
Abstract
No curative treatment exists for glioblastoma, with median survival times of less than 2 years from diagnosis. As an approach to develop immune-based therapies for glioblastoma, we sought to target antigens expressed in glioma stem cells (GSCs). GSCs have multiple properties that make them significantly more representative of glioma tumors than established glioma cell lines. Epidermal growth factor receptor variant III (EGFRvIII) is the result of a novel tumor-specific gene rearrangement that produces a unique protein expressed in approximately 30% of gliomas, and is an ideal target for immunotherapy. Using PCR primers spanning the EGFRvIII-specific deletion, we found that this tumor-specific gene is expressed in three of three GCS lines. Based on the sequence information of seven EGFRvIII-specific monoclonal antibodies (mAbs), we assembled chimeric antigen receptors (CARs) and evaluated the ability of CAR-engineered T cells to recognize EGFRvIII. Three of these anti-EGFRvIII CAR-engineered T cells produced the effector cytokine, interferon-γ, and lysed antigen-expressing target cells. We concentrated development on a CAR produced from human mAb 139, which specifically recognized GSC lines and glioma cell lines expressing mutant EGFRvIII, but not wild-type EGFR and did not recognize any normal human cell tested. Using the 139-based CAR, T cells from glioblastoma patients could be genetically engineered to recognize EGFRvIII-expressing tumors and could be expanded ex vivo to large numbers, and maintained their antitumor activity. Based on these observations, a γ-retroviral vector expressing this EGFRvIII CAR was produced for clinical application.
Introduction
Immunotherapy for glioblastoma has been suggested by a number of investigators (Johnson and Sampson, 2010). Among the potential targets for the development of immunotherapy for glioblastoma are variants of the epidermal growth factor receptor (EGFR). The most common EGFR mutant is the EGFR variant III (EGFRvIII), which harbors an in-frame deletion of exons 2–7. This deletion results in a truncated extracellular ligand-binding domain, and renders the protein constitutively active in a ligand-independent fashion (Chu et al., 1997). EGFRvIII expression has been shown to enhance tumorigenicity (Lal et al., 2002), promote cellular motility (Boockvar et al., 2003), and confer resistance to radiation and chemotherapy (Montgomery et al., 2000; Lammering et al., 2004). EGFRvIII expression has been reported in 24–67% of glioblastomas, but not in any normal tissues (Humphrey et al., 1990; Wong et al., 1992), making it an attractive target for immunotherapy.
Adoptive cellular immunotherapy involves the administration of autologous or allogenic tumor-reactive T cells into patients to achieve tumor regression and has been successful in transplant-related malignancies, leukemia, and melanoma. In melanoma, this process involves the identification of tumor-infiltrating lymphocytes (TILs) with high affinity for tumor antigens that can be selected in vitro, expanded, and administered to patients. The administration of naturally occurring TILs has been shown to have an objective response rate ranging from 49% to 72%, with up to 40% complete durable regressions in metastatic melanoma patients with bulky invasive tumors at multiple sites, including liver, lung, and notably brain (Hong et al., 2010; Rosenberg et al., 2011).
As an alternative approach to TILs, T-cell receptor (TCR) genes from tumor-reactive T-cell clones can be isolated and transferred to normal T cells (Park et al., 2011). An alternate approach to TCR-based gene therapy is to use a chimeric antigen receptor (CAR), constructed from the variable regions of the heavy and light chains of an antibody connected by a linker sequence and fused to T-cell signaling protein domains (from, e.g., CD28, CD3ζ, or 41BB) (Eshhar, 2010). We and others have reported that CAR-engineered T cells mediate objective tumor regression in multiple diverse cancers such as lymphoma and neuroblastoma (Pule et al., 2008; Kochenderfer et al., 2010, 2012; Porter et al., 2011).
