
Abstract
Because of their multi/pluripotency and immunosuppressive properties mesenchymal stem/stromal cells (MSCs) are important tools for treating immune disorders and for tissue repair. The increasing use of MSCs has led to production processes that need to be in accordance with Good Manufacturing Practice (GMP). In cellular therapy, safety remains one of the main concerns and refers to donor validation, choice of starting material, processes, and the controls used, not only at the batch release level but also during the development of processes. The culture processes should be reproducible, robust, and efficient. Moreover, they should be adapted to closed systems that are easy to use. Implementing controls during the manufacturing of clinical-grade MSCs is essential. The controls should ensure microbiological safety but also avoid potential side effects linked to genomic instability driving transformation and senescence or decrease of cell functions (immunoregulation, differentiation potential). In this rapidly evolving field, a new approach to controls is needed.
Introduction
Because of their multi/pluripotency, immunosuppressive properties, and production of cytokines or growth factors, MSCs are important tools in treating immune disorders and in tissue repair. The increasing use of MSCs has led to the development of production processes that need to be in accordance with Good Manufacturing Practice (GMP). GMP regulations first promulgated by the U.S. Food and Drug Administration are implemented in Europe (Commission Directive 2003/94/EC). These regulations require manufacturers to ensure that products are safe, pure, and effective. To achieve these goals, all aspects of manufacturing should be monitored: quality assurance management, personnel, premises and equipment, documentation, production, quality control, contract manufacture and analysis, complaints. and product recall.
In Europe, MSCs are considered advanced therapy medicinal products (ATMPs), as defined by European Regulation (European Commission [EC]) 1394/2007. Under this regulation, MSCs are considered somatic cell therapy products or tissue-engineered products depending on the source, manufacturing process, and proposed indications. The regulation contains rules for authorization, supervision, and technical requirements regarding the summary of product characteristics, labeling, and package leaflets of ATMPs that are prepared according to industrial methods and in academic institutions. The regulation refers to the European GMP rules. In the United States, MSCs are considered in the context of human cells, tissues, or cellular and tissue-based products (HCT/Ps). As for any other HCT/Ps, the production of MSCs must comply with current Good Tissue Practice requirements, under the Code of Federal Regulations (CFR).
Facilities (Part 1271.190a and b)
Environmental control (Part 1271.195a)
Equipment (Part 1271.200a)
Supplies and reagents (Part 1271.210a and b)
Recovery (Part 1271.215)
Processing and process controls (Part 1271.220)
Labeling controls (Part 1271.250a and b)
Storage (Part 1271.260a–d)
Receipt, predistribution shipment, and distribution of an HCT/P (Part 1271.265a–d)
Donor eligibility determinations, donor screening, and donor testing (Parts 1271.50, 1271.75, 1271.80, and 1271.85)
MSCs have been produced in research for more than 40 years, but the translation of research-based protocols into procedures for large-scale production of clinical-grade MSCs according to GMP requires a deep analysis of all critical aspects (Sensebé and Bourin, 2008). Most requirements are common to the culture of all other cells for clinical application; the facilities should be in accordance with Part 1271.190a and b of the CFR and consequent European regulations. As an example, the use of an open system for culturing or processing requires work in a class A environment inside a class B room. Therefore, the environment must be qualified and controlled to ensure that these levels are reached. As well, supplies and reagents, recovery, labeling controls, storage, receipt, predistribution shipment, and distribution are common to the culture of other human cells, tissues, or cell- and tissue-based products. However, culture of MSCs involves several specific parameters that should be considered. This review gives an overview of GMP requirements for the manufacture of MSCs. In cellular therapy, safety remains a main concern, and therefore this review focuses on the safety aspects of the GMP production of MSCs. Aspects of safety refer to the processes and the media, the systems (closed), and the controls that should be implemented.
Donor, Cell Sources, and Culture Processes
The determination, screening, and testing of donor eligibility for MSCs (e.g., viral testing or age) is similar to that for all cell- and tissue-based products; however, for MSCs, determining who will be a good allogeneic donor is difficult. In addition, the possibility of using a unique donor for a high number of recipients reinforces the necessity of strict selection criteria. The age of the donor is important, and BM from children contains a higher concentration of CFU-Fs than that from adults (Baxter et al., 2004). Moreover, the age of the donor seems to be directly associated with detrimental effects in terms of proliferation and multipotency of MSCs (Stolzing et al., 2008). The donor should have no abnormalities or risk of abnormalities possibly involving MSCs that to date is difficult to assess. No specific regulatory requirement exists for this matter, but the issue should be considered carefully in the use of unique donors for many patients.
