
Abstract
Neointimal hyperplasia causing recurrent stenosis is a limitation of the clinical utility of percutaneous transluminal coronary interventions (PCI). Nitric oxide (NO) inhibits smooth muscle cell proliferation, platelet activation, and inflammatory responses, all of which have been implicated in the pathogenesis of restenosis. In animals, neointimal proliferation after balloon injury has been shown to be effectively reduced by gene transfer of the inducible NO synthase (iNOS). The primary objective of this first multicenter, prospective, single-blind, dose escalation study was to obtain safety and tolerability information of the iNOS lipoplex (CAR-MP583) gene therapy for reducing restenosis following PCI. Local coronary intramural CAR-MP583 delivery was achieved using the Infiltrator balloon catheter. A total of 30 patients were treated in the study (six patients, 0.5 μg; six patients, 2.0 μg; six patients, 5.0 μg; and 12 patients, 10 μg). There were no complications related to local application of CAR-MP583. In one patient, PCI procedure-related transient vessel occlusion occurred with consecutive troponin elevation. There were no signs of inflammatory responses or hepatic or renal toxicity. No dose relationship was seen with regard to adverse events across the dose groups. Thus, coronary intramural lipoplex-enhanced iNOS gene therapy during PCI is feasible and appears to be safe. These initial clinical results are encouraging to support further clinical research, in particular in conjunction with new local drug delivery technologies.
Introduction
Mechanical injury to the endothelium plays a pivotal role in the pathogenesis of vasculoproliferative disorders, because the protective physiologic production of nitric oxide (NO) is reduced. NO exerts an important regulatory function in maintaining vascular homeostasis (Vane et al., 1990; von der Leyen and Dzau, 2001), and inhibition of neointimal lesion formation in various animal models (von der Leyen et al., 1995; Cooney et al., 2007) has been demonstrated. Enhancing NO synthase (NOS) activity by means of delivering NOS gene (called “iNOS lipoplex gene therapy”; CAR-MP583) in diseased vessels prior to final treatment (balloon angioplasty and/or stenting) had beneficial effects on clinical and angiographic restenosis rates and endothelialization (Muhs et al., 2003). Subsequently, a clinical inducible NOS (iNOS) gene therapy development program was initiated by the former biotech company Cardion AG (Düsseldorf, Germany) and included a first-in-man phase IIa clinical study program, as described in this article. The gene transfer was achieved by using an intramural injection catheter in conjunction with cationic liposomes. After the study was successfully completed, however, the gene therapy program was discontinued because of the closedown of the company.
Materials and Methods
For this study, a nonviral, liposome-based gene transduction method was developed. The lipoplex CAR-MP583 contains the therapeutic iNOS expression vector (pAH9) packaged with a cationic lipid (DC-30). The plasmid pAH9 is derived from pcDNA3 (Invitrogen, Carlsbad, CA) and carries the human iNOS gene under the control of a constitutive promoter [cytomegalovirus (CMV) immediate-early promoter/enhancer]. DC-30 is a mixture of the monocationic lipid DC-Chol [3N-(N′,N′-dimethylaminoethane)carbamoylcholesterol] and the neutral colipid 1,2-dioleoylphosphatidylethanolamine. All lipids were produced at Avanti Polar Lipids, Inc. (Alabaster, AL). Clinical grade lipoplexes were produced at Boehringer Ingelheim Austria (Vienna, Austria). Preclinical evaluation of CAR-MP583 was performed in femoral and coronary artery stent models (Wiktor stent) in Göttingen minipigs (20–25 kg) as described elsewhere (Muhs et al., 2003). CAR-MP583 did not show notable toxicity related to the iNOS lipoplex formulation (von der Leyen and Chew, 2005).
Objectives of the REGENT I clinical study
The primary objective was to obtain safety and tolerability information of the iNOS lipoplex (CAR-MP583) gene therapy. The intramural drug delivery device Infiltrator (Interventional Therapeutics, San Diego, CA) was used to deliver the lipoplex at the lesion site. The primary combined clinical endpoint of major adverse cardiac events (MACE) by 30 days after treatment with CAR-MP583 and stent placement or balloon angioplasty was investigated. MACE events were defined as death, Q-wave or non–Q-wave myocardial infarction (MI), emergent coronary artery bypass graft surgery, and repeat target lesion revascularization.
