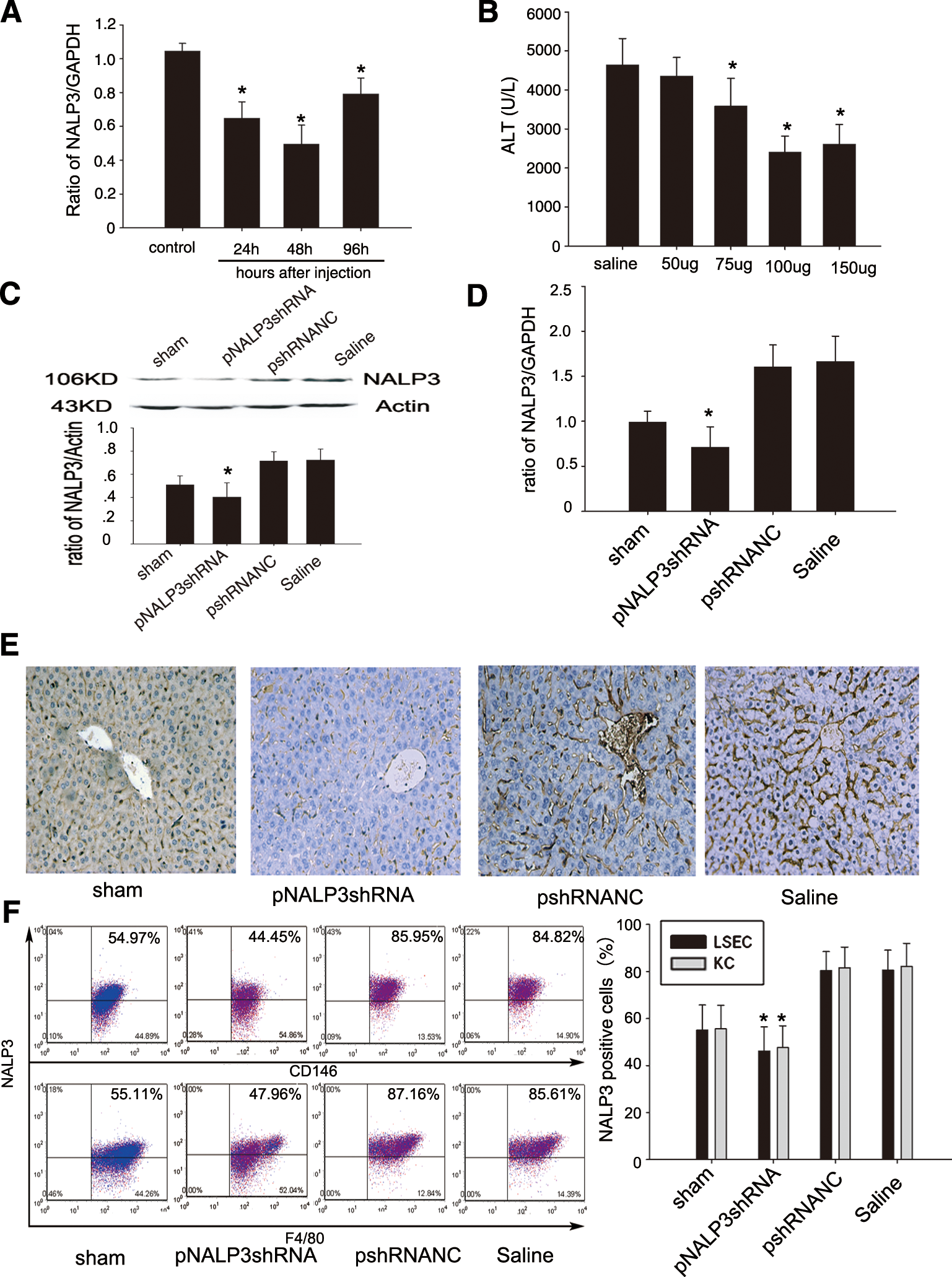
pNALP3shRNA inhibits target gene expression in vivo. (
We next checked the silencing of NALP3 after 6 hr of reperfusion in vivo. The results showed that hydrodynamic injection of 100 μg of pNALP3shRNA significantly inhibited the expression of NALP3 not only at the protein level, as determined by Western blotting (Fig. 3C; p < 0.05) and immunohistochemical staining (Fig. 3E), but also at the mRNA level, as assessed by RT-PCR (Fig. 3D; p < 0.05), whereas there was no significant difference in mice pretreated with pshRNANC and saline.