Many sources of MSCs have been described and used (Fig. 1). The main candidates for clinical application are BM, AT, and cord blood (Kern et al., 2006). BM and AT are widely available and easy to collect by standardized procedures. MSCs are more abundant in AT than in BM. For cord blood, various collection methods result in variable cell yield and viability of the MSCs obtained; the success rate in isolating and further expanding MSCs depends on the time between collection and processing, the volume of blood collected, and the cell content (Bieback et al., 2004), which highlights the need for a short delay between delivery and harvesting. The other sources envisioned for clinical applications are related to fetal or neonatal tissues that contain large populations of MSCs in amniotic membrane and Wharton's jelly from umbilical cord. The interest in these sources relates to obtaining more primitive MSCs.

Potential sources for the production of mesenchymal stem cells (MSCs) for clinical uses. The most commonly used tissue is the bone marrow (BM). Total nucleated BM cells or mononucleated cells obtained by density gradient can be used. Various antibodies can also be used to enrich colony-forming unit-fibroblasts (CFU-Fs) in the seeded fraction. For adipose tissue (AT), a first step of enzymatic digestion and centrifugation should apply to obtain the stromal vascular fraction (SVF) containing AT MSCs. The other potential sources are still far from clinical-grade production. ADSC, adipose-derived stem cells.
In addition to the important issues of choice of donor and source of MSCs, the process of culturing MSCs is a major consideration. One could start with unfractionated or fractionated cells from tissue. MSCs are present in the mononuclear fraction of BM cells after density-gradient separation, and AT should be dissociated by use of collagenase and centrifuged to obtain the stromal vascular fraction containing MSCs or AT-derived stem cells. BM can be further enriched for MSCs with immunomagnetic devices or by fluorescence-activated cell sorting. Various antigens allow for good enrichment of MSCs; fluorescence-activated cell sorting of the STRO-1bright fraction leads to a 950-fold enrichment of CFU-Fs, and STRO-1+CD106+ cells represent a highly purified MSC population (Gronthos et al., 2003). Other cell antigens, such as α1-integrin subunit (CD49a) or CD271 (LNGFR) (Quirici et al., 2002), can be used to enrich the BM mononuclear cell fraction. CD49a is expressed in MSCs, unlike in CD34+ hematopoietic stem cells, and all CFU-Fs are present in the CD49a+ fraction (Deschaseaux and Charbord, 2000). However, these procedures lead to enriching MSCs rather than truly selecting pure MSCs. Therefore, these procedures should be demonstrated to drive a real enrichment without the loss of some functional or stem subpopulations (Colter et al., 2001). Despite numerous antibodies for experimental selection procedures, “clinical-grade” antibodies for MSC enrichment should be developed. Whatever the cell source, cell population, fractionated status, or separation by density gradient, the MSC populations are isolated by their physical property of adherence to plastic.
Because MSCs are adherent cells, cell-plating density is a critical parameter to ensure a good expansion rate and to maintain the required functions of MSCs. The cell-plating density is extremely variable; some clinical trials have involved a high cell density, such as 170 × 103 mononuclear cells/cm2, and others a low density, such as 50 × 103 nucleated cells. After the first seeding, the plating density should be decreased (Koç et al., 2000). The choice of cell density remains pivotal, and use of a low or very low plating density could better maintain a high proliferation rate and multipotentiality (Colter et al., 2001). For clinical-scale production of MSCs, use of a very low plating density is difficult and leads to wide culture surfaces and nonoptimal cell production. After the first culture phase, for the following passages, a plating density of 1000 cells/cm2 seems a reasonable compromise and allows for a high number of harvested cells (Sekiya et al., 2002). The possibility of some resting hematopoietic cells within the MSC layer after the first step and the normal growth inhibition at confluence lead to the culture of successive passages to obtain a large amount of pure MSCs. However, sequential passages may affect the quality of MSCs. Cultured MSCs are committed cells (Delorme et al., 2009); successive passages slow the proliferation rate, and cells progressively show loss of multipotentiality, retaining only an ability to differentiate into osteoblasts (Banfi et al., 2000; Muraglia et al., 2000). After 3 weeks of culture and 12–15 doublings, the proliferation rate slows, and MSCs lack some of their multipotency. Therefore, for efficiency and safety reasons (see the following discussion on safety), the number of population doublings should be reasonable (<20).