Secondary endpoints were defined as follows: (a) MACE by 6 months after treatment with CAR-MP583 and stent placement or balloon angioplasty; (b) target vessel failure, defined as the combined clinical endpoint of cardiac death, Q-wave MI, or clinically driven repeat revascularization of the target vessel, by 6 months after treatment with CAR-MP583 and stent placement or balloon angioplasty; (c) vascular and bleeding complications, defined as the occurrence of any of the following resulting from the index procedure: hematoma at access site >5 cm, false aneurysm, arteriovenous fistula, retroperitoneal bleed, peripheral ischemia/nerve injury, vascular surgical repair; (d) transfusions of blood products due to blood loss resulting from the index procedure; and (e) drug delivery success, defined as correct placement and inflation of the Infiltrator balloon at the target lesion and successful drug infusion.
Design of the REGENT I study
This clinical trial was designed by Harvard Clinical Research Institute (HCRI) as a prospective, single-blind, multicenter, dose escalation study with an intention-to-treat sample. Patients were enrolled to receive iNOS lipoplex (CAR-MP583) gene therapy with an escalating dose treatment of 0.5, 2.0, 5.0, or 10.0 μg. After treatment of six patients in the first group and no unexpected toxicities within 7 days of the dose treatment, the dose was escalated. CAR-MP583 was administered into the target lesion prior to final coronary intervention. Clinical follow-up was performed at 7 days, 30 days, and 6 months. According to standard procedures at a given site, a control angiography was performed at 6 months.
Population
Patients had de novo or restenotic lesions in native coronary artery segments that were amenable to percutaneous treatment with balloon angioplasty and/or stent and that could be accessed with the Infiltrator. The sample size was between six and 48 patients (minimum of six and maximum of 12 patients per dose level).
Treatments
The investigational product, iNOS lipoplex (CAR-MP583), was applied with the Infiltrator. Patients received up to three applications (depending on the length of the lesion) of 0.5, 2.0, 5.0, or 10.0 μg of CAR-MP583.Each patient received only one dose level. The study started with the lowest dose level and advanced to the next higher dose level in accordance with a predefined dose escalation scheme based on the occurrence of unexpected toxicities (i.e., toxicity of grade 2 or higher on the World Health Organization Modified Toxicity Scale), which were adjudicated by a Clinical Events Committee (CEC; see below).
Data collection
The following data were collected: (a) efficacy parameters: lesion success, procedural success, and drug delivery success; (b) angiographic parameters (post hoc analyses): reference vessel diameter, minimal luminal diameter (MLD), degree of stenosis, late MLD loss, and presence of binary restenosis; (c) safety data: adverse events (AEs), such as MACE, ischemic events, hemorrhagic/vascular events, hematological dyscrasia, and other adverse events; (d) laboratory data: hematology, clinical chemistry, urinalysis, total creatine kinase, and creatine kinase-MB isoenzyme; and (e) clinical data: blood pressure, pulse, ECG, physical examination, anginal status, repeat procedures, and rehospitalizations.
Safety evaluation and regulatory compliance
The clinical study protocol and informed consent document were reviewed and approved by the respective local independent ethics committees and by the Committee on Somatic Gene Therapy of the German Federal Association of Physicians (“Kommission für somatische Gentherapie, Bundesärztekammer”). Each patient gave written informed consent prior to the conduct of any study-related procedures. The CEC was responsible for ongoing adjudication of (severe) AEs. CEC-adjudicated events were reviewed by the study's independent Data and Safety Monitoring Board, which had to approve each dose escalation step within the study's dose escalation scheme. Safety reporting obligations were fulfilled by Drug Safety Alliance (Durham, NC) and Regulatory Affairs North America LLC (Durham, NC), in full compliance with the applicable German regulations and the U.S. Code of Federal Regulations. The study was monitored on behalf of the sponsor by Parexel (Berlin, Germany). Data management and statistical analysis of the study were performed by HCRI (Boston, MA). Routine laboratory safety assessments were performed according to standard laboratory procedures at each study center's local accredited laboratory. All ECGs were centrally evaluated by HCRI's ECG core laboratory. All angiograms were quantitatively evaluated by the Angiographic Core Laboratory at Department of Cardiology (Frankfurt, Germany). The study took place between 20 March 2001 and 5 September 2002.