NALP3 fluorescence in cells was measured by fluorescence-activated cell sorting (FACS), which showed significantly decreased fluorescence in both CD146+ liver sinusoidal endothelial cells (LSECs) and F4/80+ Kupffer cells in the pNALP3shRNA group compared with the pshRNANC and saline groups (Fig. 3E; p < 0.05).
Pretreatment with pNALP3shRNA ameliorates liver I/R injury
To evaluate the effects of hydrodynamic injection on liver, the serum ALT and AST levels in normal mice and mice treated by hydrodynamic injection of saline or pNALP3shRNA at 24 and 48 hr after injection were analyzed. We observed that sALT and sAST increased 24 hr after injection of both saline and pNALP3shRNA (Supplementary Fig. S2A and B; p < 0.05), but recovered to the baseline of normal mice by 48 hr after injection. There was no significant difference between the saline group and the pNALP3shRNA group at either 24 or 48 hr after injection (Supplementary Fig. S3A and B). The mRNA of NALP3 was also determined by RT-PCR in normal mice and mice treated by hydrodynamic injection of saline at corresponding time points (Supplementary Fig. S2C). Our data suggested that hepatocellular function recovered to near normal levels by 48 hr after hydrodynamic injection.
To investigate the functional significance of NALP3, mice pretreated with pNALP3shRNA, pshRNANC, or saline were subjected to liver I/R. Serum and liver samples were harvested 6, 12, and 24 hr after reperfusion. Serum ALT and AST levels were analyzed as a measure of hepatocellular injury. As shown in Fig. 4A, 1 hr of