The medium is crucial for efficacy and safety of MSC culture. The media used in culture processes should maintain the phenotype, genotypic stability, and functions of MSCs during multiple passages. Therefore, culture conditions must be optimized. Numerous growth factors and cytokines are useful, but the complete growth factor needs for MSC culture are still unknown and could depend on the target, the amount of cells, and the type of MSCs, undifferentiated or committed. Whatever the target, some growth factors or cytokines are crucial and include at least platelet-derived growth factor (PDGF), epidermal growth factor (EGF), transforming growth factor (TGF)-β, insulin-like growth factor, and fibroblast growth factor-2 (FGF2) (Sensebé et al., 2006; Ng et al., 2008). In particular, FGF2 can give excellent expansion efficiency and preserve MSC differentiation potential in part. Although consensus is lacking on the ideal medium for MSC culture, standard Dulbecco's modified Eagle's medium (DMEM) or α-minimal essential medium is commonly used, with the addition of animal serum, fetal bovine serum (FBS) or human serum or plasma (Müller et al., 2006; Kocaoemer et al., 2007), and growth factors. The standard culture condition for MSC expansion has been FBS-supplemented medium with or without added growth factors, with a concentration of 10% typically used. FBS shows significant batch-to-batch variability, which may negatively affect production efficiency; the FBS should be carefully tested before use. Some regulatory authorities, such as PEI in Germany, prohibit the use of FBS, but FBS can still be used for GMP production of MSCs. A Transportation Security Administration (TSA) certificate should be obtained to ensure safety in terms of the risk of infectious disease transmission. As well, during culture, MSCs can retain in their cytoplasm proteins present in FBS, which incurs risk of sensitization (Spees et al., 2004). Safety requirements imply replacing FBS with more secure products such as human components or a fully defined serum-free medium. Human products, such as human AB serum or “platelet lysates” from blood transfusion-secured sources, have been tested. The most promising and most often used product is the platelet lysate corresponding to human plasma enriched by growth factors from platelets. The platelet concentrates are obtained from blood transfusion centers that ensure microbiological safety; they are subjected to freeze–thaw cycles to disrupt platelets and allow the release of growth factors. After a final filtration, the platelet lysate can be used or stored frozen. As demonstrated by many teams, the use of platelet lysates represents an efficient alternative to FBS (Müller et al., 2006; Kocaoemer et al., 2007). However, the functionality of MSCs obtained when changing the medium should be tested. For example, the differentiation potential of MSCs may be influenced by the use of platelet lysate-containing medium, as was shown for osteogenic differentiation potential (Kasten et al., 2008). The proliferation potential of the platelet lysate-based system seems at least identical to or even higher than that of the classic FBS-based culture. Different serum-free media based on cocktails of growth factors (e.g., FGF2, PDGF, TGF-β) have been developed for research purposes; these media can maintain the main phenotypic and functional characteristics of cultured MSCs (Chase et al., 2010). However, serum-free medium of clinical grade is not available for producing GMP-grade MSCs. Various cocktails of growth factors may be efficient, but a central problem is the need for a specific protein (e.g., fibronectin) to ensure the first attachment of the cells to the culture surface and that the protein is produced under conditions avoiding xenogenic contaminants.