Periprocedural medication
Patients were treated with intravenous heparin to achieve an activated clotting time of approximately 300 sec and with combined antiplatelet therapy, consisting of a chronic aspirin therapy (100 mg/day) and clopidogrel (75 mg/day), which started immediately after the procedure with an initial single loading dose of 300 mg and was maintained for at least 4 weeks after the procedure.
Clinical and angiographic follow-up
Patients were screened before the planned procedure and were seen at days 1, 7, 14, and 28 for an interview, check of hematological parameters, and ECGs. After 6 (4–7) months, these evaluations and the stress test used before the index procedure were repeated, followed by control angiography. If repeat angiography was performed before 4 months for recurrence of symptoms, it served as control angiography only if restenosis was present; otherwise it was repeated after 6–7 months. After 12 ± 1 months, a final interview and ECG were obtained.
Angiographic analysis
Three angiograms in identical projections were obtained before and after injection of nitroglycerin immediately before and after the application of CAR-MP583 and at follow-up. All angiograms were analyzed at the Frankfurt University cardiology core laboratory using a validated computer-assisted quantitative angiographic system (QAngio XA 7.1 system; Medis, Leiden, The Netherlands). Angiographic measurements were performed on the entire artery segment covered by the therapy. Late loss (in millimeters) was defined as the change in lesion MLD from the final to the follow-up angiogram. Lesion success was defined as the attainment of <50% residual stenosis (by quantitative coronary angiography) after final treatment. Angiographic binary restenosis at the 6-month follow-up was defined as ≥50% diameter narrowing.
Statistical procedures
The primary analysis variable was the occurrence of MACE by 30 days (see the primary objective above). The analyses were performed on all patients who were enrolled in the trial. All analyses were based on available data, and no imputations were made. The analyses are presented using descriptive statistics only.
Results
Study subjects and conduct
The patient characteristics are summarized in Table 1. Thirty-two patients were enrolled in total. Thirty patients were treated with CAR-MP583 (six in each of the 0.5-, 2.0-, and 5.0-μg groups and 12 in the 10.0-μg group), and two patients could not be treated due to technical problems. All 32 patients completed the 6-month follow-up. Nearly all patients of each dose group had a history of angina; about half of the patients in each dose group had previously had an MI. With the exception of the 2.0-μg dose group, about two thirds of the patients in each group had undergone percutaneous revascularization prior to enrollment in the study. The target lesion was located in the left anterior descending artery in half or more patients of each dose group. Overall, 18 patients had de novo target lesions, two patients had non–in-stent restenotic lesions, and 10 patients had in-stent restenotic lesions. Most of the patients with restenotic lesions were treated in the 5.0- and 10.0-μg dose groups. All 20 patients with de novo lesions and non–in-stent restenotic lesions received one or more stents after dosing with CAR-MP583.
ACE-I, angiotensin-converting enzyme inhibitor; ASA, acetylsalicylic acid; ATRB, angiotensin II receptor blockers; CABG, coronary artery bypass graft; MI, myocardial infarction; PCI, percutaneous coronary intervention.
Analyses of procedural outcomes
Drug delivery by the Infiltrator catheter was assessed as successful in all 30 treated patients (Fig. 1). Lesion success was achieved in all 30 treated patients. The procedure was rated as successful in 29 of the 30 treated patients. For one patient in the 0.5-μg group, the procedure was rated as unsuccessful.