warm hepatic ischemia followed by 6, 12, and 24 hr of reperfusion significantly increased sALT and sAST levels in saline- and pshRNANC-pretreated mice subjected to I/R. Pretreatment with pNALP3shRNA resulted in significant protection from hepatic injury (p < 0.01). Liver histology confirmed the sALT and sAST estimation of liver damage at 6 hr of reperfusion (Fig. 4B). Large areas of necrosis were present in liver tissue from saline- and pshRNANC-pretreated mice whereas minimal damage was noted in samples from pNALP3shRNA-pretreated mice. In addition, in situ TUNEL staining of livers subjected to 1 hr of ischemia and 6 hr of reperfusion showed a significantly decreased frequency of apoptotic cells in the pNALP3shRNA group compared with the pshRNANC and saline groups (Fig. 4C and D; p < 0.01). These results suggested that pretreatment with pNALP3shRNA demonstrated net protection from liver I/R injury.
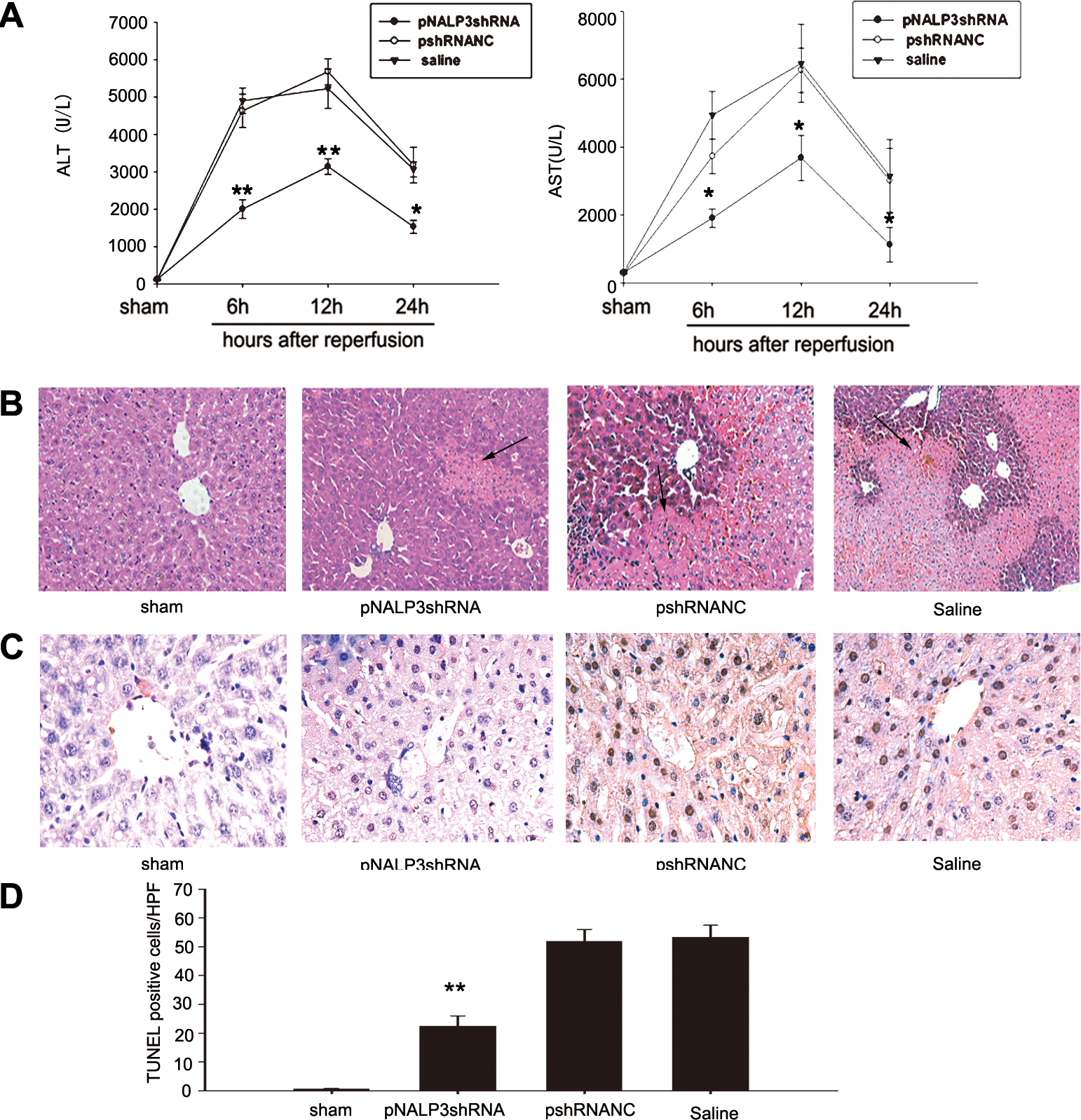
Pretreatment with pNALP3shRNA plasmid protects against liver I/R injury. (
Inhibition of NALP3 reduces IL-1β and IL-18 maturation in liver I/R
To determine the importance of NALP3 signaling in liver I/R injury, we measured serum IL-1β in sham mice and mice pretreated with pNALP3shRNA, pshRNANC, or saline followed by 6 hr of reperfusion. Mice pretreated with pNALP3shRNA exhibited decreased circulating IL-1β and IL-18 levels compared with saline- or pshRNANC-pretreated mice (Fig. 5A; p < 0.05), whereas there was no difference in mice pretreated with pshRNANC compared with saline-pretreated mice (Fig. 5A).