A problem in the study of glioblastoma-reactive lymphocytes is the identification of target cells (e.g., glioma cell lines) that are representative of primary glioblastoma. Detailed molecular analysis of many cancer cell lines has now demonstrated that established lines often do not mirror the molecular characteristics of primary human cancers, and this is the case for glioma lines (Venere et al., 2011). Alternatives to the use of established glioma cell lines are the analysis of tumor stem cell (TSC) lines. The TSC paradigm proposes that a subpopulation of cells exists in cancer that gives rise to all the cells in a differentiated tumor (Dirks, 2006). Several laboratories, including ours, have demonstrated that primary glioma-derived cells grown as neurospheres share properties not found in glioma cell lines, and harbor features consistent with TSCs (Lee et al., 2006; Venere et al., 2011). TSCs derived directly from primary glioblastomas [glioma stem cells (GSCs)] harbor extensive similarities to primary glioblastomas and are likely to be a more accurate in vitro model for drug development. We thus sought to develop immune-based approaches targeting GCSs as a potential treatment for glioblastoma and report, for the first time, that EGFRvIII is expressed in GSC lines and that EGFRvIII CAR-engineered T cells effectively target these lines.
Materials and Methods
GSC lines, tumor cell lines, and human peripheral blood lymphocytes (PBLs)
GSCs (0308, 1228, and 0822) were cultured as previously described (Lee et al., 2006). In brief, tumor tissue was enzymatically dissociated into single cells. Red blood cells and cellular debris were removed by ACK lysis buffer and differential centrifugation. Cells were grown as spheroids in NBE medium comprised of Neurobasal-A medium (Invitrogen, Carlsbad ,CA), N2 and B27 supplements (0.5×concentration each; Invitrogen), and recombinant human basic fibroblast growth factor (bFGF) and epidermal growth factor (EGF) (25 ng/mL each; R&D Systems, Minneapolis, MN) and maintained at 37°C with 5% CO2.
Cells lines (NIH-3T3, BHK, 293GP, U87, and U251) were cultured in RPMI 1640 medium plus 10% fetal bovine serum (FBS) (Biofluids, Rockville, MD) and penicillin/streptomycin (Invitrogen). All of the PBLs used in this study were obtained from patients treated at the Surgery Departments, National Cancer Institute (NCI)/Duke on Institutional Review Board–approved protocols that conform to the Declaration of Helsinki protocols, and all patients gave written informed consent. Human lymphocytes were cultured in AIM-V medium (Invitrogen) supplemented with 5% human AB serum (Valley Biomedical, Winchester, VA), 50 units/mL penicillin, 50 μg/mL streptomycin (Invitrogen), and 300 IU/mL interleukin-2 (IL-2) and maintained at 37°C with 5% CO2.
Retroviral vector containing the anti-EGFRvIII CAR gene
Based on a review of scientific literature and publicly available databases, the amino acid sequences for seven monoclonal antibodies (mAbs) (four murine and three human) specific for EGFRvIII were obtained and used to synthesize single-chain variable fragment (scFv) genes (Wikstrand et al., 1995; Lorimer et al., 1996; Reist et al., 1997; Nakayashiki et al., 2000; Weber, 2005). The amino acid sequences for variable light and heavy chains (VL and VH, respectively) of the antibodies were combined in the following order: amino acid leader sequence (from the human GM-CSFR protein), VL, a peptide linker peptide sequence (GSTSGSGKPGSGEGS), and VH, which were reverse-translated and produced as a codon-optimized synthetic DNA (Invitrogen). These scFv genes were inserted into MSGV1-based γ-retroviral vector MSGV-4D5-28Z or MSGV-4D5-28BBZ (Zhao et al., 2009) using T-cell signaling domains from CD28-CD3ζ (28Z) or CD28-41BB-CD3ζ (28BBZ) to produce the CAR-expression vectors. γ-Retroviral vector supernatants were generated by transfecting the respective vector DNA from each of the constructs with a plasmid encoding RD114 envelope into 293-GP cells using the Lipofectamine 2000 reagent (Invitrogen) in Optimem medium (Invitrogen).