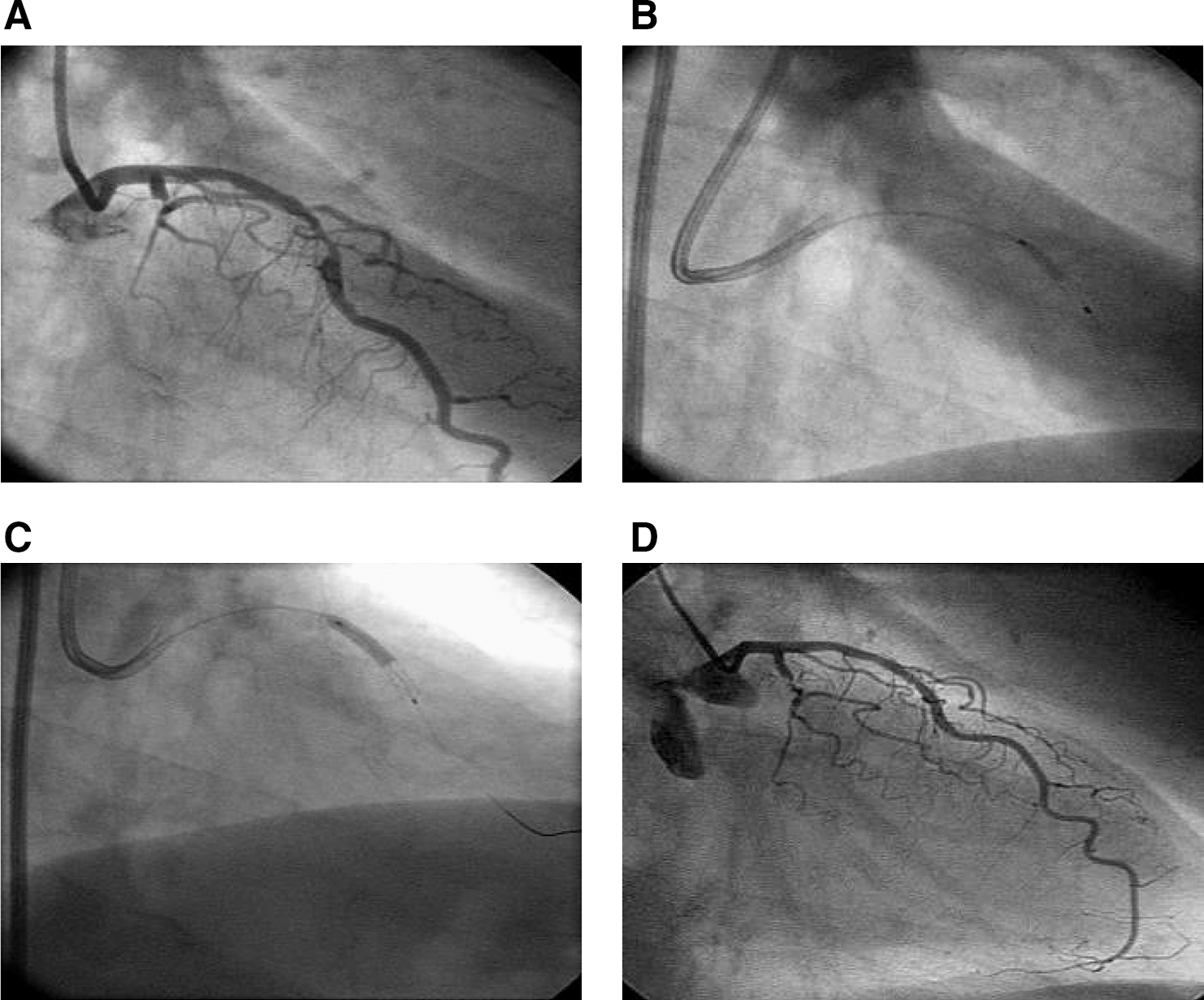
Local intramural gene transfer during percutaneous coronary intervention (PCI). (
Analyses of safety
CAR-MP583 was well tolerated at all doses investigated. No notable differences were observed between the dose groups with regard to the primary analysis variable (occurrence of MACE up to 30 days) or any of the predefined secondary analysis variables. Only one patient (0.5-μg group) had an event classified as MACE in the first 30 days of the study (non–Q-wave MI and repeat target lesion revascularization on day 1; also classified as target vessel failure).
In the period between 30 days and 6 months, the above patient and four other patients had events classified as MACE. All events were repeat target lesion revascularizations that occurred ≥120 days after administration of study medication and were also classified as target vessel failure. Importantly, four of these five patients had presented with in-stent restenotic lesions at baseline, and one patient with a de novo lesion.
No dose relationship was seen with regard to AEs across the dose groups (both for the total numbers of AEs and the individual AEs themselves). The AE profile was characteristic of the population. Cardiac disorders, vascular disorders, and associated procedures accounted for the majority of the AEs.
No relevant changes were observed in laboratory safety variables or pulse. Blood pressure values were consistently decreased after the procedure. This was not unexpected in a population with a high proportion of patients with a history of hypertension.