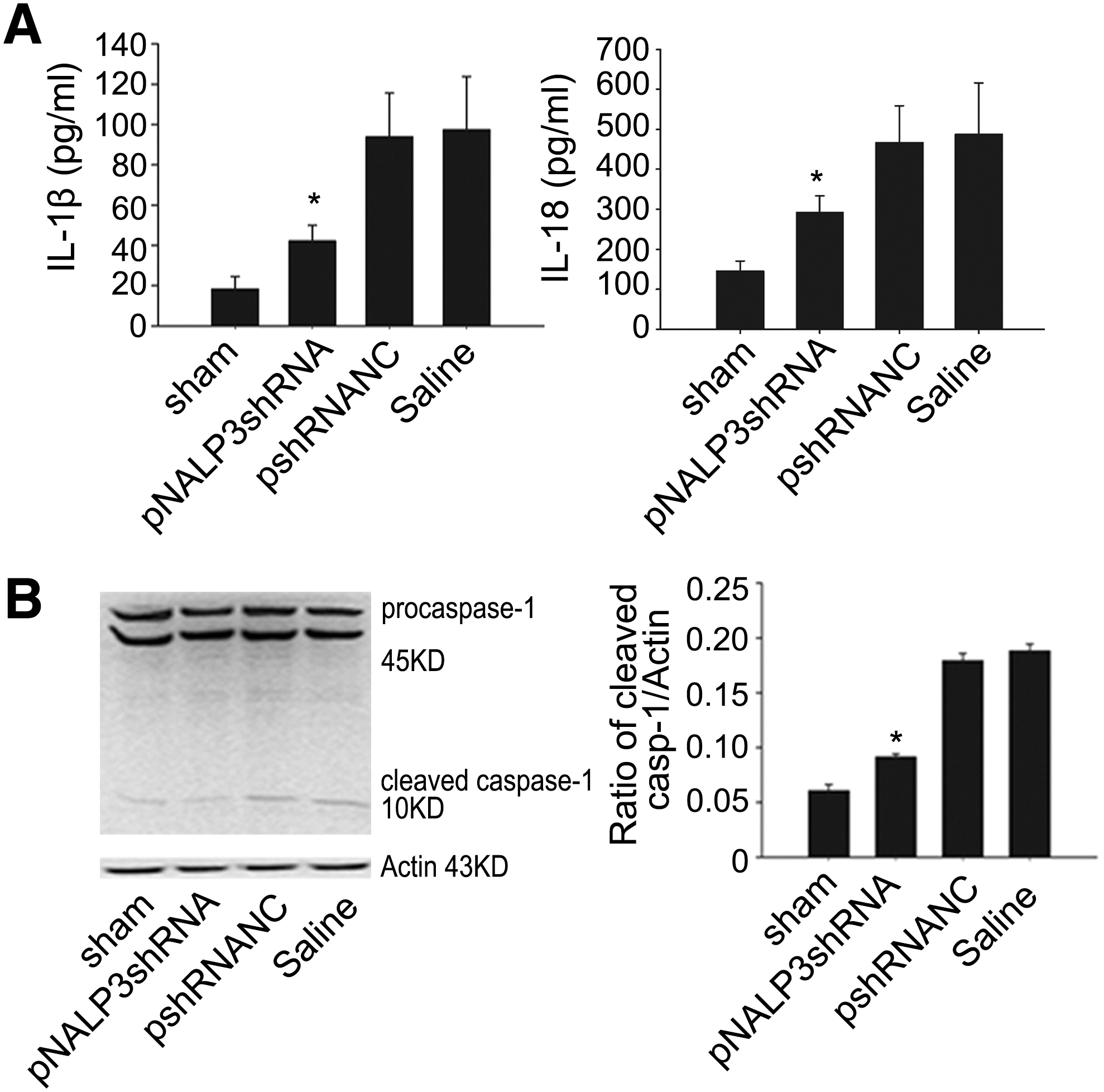
Interference with NALP3 suppresses serum IL-1β and IL-18 upregulation in liver after I/R. (
Both IL-1β and IL-18 are produced as inactive cytoplasmic precursors (pro-IL-1β, pro-IL-18), which are cleaved by caspase-1 (IL-1β-converting enzyme [ICE]) to their mature active forms (Dinarello, 1998). To determine the effect of pNALP3shRNA plasmid on the activation of caspase-1, we examined cleaved caspase-1 by immunoblotting with antibodies against the p10 subunit of caspase-1. Mice pretreated with pNALP3shRNA had lower expression of cleaved caspase-1 after 6 hr of reperfusion compared with mice pretreated with pshRNANC or saline (Fig. 5B; p < 0.05). However, there was no significant difference in cleaved caspase-1 expression level between saline- and pshRNANC-pretreated animals (Fig. 5B).
Gene silencing of NALP3 downregulates HMGB1, proinflammatory cytokines TNF-α and IL-6
On the basis of the previously described observations, we next investigated whether silencing of NALP3 could influence Toll-like receptor (TLR) ligands. HMGB1 is an important TLR ligand (TLR4, TLR2) and can be either released passively from injured or necrotic cells or secreted by activated immune cells such as monocytes, macrophages, and dendritic cells (Bianchi and Manfredi, 2007). Western blot analysis of HMGB1 expression was performed on Kupffer cells from mice that had been subjected to liver I/R and from sham mice. HMGB1 protein expression was upregulated in the liver of saline- or pshRNANC-pretreated mice. In contrast, mice pretreated with pNALP3shRNA exhibited downregulated HMGB1 expression compared with pshRNANC- or saline-pretreated animals (Fig. 6A; p < 0.05). There was no significant change in HMGB1 expression in pshRNANC-pretreated mice compared with saline-pretreated mice (Fig. 6A).