Production of clinical-grade anti-EGFRvIII mAb 139-CAR vector
A PG13 packaging cell line encoding the 139-28BBZ CAR was produced by transinfection and, following limiting dilution, cloning and screening via RNA dot blot. Clone F10 was selected for production of the Current Good Manufacturing Practices (cGMP) master cell bank (MCB), as it had the highest signal in the RNA dot blot and was able to efficiently transduce human PBL as well as elicit IFN-γ release in an antigen-specific manner. γ-Retroviral vector was produced in the NCI Surgery Branch Vector Production Facility (National Cancer Institute, Bethesda, MD). In brief, CellBIND expanded surface roller bottles (Corning, Acton, MA) were seeded on day 0 with PG13-139-28BBZ (F10) at a cell density of 4×104 cells/cm2 in 200 mL of D10 medium consisting of high glucose (4.5 g/L), Dulbecco's modified Eagle medium (Invitrogen) supplemented with 10% FBS (Hyclone, Logan, UT) and 6 mM glutamine (final concentration; Invitrogen). On day 3, the medium was exchanged and replaced with 120 mL of D10 medium. Medium containing the γ-retroviral vector was then harvested daily with roller bottles being refed with 120 mL of medium. If glucose levels dropped below 2 g/L, the volume of the medium exchanged was doubled to 240 mL per roller bottle, at which point the bottles were divided into two sets of 13 bottles for subsequent harvests. Each of the six harvests consisted of 3 L of bulk vector supernatant, which was clarified via modified step filtration and stored at −80°C until further use. An aliquot from each harvest was tested for transduction efficiency and cytokine release as described. The cGMP-quality MCB and γ-retroviral vector supernatant were subjected then to an extensive biosafety testing program in accordance with current U.S. Food and Drug Administration (FDA), Center for Biologics Evaluation and Research regulatory guidelines.
EGFRvIII CAR vector transduction and analysis
Peripheral blood mononuclear cells (PBMCs) from healthy donors and glioblastoma patients (post resection, prior to treatment) were transduced as previously described (Johnson et al., 2009). In brief, frozen PBMCs were thawed and cultured in AIM-V medium (Invitrogen) supplemented with 5% human AB serum, plus antibiotics, 300 IU/mL IL-2, and 50 ng/mL OKT3. Two days later, cells were transduced at 0.25×106/mL with retroviral supernatant spun onto RetroNectin (Takara Bio, Otsu, Japan) coated nontissue culture treated six-well plates as described by the manufacturer. Transduced cells were allowed to expand in AIM-V medium as above, without OKT3.
Rapid expansion protocol (REP)
Transduced (or untransduced control PBLs from the same donor) PBLs were expanded in vitro using REP (Riddell and Greenberg, 1990). In brief, T cells were cultured in complete AIM-V plus 10% human AB serum plus 300 IU/mL IL-2 and 50 ng/mL OKT3 in the presence of 100-fold excess 5,000 Rad irradiated allogeneic PBMC feeder cells, and allowed to expand 10–14 days.
Flow cytometry of EGFRvIII CAR receptor on T cells
Cells were stained for surface EGFRvIII CAR expression using goat anti-human F(ab’) 2-biotinylated antibody (Jackson Immunotech, West Grove, PA) or biotinylated protein L (GenScript, Piscataway, NJ), with secondary detection by streptavidin-coupled phycoerythrin as previously described (Zheng et al., 2012). Negative staining controls were conducted by staining untransduced cells from the same donor. For intracellular staining of T cells cocultured with tumors 1:1 over 18 hr, with monensin in RPMI 1640 medium plus 10% FBS. Post coculture, cells were stained for CD3, CD8, and intracellularly for IFN-γ. FACS plots are shown gated on CD3+ (BD Biosciences, San Jose, CA).
Cytokine release assay
CAR-engineered PBLs were tested for antigen-specific reactivity in cytokine release assays using GSC lines and tumor cells. In these assays, effector cells (1×105) were cocultured with an equal number of target cells in AIM-V medium in a final volume of 0.2 mL in duplicate wells of a 96-well U-bottom microplate. Culture supernatants were harvested 18–24 hr after the initiation of coculture and assayed for IFN-γ by ELISA (Thermo Scientific, Rockford, IL) or using BD Biosciences Cytometric Bead Array system and flow cytometry. IFN-γ ELISpot was conducted using EGFRvIII CAR T cells cultured 2:1 with tumor targets as described (Johnson et al., 2009).
51Cr release assay
The ability of the transduced PBLs to lyse target cells was measured using a 51Cr release assay as previously described (Johnson et al., 2009). In these assays, CAR-engineered PBLs were coincubated with decreasing ratios of 51Cr-labeled target cells [effector-to-target ratio (E:T)] in AIM-V medium in 96-well U-bottom plates at 37°C for 4 hr. Lysis was measured by 51Cr release in the medium: percent lysis=(sample release – minimum release)/(maximum release – minimum release)×100, average of duplicate samples.