Analyses of angiographic parameters
All data reported were based on the central evaluation of the study angiograms (Table 2). All patients had pre- and postprocedure angiograms performed. A follow-up angiogram at the 6-month follow-up was available in 22 (73%) of the treated patients. Although there were only small differences between the dose groups with regard to minimal reference vessel diameter (RVD) at any time point, preprocedure maximal RVD reached 3.6–3.8 mm in the 0.5-, 2.0-, and 10.0-μg groups, as compared with only 3.0 mm in the 5.0-μg group. There were no appreciable differences within dose groups across the time points. The median degree of stenosis was above 50% at the preprocedure time point and below 15% at the postprocedure time point in all dose groups. Consistent with the findings for MLD and late MLD loss, the degree of stenosis was higher again in the 0.5-, 2.0-, and 5.0-μg groups at the 6-month follow-up, with the median values ranging from 40% to 51%. In contrast, in the 10.0-μg group, there was only a slight re-increase to 21% in the median degree of stenosis. This was also reflected in the binary restenosis rate, which tended to be lower in the 10.0-μg group than in the 2.0- and 5.0-μg groups (p = 0.03; Fig. 2).
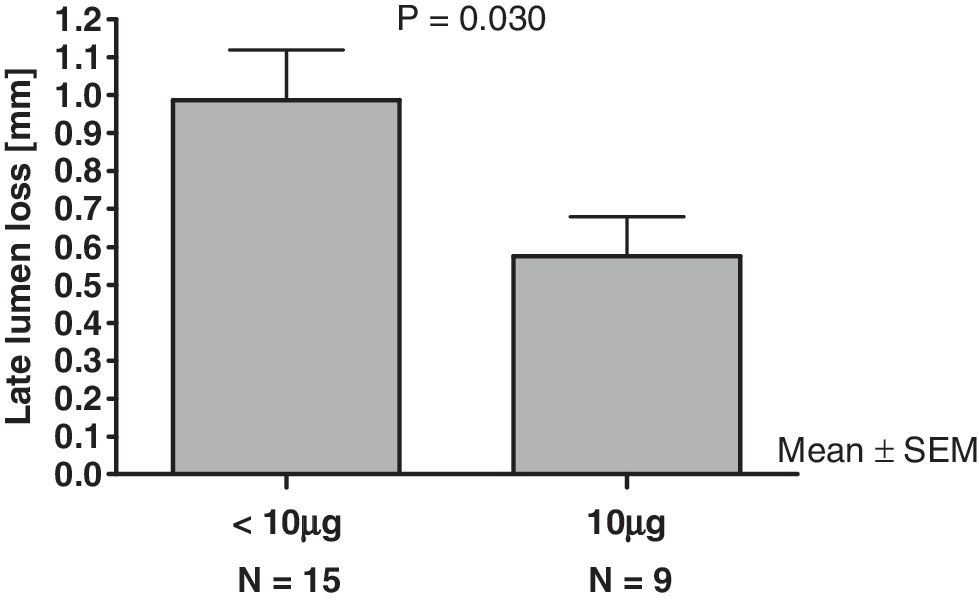
Stratifying patients according to the applied dose, late lumen loss was significantly lower in the “high dose” group (10 μg CAR-MP583; n = 9) compared to patients who received lower dosages (0.5, 2.0, 5.0 μg; n = 15). Since this study was conducted to prove safety and feasibility, not all patients could be reinvestigated after 6 months.
GP, glycoprotein; LAD, left anterior descending artery; LCX, left circumflex artery; RCA, right coronary artery.
The study was performed primarily for safety and feasibility. However, analysis of the angiograms is shown in Table 3, giving all angiographic details obtained. Follow-up angiograms were achieved in only 73% of the treated patients. Interestingly, the observed late lumen loss in patients receiving the maximal dose of CAR-MP583 (10 μg) was significantly lower compared with those who received doses from 0.5 to 5 μg.