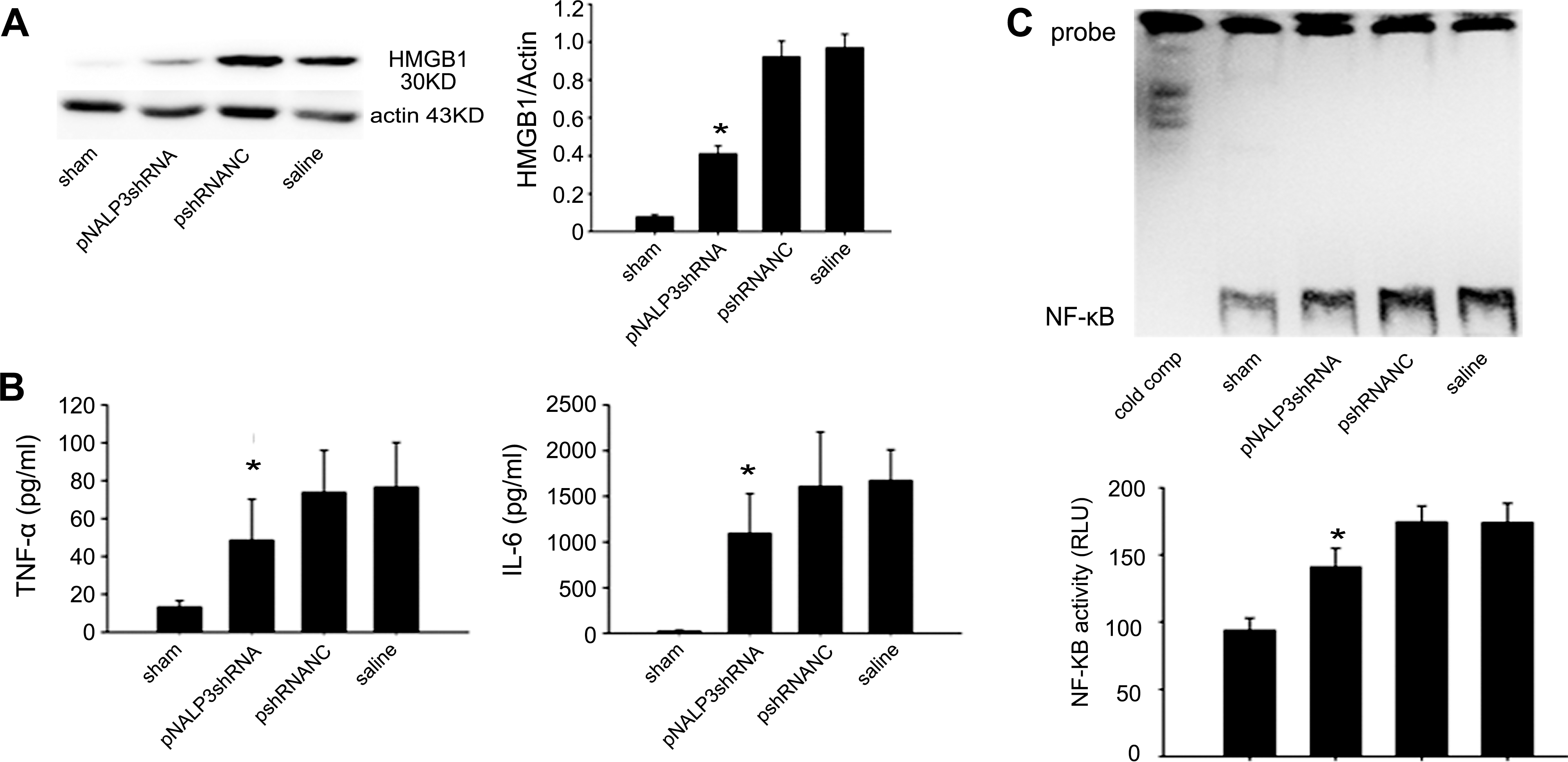
pNALP3shRNA pretreatment inhibits HMGB1, TNF-α, and IL-6 upregulation after liver I/R. (
Because proinflammatory cytokines, such as TNF-α and IL-6, have been shown to play key roles in the pathophysiology of liver I/R injury (Camargo et al., 1997; Lentsch et al., 2000), we measured serum levels of TNF-α and IL-6 after I/R. Compared with the sham group, pshRNANC- and saline-pretreated mice undergoing I/R presented increased levels of TNF-α and IL-6. However, mice pretreated with pNALP3shRNA exhibited lower levels of serum TNF-α and IL-6 compared with pshRNANC- and saline-pretreated mice (Fig. 6B; p < 0.05). To explain which mechanism might be involved in the decrease in these proinflammatory cytokines, we next examined NF-κB, a pivotal transcription factor responsible for proinflammatory cytokine expression, by analysis of its DNA-binding activity in liver. As shown in Fig. 6C, pNALP3shRNA pretreatment significantly suppressed the activation of NF-κB. Taken together, our results suggest that pretreatment with pNALP3shRNA may inhibit the production of serum TNF-α and IL-6 by suppressing NF-κB activity.
Knockdown of NALP3 decreases inflammatory cell infiltration in liver I/R