Results
Development of a CAR targeting EGFRvIII
GSC lines (Supplementary Fig. S1; Supplementary Data are available online at
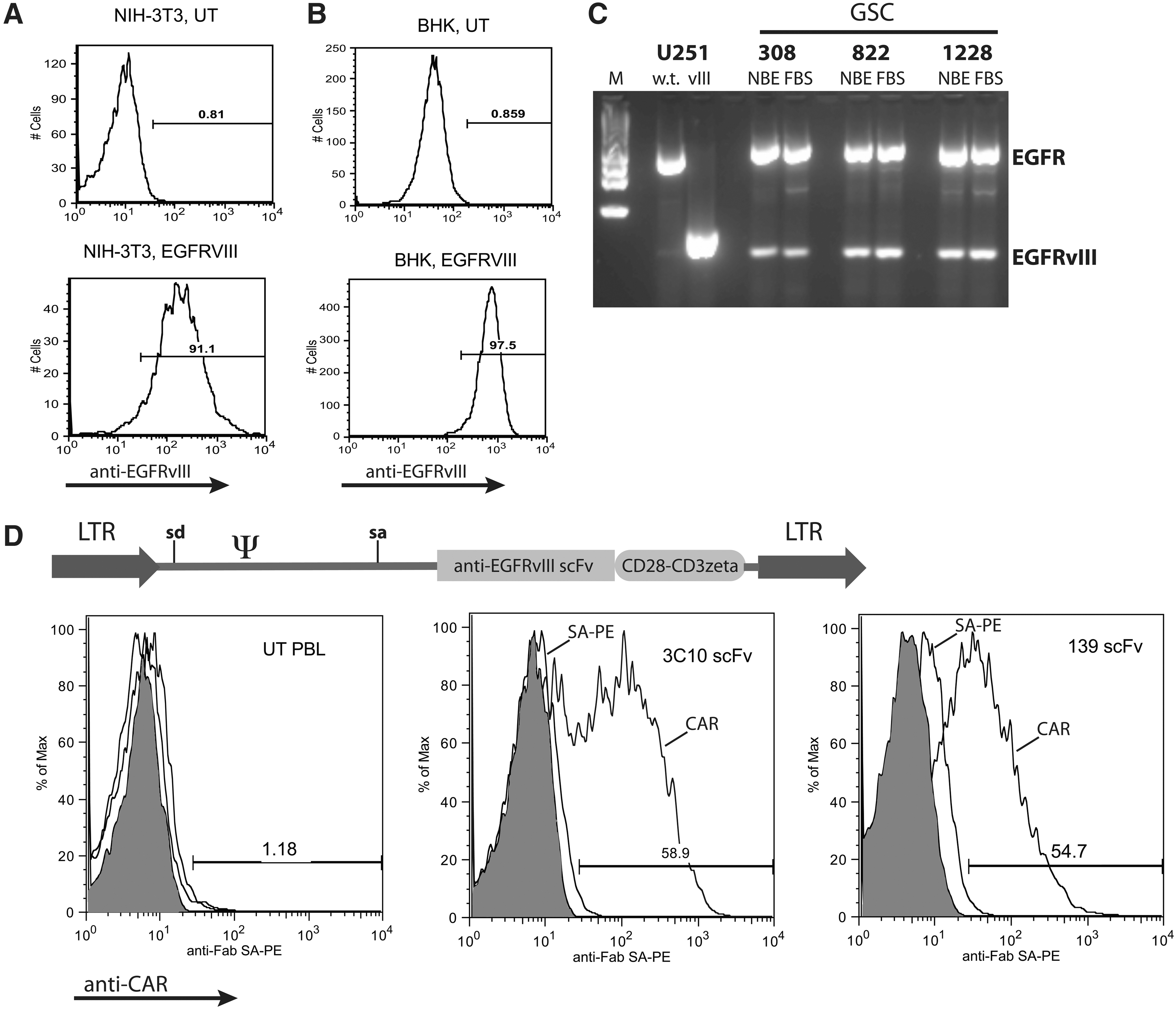
Development of CARs targeting EGFRvIII. NIH-3T3 cells
CARs targeting EGFRvIII were produced by combining single-chain antibody sequences (scFv) from seven different anti-EGFRvIII antibodies to the T-cell signaling domains of CD28 and CD3ζ. A total of nine different constructs were assembled (in two constructs, the order of the VL and VH were alternated) based on murine antibodies 3C10, MR-1, Y10, and L8A4 and human antibodies 131, 139, and 13.1.2, which were inserted into the γ-retroviral vector MSGV1 (Fig. 1D). The expression of each construct was tested by staining transduced PBLs using an anti-Fab–specific reagent (or protein L in later experiments), followed by flow cytometry analysis. Three of the nine vectors constructed demonstrated reproducible CAR expression in transduced PBLs, specifically those CARs based on antibodies 3C10, L8A4, and 139 (Fig. 1D; L8A4-CAR not shown).