Discussion
The REGENT I Extension study was the first study to use iNOS lipoplex (CAR-MP583) gene therapy in humans. Originally, REGENT I Extension was planned as the European counterpart of REGENT I, which was scheduled to be performed in the United States. The overall design of the two studies was similar. After a FDA Investigational New Drug application and a public hearing at the Recombinant Advisory Committee of the National Institutes of Health were successfully completed, REGENT I had to meet some local Institutional Ethics Committee requirements, such as a 72-hr window between informed consent and coronary intervention, and a 6-week follow-up angiogram for all patients. These requirements proved to be prohibitive for successful patient recruitment in routine cardiology practice. Thus, REGENT I Extension was the only study performed. REGENT I Extension was a phase I dose escalation study to obtain safety and tolerability information on CAR-MP583 gene transfer when applied after PTCA, but prior to final percutaneous treatment in patients with clinically significant coronary artery disease.
The CAR-MP583 clinical development program was initiated and sponsored by the former German biotech company Cardion AG. Due to its corporate policy and strategic reasoning, it was not possible to publish the data directly after completion of the clinical study. Eventually, the rapidly evolving role of DES led to the discontinuation of the gene therapy program at Cardion AG. However, the authors involved in planning, management, and execution of this gene therapy trial strongly believe that the data of this clinical study should be made available to the scientific community.
The present study was a dose escalation study that evaluated the safety of various dose levels of the iNOS lipoplex (CAR-MP583) gene therapy applied intramurally for reduction of restenosis in patients with de novo or restenotic lesions in native coronary arteries.
The study enrolled a total of 32 patients in four study sites in Germany. Thirty of the 32 patients were treated with CAR-MP583 in accordance with the dose escalation scheme. Six patients received 0.5 μg, six received 2.0 μg, six received 5.0 μg, and 12 received 10.0 μg of CAR-MP583. The remaining two patients did not receive CAR-MP583 due to technical problems with the Infiltrator. All 32 patients completed the 6-month follow-up as planned in the study protocol. CAR-MP583 was well tolerated at all doses investigated. Notably, no differences were observed between the dose groups with regard to the primary analysis variable (occurrence of MACE up to 30 days) or any of the predefined secondary analysis variables. As assessed by the almost 100% drug delivery and lesion success rates, treatment with the Infiltrator and CAR-MP583 was feasible. Although based on small numbers and incomplete 6-month follow-up data, the angiographic results are suggestive of a dose-response relationship, with the best results observed in the 10.0-μg group. The results in the 10.0-μg group are particularly interesting considering that this group enrolled seven of the 12 patients who had presented with restenotic lesions at baseline, and had the highest proportion of such patients among all groups.
A key advantage of using NOS, the therapeutic gene, is the fact that NO generated by the transgene product can diffuse easily to neighboring cells. This “bystander” effect allows therapeutic effects not only in transduced cells of the vascular wall, but also in cells directly adjacent to the cells expressing the transgene. Via multiple pathways, NO can either stimulate or inhibit cellular growth, depending on the cell type. It is a potent inhibitor of smooth muscle cell growth, mediated by both cyclic GMP-dependent and -independent mechanisms. Furthermore, NO can enhance the mitogenic effects of vascular endothelial growth factor and basic fibroblast growth factor during endothelial wound healing and angiogenesis, thereby stimulating the growth of endothelial cells. Both inhibition of smooth muscle cell proliferation and propagation of endothelial regrowth are therapeutic effects of NO important for successful inhibition of restenosis.
As there was no established process available for coating stents with DNA vectors with subsequent efficient local deposition of the vector to the target cells, gene therapy was performed by using a local drug delivery device. The Infiltrator catheter is a triple lumen catheter incorporating a low-pressure positioning balloon with a series of microminiaturized injector ports mounted on its surface (Barath et al., 1997). Microliter quantities of drug (typically 400 μl) containing sufficient quantities of the therapeutic product can be delivered directly into the tunica media of the arterial wall in a precise fashion. Injection of the drug can be accomplished in less than 10 sec, without substantial intimal damage, with efficient deposition along the length of the delivery ports (15 mm), and with near-zero luminal washout. The Infiltrator has been studied extensively in several animal models. The use of the Infiltrator device in two human clinical studies has been reported (Pavlides et al., 1997; Reimers et al., 1998). A total of 41 patients and 57 restenotic lesions were successfully treated with either low-molecular-weight heparin or methylprednisolone acetate, respectively.In both studies, there were no complications (prolonged chest pain, persistent ST segment changes, vessel occlusion, vessel dissection, or slow flow) from the use of the Infiltrator. Both reports support our finding that the device was safe and feasible to use.