To explore other mechanisms that may be involved in protection against liver injury of mice pretreated with pNALP3shRNA, we investigated macrophage and neutrophil infiltration in liver I/R by immunohistochemical staining. As show in Fig. 7, infiltrated macrophages and neutrophils were significantly decreased in pNALP3shRNA-pretreated mice than in pshRNANC- and saline-pretreated mice (p < 0.01).
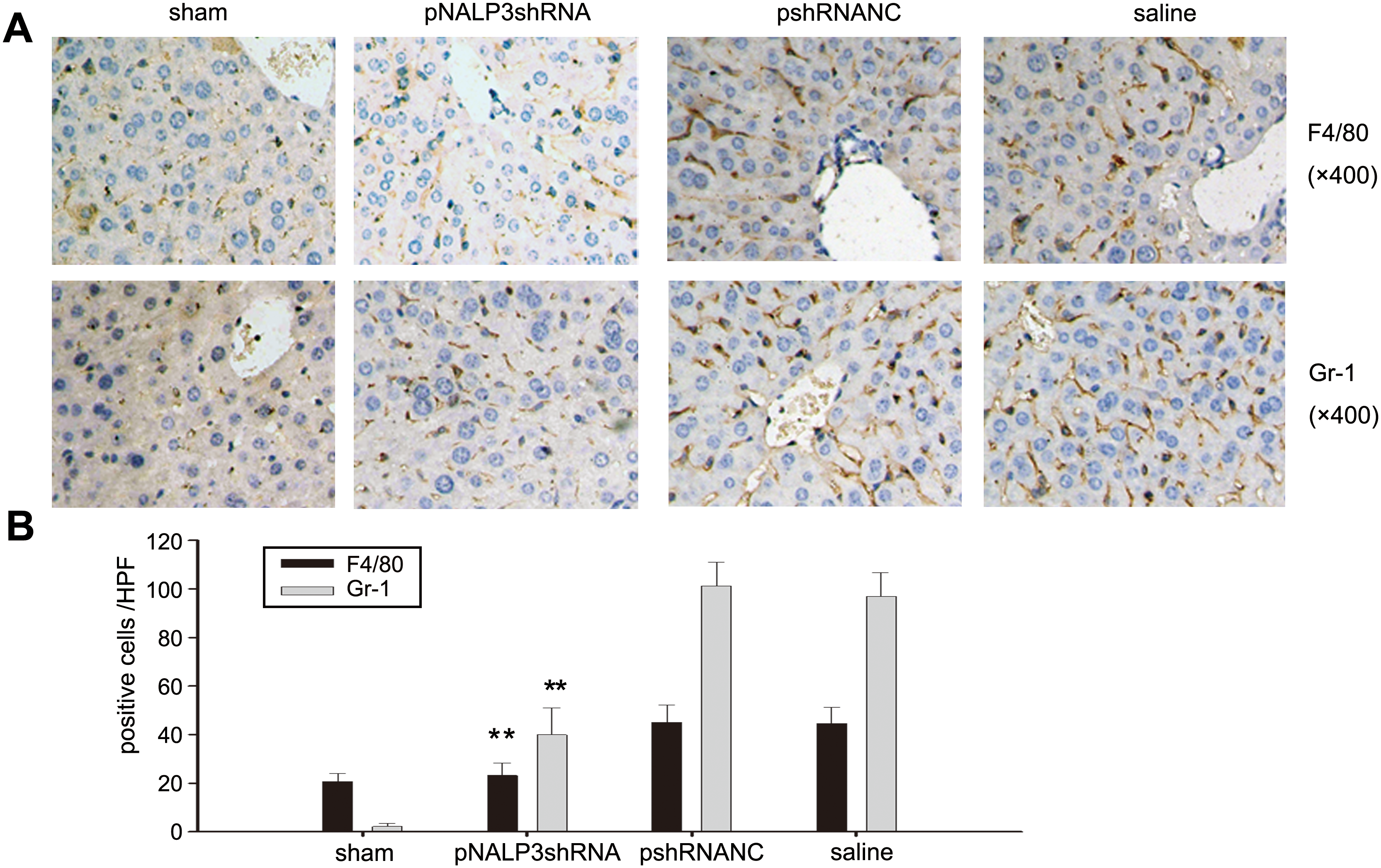
Silencing NALP3 diminishes inflammatory cell infiltration after liver I/R. (
Discussion
Warm liver I/R injury is characterized as a cascade of prominent inflammatory events. Prompt inflammatory response in liver was evident in our model of warm liver I/R injury by the elevation of proinflammatory mediators such as ROS, IL-1β, IL-18, TNF-α, IL-6, and HMGB1, which could activate the NALP3 inflammasome. Consistent with other studies (Sutterwala et al., 2006; Bauernfeind et al., 2009) suggesting that NALP3 expression could be increased under some conditions, we also found that NALP3 expression in NPCs increased during liver I/R, suggesting that NALP3 signaling may play an important role in liver I/R. Taken together, we propose that inhibition of NALP3 may offer an important new therapeutic approach in liver I/R injury.
The central effector molecule of the NALP3 inflammasome is the cysteine protease caspase-1 that, on activation, cleaves cytosolic pro-IL-1β, pro-IL-18, and pro-IL-33 to their active forms IL-1β, IL-18, and IL-33, which are then secreted (Pedra et al., 2009). In this study, we found that the activities of caspase-1 as well as NALP3 were increased in liver I/R. Consistently, gene silencing of NALP3 dramatically suppressed the activation of caspase-1 in liver I/R, resulting in impairment of mature IL-1β and IL-18 production. Our finding is well supported by previous studies of transfer of the IL-1 receptor antagonist gene into rat liver, or administration of IL-18 antibody abrogating liver I/R injury (Shito et al., 1997; Harada et al., 2002; Takeuchi et al., 2004). In congruence with reports by Kato and colleagues (2002), who used IL-1 receptor (IL-1R) knockout mice to show that IL-1β could augment neutrophil accumulation, we also found that influx of macrophages and neutrophils was greatly reduced by inhibition of NALP3. Notably, because the NALP3 inflammasome signaling pathway is essential not only for IL-1β and IL-18 production but also for dampening the deleterious innate immunity-dominated response cascade activated by reactive oxygen species (ROS) generated subsequent to reoxygenation in liver I/R, the current work represents an improvement over the other existing approaches against liver I/R injury such as IL-1R and IL-1 blockade (Supplementary Fig. S4). Thus our results have documented the NALP3 inflammasome as a novel target for putative potent therapeutic intervention against liver I/R injury.