To test the biological activity of these three anti-EGFRvIII CAR constructs, γ-retroviral vector supernatant was produced and used to transduce PBLs, which were cocultured with EGFRvIII-expressing target cell lines. Specific IFN-γ production was determined for all three constructs by coculture with EGFRvIII gene-engineered cell lines (Fig. 2A; representative data for cocultures with NIH-3T3 and BHK). In these coculture assays, the CARs based on mAbs 3C10, L8A4, and 139 yielded specific IFN-γ production when exposed to EGFRvIII-expressing target cells, but not cells engineered to overexpress the wild-type EGFR gene. Based on the observation that the 139 CAR was slightly more reactive and is of human origin, and therefore less likely to be immunogenic in patients, all subsequent assays were done with the 139 scFv-based CAR construct. T cells transduced with the 139-CAR were sorted into CD8 and CD4 T-cell populations and independently tested for reactivity. Both CD4 and CD8 T cells specifically produced IFN-γ in coculture with EGFRvIII target cells (Fig. 2B), and CD4+ T cells also produced IL-2 (Fig. 2C).
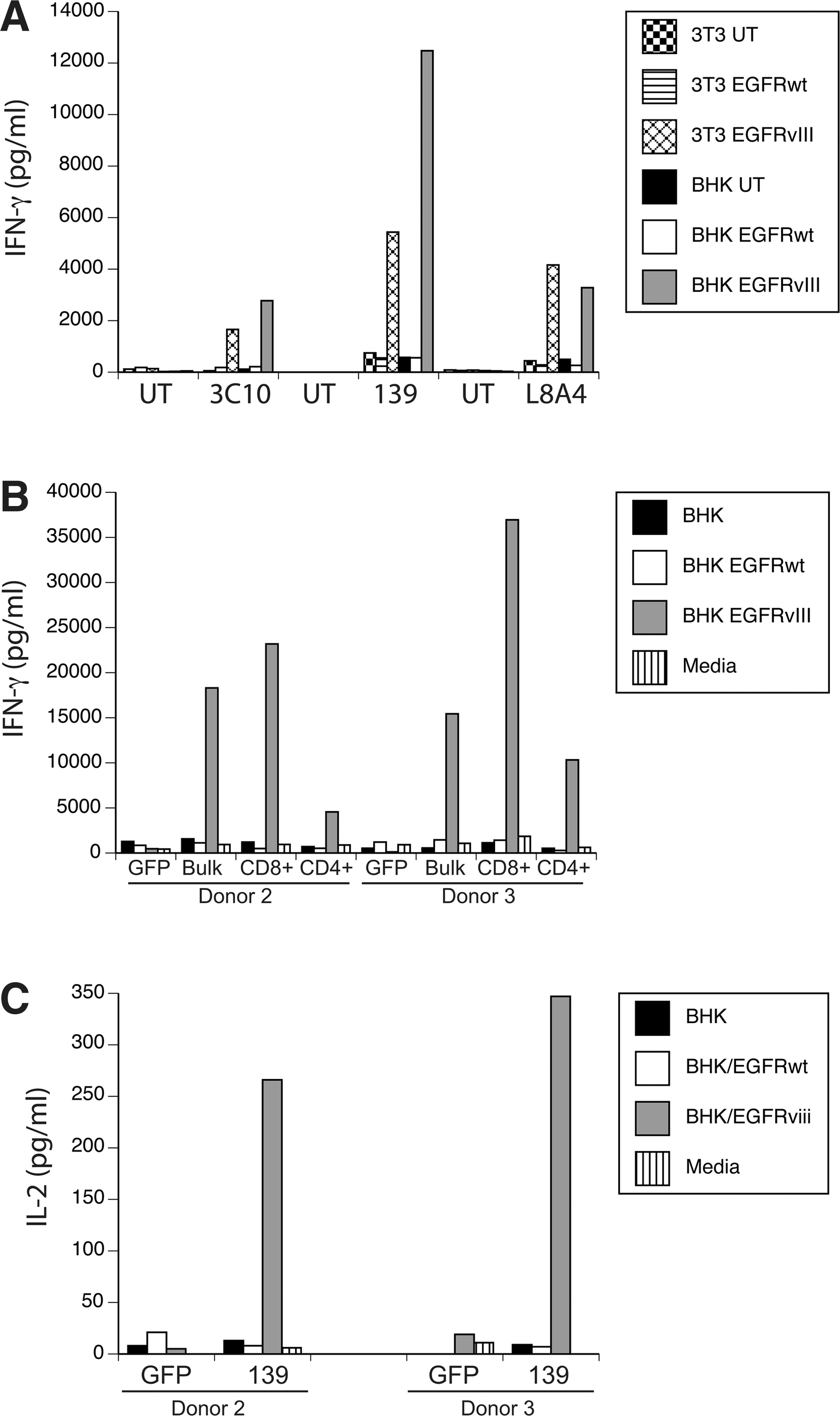
Anti-EGFRvIII CAR vector-engineered T cells specifically recognize EGFRvIII-expressing cells.