Closed Systems and Bioreactors for MSC Culture
For MSCs, as for any other cellular products needing a long culture period, attention must be paid to aseptic conditions (as stated in Annex 1 of the current European GMP regulations) and their validation. A full validation of aseptic techniques should be considered a crucial part of any validation process for MSC production, and closed systems instead of classic flask-based cell manipulations are preferred. Therefore, automated, closed devices are of great interest in facilitating further development of a GMP-grade system for MSC expansion. For the culture of a high number of cells, MSCs require a wide surface area that could lead to the use of large number of simple culture flasks; therefore decreasing the number of culture vessels is important. Companies such as Nunc and Corning have produced large, multilayered systems (from 1 to 10 layers or more) that could be stacked in incubators. These culture vessels can be used to easily produce MSCs in a nearly closed system, thus greatly reducing the number of flasks needed, the most complicated step being the recovery of MSCs by a proteolytic enzyme. Decreasing the number of flasks greatly improves the microbiological safety, traceability, staff workload, and costs. Such a method with use of two-layers CellSTACKs (Corning), and specific bags and tubing (Macopharma), allows for producing half a billion pure MSCs in 3 weeks with a minimal number of interventions (Tarte et al., 2010). Other parameters could be of importance, as reported for antiinflammatory properties (Bartosh et al., 2010); changing from two- to three-dimensional culture could increase or improve some function(s).
However, this stacking process is not fully GMP compliant and not fully closed; a class A cabinet is needed for each manipulation. To increase the GMP compliance, safety, and traceability, a fully closed and automated bioreactor is needed. Bioreactors allow for translating research-based experimental processes into scalable cell-production processes. The main criteria for growing MSCs in bioreactors include a large ratio of surface area to volume, a closed system, and automated inoculation and harvesting. Different designs of bioreactors, such as parallel-plate, hollow-fiber, or microfluidic bioreactors (Godara et al., 2008), could improve these criteria. Companies such as CaridianBCT have developed fully automated bioreactors based on hollow-fiber technology that can allow for a large expansion of MSCs in a GMP-compliant system (Rojewski et al., 2010). Linked with animal- or serum-free media, these technologies can provide optimal tools to deliver MSCs of clinical grade that were produced according to GMP.
Controls for Cultured MSCs
The safety of the clinical use of MSCs is directly linked to the controls implemented during the development process and at batch release. During culture expansion processes, MSCs, like other cells, are exposed to different risks, bacterial contamination, xenogenic risk, and finally, genomic instability. Controls entail two aspects: (1) the first set of controls is linked to the development of the process and should allow for the qualification of the process itself; during development, functional controls should demonstrate efficacy, particularly in terms of differentiation or immunoregulation; and (2) for cell release in the clinical phase, controls should ensure safety and efficacy and should allow for rapid release of cells. Whatever the type of controls, they must be standardized. A major concern is that no quantitative functional parameter has been directly correlated to clinical efficiency to date and the identification of such efficiency-related controls is urgently needed, in relation with our increasing knowledge of the biology of MSCs.
The controls for cell release must include bacteriological tests (mainly in liquid medium), phenotype controls (absence of hematopoietic contamination, expression of mesenchymal markers), a visual follow-up of cultures (morphology, evolution of cellular density), and, if possible, functional analyses (clonogenic testing, secretion of specific molecules). However, results of these latter tests, because of the time they take, are not usable without prolonged conservation of the graft (cryopreservation). It should be noted that MSCs are easily cryopreserved and thawed without impairment of their functions. The final quality controls must include viability and phenotype tests, for example, tests that are compatible with a rapid release of the graft. All these controls are mandatory and reflect standard controls of all cellular products obtained after culture amplification.
For clinical use of MSCs, a major safety concern is the genomic stability of MSCs: the risk of cell transformation and risks related to the use of senescent cells. Use of immortalized MSCs transduced by human telomerase reverse transcriptase revealed that transformation of human MSCs is a long, multistep process involving the deletion of p16 ink4a (Serakinci et al., 2004). A few studies have shown cultured human MSCs with chromosome abnormalities that finally transform (Rubio et al., 2005; Rosland et al., 2009); however, these results were retracted because they were related to contamination by exogenous cancer cell lines (Garcia et al., 2010; Torsvik et al., 2010). Using karyotype and comparative genomic hybridization, other authors have shown the genetic stability of human MSCs, unlike mouse MSCs, during a long culture process. We have shown that clinical grade-cultured human MSCs, regardless of the presence of aneuploidy (mainly duplication of chromosomes 5 and 8), reached senescence and never transformed (Tarte et al., 2010). However, in our study and in some others, deletion/mutation analyses revealed that rare productions of MSCs reached senescence without expressing p16 ink4a (Shibata et al., 2007).