Restenosis occurs in response to arterial injury. The loss of the protective function of the endothelium (and endothelium-derived NO) after intra-arterial balloon trauma is one of the hallmarks in restenotic pathology (Myers et al., 1996; Ross, 1999). Besides its long known vasodilatory capacity, NO is a potent inhibitor of platelet activation (Radomski et al., 1990), vascular smooth muscle cell proliferation (Garg and Hassid, 1989; von der Leyen et al., 1995), cell migration (Dubey et al., 1995), and extracellular matrix synthesis (Kolpakov et al., 1995). These complex, potentially beneficial therapeutic effects of NO were the rationale for using NO as a therapeutic agent to influence the pathological course of restenosis following vascular injury. As there are limitations associated with the systemic administration of NO (donors), the prospect of local vascular gene transfer provides a medical avenue by overexpressing the therapeutic gene at the site of vascular injury, thereby avoiding unwanted systemic side effects.
Increased synthesis of the bioregulatory molecule NO is reported to be a part of the cellular and biochemical events activated during wound repair (Schaffer et al., 1996). Transient iNOS expression achieved with adenoviral-mediated gene transfer using a simple, brief topical exposure to low-titer virus was adequate to reverse the delayed wound closure in iNOS-deficient mice (Yamasaki et al., 1998). Thornton et al. (1998) demonstrated that transient expression of iNOS with increased NO synthesis within the wound milieu was followed by increased collagen accumulation. The results in iNOS-deficient mice support the hypothesis that an increase in NO production resulting from up-regulation of iNOS expression may be an important part of an endogenous physiological repair mechanism, thereby stabilizing the vascular remodeling process (Assoian and Marcantonio, 1996; McCarthy et al., 2008).
To date, the restenosis rate after percutaneous coronary intervention can be neglected because DES, as well as drug-eluting balloons, have been introduced in the clinical routine of interventional cardiology (Garg and Serruys, 2010a). Therefore, it seems to be rather difficult to introduce new strategies into the current concepts of interventional therapy. A cardiologist might keep a critical watch over “gene therapy,” but, on the other side, toxic agents are applied routinely in the coronaries.
However, as dual prolonged antiplatelet therapy, as well as stent thrombosis, is a concern with respect to the long-term safety of DES, new technologies including new stents, as well as advanced new therapeutic substances, are warranted (Garg and Serruys, 2010b; Holmes et al., 2010). The continuous release of an antithrombotic agent with additional antiproliferative activity may actually be an interesting new avenue for future vascular stenting. NO is such an endogenous substance. New coating technology might provide stents with continuous release of an iNOS gene therapy vector into the vascular wall. Zhong et al. (2009) described a hydrogen-coated polyester stent graft impregnated with a LacZ-expression vector and demonstrated transduction of the vascular wall. Fishbein et al. (2006) published a vascular gene therapy concept that employed local delivery of gene vectors from bare-metal stents by use of a biodegradable synthetic complex, resulting in inhibition of in-stent restenosis in rat carotid arteries. Also, Sharif et al. (2008) showed accelerated re-endothelialization and inhibition of restenosis after adenovirus-mediated eNOS gene transfer from a gene-eluting stent. Thus, there are concepts available that might be able to combine an iNOS gene therapy vector together with a stent system. It is hoped that this article will encourage the scientific community to test this promising hypothesis in more detail.
Overall, it can be concluded that the iNOS gene therapy approach presented here by employing CAR-MP583 was well tolerated at all doses investigated. It appears to be a safe therapeutic principle for the prevention of coronary restenosis, provided it can be applied easily to the coronary artery wall with sufficient cellular transfer capacity. Further developments in NOS-eluting stent or balloon technology may provide such a tool for gene delivery in the future.
Footnotes
Acknowledgments
The authors gratefully acknowledge the collegial cooperation with Harvard Clinical Research Institute (Richard Kuntz, MD; Carolina Karam, MD), Drug Safety Alliance (Catherine C. Stokes), and Regulatory Affairs North American (Nancy Chew). The authors gratefully acknowledge Astrid Schiebeling and Beate Mantz for expert technical assistance.
Author Disclosure Statement
This study was sponsored by Cardion AG (Erkrath, Germany). Heiko E. von der Leyen was Medical Director for Cardion AG until 2001. The clinical investigators received financial reimbursements for contributing to the study, but no competing financial interests exist.