The NALP3 inflammasome is implicated in the production of mature IL-1β and IL-18 in response to a variety of signals, including the pathogen-associated molecular pattern (PAMP) and endogenous stress-associated danger signals (DAMP) such as ATP and HMGB1 (Mariathasan et al., 2006; Bamboat et al., 2010b). It has been shown that HMGB1 is an early mediator of injury and inflammation in liver I/R (Tsung et al., 2005b). Inhibition of HMGB1 activity with neutralizing antibody or cisplatin significantly decreased liver damage after I/R, whereas administration of recombinant HMGB1 worsened I/R injury (Tsung et al., 2005b; Cardinal et al., 2009). Our results show that the expression of HMGB1 is decreased by gene silencing of NALP3. This may be due to the reduction of IL-1β and IL-18, stimulators for hepatocyte necrosis, and release of HMGB1. In line with our finding, it has been shown that IL-1 could stimulates beta cell necrosis and release of HMGB1 (Steer et al., 2006), and NALP3 facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways (Willingham et al., 2009). More recently, Lamkanfi and colleagues demonstrated that HMGB1 release is dependent on inflammasome in endotoxemia (Lamkanfi et al., 2010). Further study is needed to determine the exact mechanisms by which suppression of NALP3 expression leads to reduction of the release of HMGB1.
Proinflammatory cytokines, including TNF-α and IL-6, have been shown to play a crucial role in the pathogenesis of liver I/R injury (Lentsch et al., 2000). NF-κB is an important regulator of gene expression for a large number of proinflammatory cytokines, CXC chemokines, and vascular cell adhesion molecules, and is activated during I/R injury of liver (Zwacka et al., 1998). Activation of NF-κB in whole liver during I/R injury is an indicator of the extent of injury (Kuboki et al., 2007b). However, the precise function of NF-κB activation in liver I/R injury is controversial, and there is evidence to support both protective (Kuboki et al., 2007a) and injurious (Luedde et al., 2005) actions. Using EMSA, we found that interference with NALP3 can decrease NF-κB DNA-binding activity and serum proinflammatory cytokine TNF-α and IL-6 levels, consequently ameliorating hepatic injury.
It is well known that RNA interference by siRNA mediates efficient gene silencing. However, application of siRNA for in vivo gene therapy is limited because of instability in vivo (Morrissey et al., 2005) and the requirement for large amounts of siRNA, which is prohibitively expensive (Lee and Kumar, 2009). siRNA-expressing vector (shRNA) is more versatile and stable in vivo than siRNA (Suzuki et al., 2009), and therefore we chose hydrodynamic injection of shRNA plasmids into mice in this study to test the role of NALP3 in liver I/R.
Gene transfer into the liver by hydrodynamic injection of plasmid via the tail vein can be achieved easily (Liu et al., 1999; Maruyama et al., 2002; Zhu et al., 2006). Our study also showed that pNALP3shRNA is taken up mainly in the liver, which is in line with previous studies suggesting that the liver is the major site for transgene expression (Liu et al., 1999; Maruyama et al., 2002; Zhu et al., 2006; Jacobs et al., 2010). Moreover, our data also demonstrate that NALP3 expression was effectively inhibited by pNALP3shRNA plasmid transfection both in vitro and in vivo, which protects against warm hepatic injury through downregulation of the production of IL-1β and IL-18. Although our study did not directly identify the percentage of each cell type (hepatocytes, Kupffer cells, LSECs) taking up pNALP3shRNA in liver after hydrodynamic injection, we measured the inhibitory effects of pNALP3shRNA on NALP3 expression in Kupffer cells and LSECs. Other organs, such as the lung, heart, kidney or spleen, might also take up pNALP3shRNA slightly, and therefore further studies should be performed to improve the specific distribution of pNALP3shRNA. In addition, more valid methods should be explored because hydrodynamic delivery is unlikely to be applicable to human therapy. An efficient oral delivery vehicle (Aouadi et al., 2009), and viral vector carrier, are under consideration.
In summary, our results demonstrate that NALP3 signaling is involved in liver I/R injury, that gene silencing of NALP3 via shRNA plasmid significantly attenuates liver I/R injury, and that this protective effect is associated with reduced production of proinflammatory cytokines IL-1β, IL-18, TNF-α, and IL-6; reduced production of HMGB1; and reduced inflammatory cell infiltration. This may provide a new strategy for treatment of liver I/R in the clinic.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grants 30772039 and 81072440), a Central Universities Research grant (2010MS033), an NSFC-Guangdong Union grant (U0832003), and the Ministry of Science and Technology of China (2007CB512402). The authors thank Professor Jie Tang for providing the HMGB1 monoclonal antibody. The expert technical assistance of Ping Xiong and Yong Xu is gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.