The use of senescent cells should not be underestimated; senescent MSCs lose a part of their differentiation potential and their secretion profile changes. Since the description of the limited life span of cultured fibroblasts by Hayflick in the 1960s (Hayflick, 1963), numerous data have demonstrated that, over time, either in vitro or in vivo within tissues, cells stop dividing and become senescent. Replicative senescence arises through various mechanisms: telomere shortening, activation of the pRB pathway through the INK4a/ARF locus encoding p16 ink4a and p19 ink4a , and activation of the p53 pathway (Krishnamurthy et al., 2004; Campisi and d'Adda di Fagagna, 2007). However, senescence and transformation are tightly linked; cells becoming senescent could transform after a transient senescence crisis or the abrogation of senescence mechanisms by a re-increase in telomere length or repression of p16 ink4a and p53 activity (Serakinci et al., 2004). Senescence is associated with cell cycle arrest, resistance to apoptosis, and chromatin reorganization into senescence-associated heterochromatin foci, in which E2F target genes are silenced by pRB (Park et al., 2004; Campisi and d'Adda di Fagagna, 2007). Interestingly, cells becoming senescent present a specific senescence-associated secretory phenotype including cytokines, chemokines, growth factors, and surface molecules such as interleukin-6, matrix metalloproteinases, hepatocyte growth factor, FGF2, and GROa that could act as autocrine or paracrine factors (i.e., Wnt16B). This phenotype could reinforce the senescence arrest (Binet et al., 2009), stimulate cancer cell growth and invasion in vitro and in vivo (Coppé et al., 2008), and modulate the immune response and inflammation, although few data are available in this specific field. Moreover, injected MSCs can sustain the growth of cancer (Djouad et al., 2003), and p53-mutated MSCs can migrate to mammary tissue and form an inductive microenvironment for mammary carcinoma (Houghton et al., 2010). Few studies have focused on the senescence and genetic stability of cultured MSCs. During culture, the senescence of MSCs induces growth arrest, with normal telomere shortening (Kim et al., 2009) and decreased differentiation with, finally, predominant osteogenic potential, downregulation of genes related to tumorigenesis (e.g., myc, ras), upregulation of a gene cluster from chromosome 4, and upregulation of some microRNAs (Wagner et al., 2008). All these changes appear to be a continuous process. Senescence of MSCs is a complicated, finely tuned process at genomic, transcriptomic, epigenetic, and proteomic levels. It is now evident that the risk of transformation is low (Prockop et al., 2010), and karyotype and comparative genomic hybridization have low sensitivity and do not appear to be relevant controls, neither indicating the real risk of MSC transformation or their senescent status. Last, regarding safety issues, real relevant controls for transformation and senescence should be implemented. These controls could refer to genes or molecules involved in the senescence/transformation pathways, such as p53, p21, and p16 (Tarte et al., 2010).
Relevant efficient controls are of major importance in GMP and should ensure the safety and efficacy of cultured MSCs. For microbiological safety, controls are well described and standardized. However, as previously discussed, controls should be extended and standardized for other aspects of cultured MSCs such as risks related to transformation and senescence without excluding testing of immunomodulatory properties and differentiation potentials. For example, in a European Consortium of the 7th Framework Program of European Commission (CASCADE, no. 223236; HEALTH-F5-2009-223236), we established reproducible interlaboratory protocols that yielded comparable results with use of the same cell types in two independent laboratories (Department of Clinical and Experimental Medicine, Verona, Italy and INSERM U917, Rennes, France). These protocols will be used as quality controls of GMP-grade MSCs to test their immunomodulatory properties before their clinical use in patients.
All these data demonstrate that research teams, those involved in research and development, and clinicians performing clinical trials should coordinate to develop, test, and define relevant and efficient controls (Fig. 2). These controls should then be presented to and discussed with regulatory authorities.
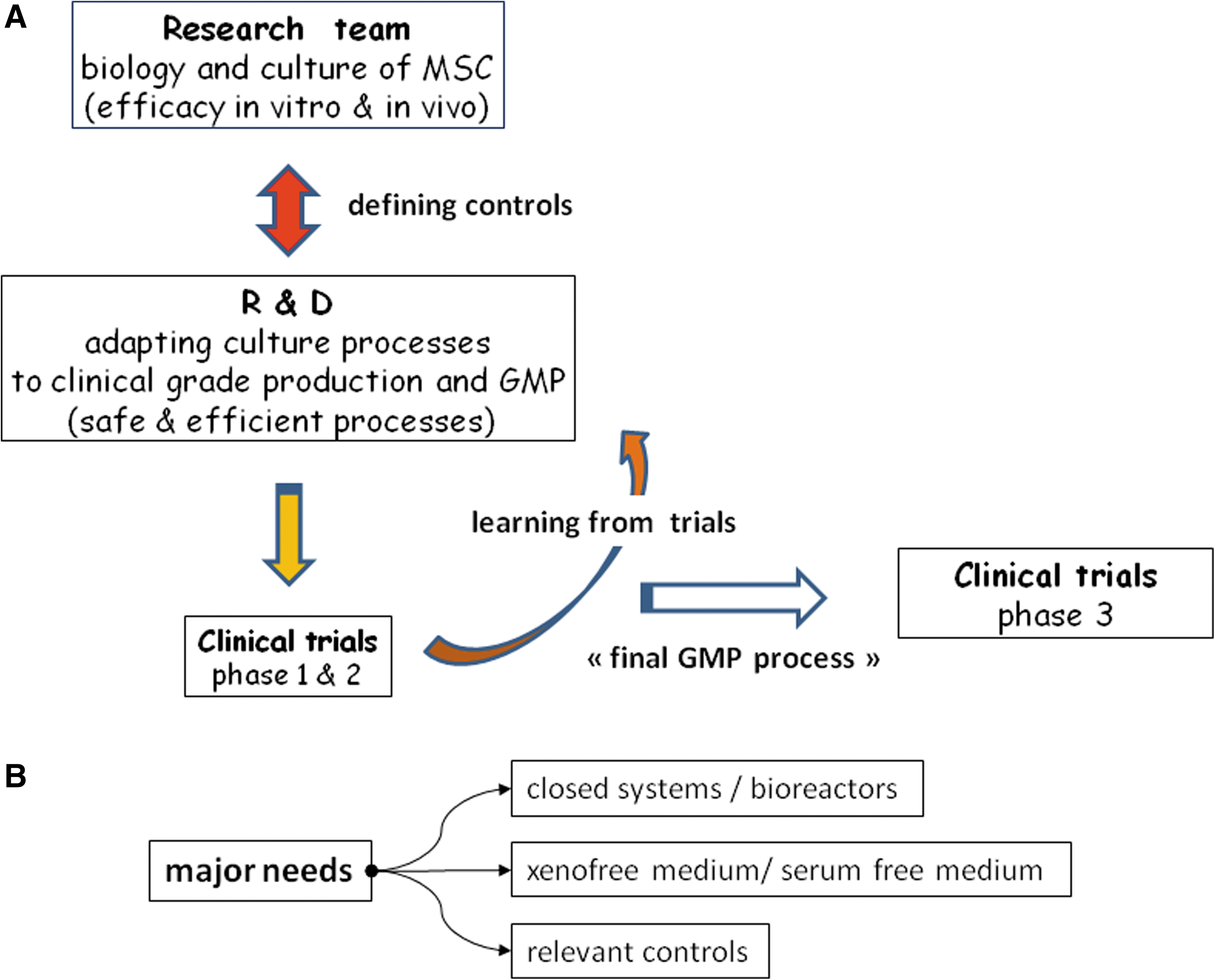
Development and needs for MSC production. (
Current Perspectives
The field of MSCs is evolving quickly, and since the first reported trial in 1995, cultured MSCs have been used in clinical situations ranging from immunological intervention, to the treatment of graft-versus-host disease during hematopoietic stem cell transplantation, and to tissue repair and bone engineering. Despite numerous registered clinical trials (125 trials registered at
Footnotes
Acknowledgments
This work was supported by a grant from the 7th Framework Program of the European Commission: CASCADE (no. 223236; HEALTH-F5-2009-223236).
Author Disclosure Statement
No competing financial interests exist for any of the authors.