A third-generation human mAb-based anti-EGFRvIII CAR specifically recognizes GSCs
A second 139-scFv-based CAR vector was assembled using T-cell signaling domains from CD28-41BB-CD3ζ (named 139-28BBZ) and compared with the original CAR vector that used CD28-CD3ζ signaling domains (named 139-28Z) (Fig. 3). These γ-retroviral vectors were used to transduce human PBLs, and T cells were tested for reactivity in coculture assays using as targets glioblastoma cell lines engineered to express EGFRvIII. Biological activity, as determined by IFN-γ release, demonstrated that the two different vector-transduced T cells were equally reactive against EGFRvIII-expressing glioma cell lines, U87 and U251. We next determined the ability of EGFRvIII CAR-engineered T cells to lyse glioma target cells in a standard 51Cr release assay. As shown in Fig. 3, both vectors specifically lysed only cell lines engineered to express the mutant EGFRvIII and not to control or wild-type EGFR engineered cell lines. Based on our experience, and in publications reported by others, the presence of additional signaling domains from the 4-1BB protein (this CAR design is referred to as a third-generation CAR) is associated with a better survival of CAR-engineered cells in animal models (Zhao et al., 2009; Zhong et al., 2010; Song et al., 2011). Therefore, the construct containing the CD28 and 4-1BB cosignaling elements was chosen for further analysis in cocultures targeting GSC lines.

Equivalent function of second- and third-generation anti-EGFRvIII CAR vectors. Using anti-EGFRvIII human mAb 139, a third-generation CAR vector was assembled using CD28, 4-1BB, and CD3ζ T-cell signaling domains (139-28BBZ, diagram at top of figure). This vector was compared with the original vector design containing CD28 and CD3ζ T-cell signaling elements (139-28Z, diagram at top of figure). Primary human T cells were transduced with both vectors and evaluated for IFN-γ cytokine production following overnight coculture (imbedded tables) and in 4-hr 51Cr release assays to determine cell lysis activity (shown as percent lysis at the indicated E:T). Target cells included: U87 cells transduced with a GFP vector (GFP), wild-type EGFR (EGFRwt), or EGFR variant III (EGFRvIII); and U251 cells transduced with wild-type EGFR (EGFRwt) or EGFR variant III (EGFRvIII). A vector expressing an anti-ERBB2 CAR (ERBB2) was used as a control along with GFP and untransduced (UnTd) cells. Data are representative of three independent experiments. IFN-γ values were determined by ELISA (shown in pg/mL, mean of triplicate determinations), and percent lysis was plotted as the mean of quadruplicate determinations. Differences between recognition of EGFRvIII-transduced versus control lines were statistically significant (p<0.05, Student's t test).
PBLs from two donors were engineered with the third-generation (139-28BBZ) EGFRvIII CAR vector, as well as a control CAR against ERBB2. These engineered T cells were cocultured with GSC lines and control EGFRvIII-expressing cell lines (Fig. 4). EGFRvIII CAR-engineered T cells demonstrated specific recognition of the U251 EGFRvIII, when compared with the U251 EGFR wild-type gene-engineered cells, and recognized all three GSC lines tested (308, 822, and 1228). Data shown in Fig. 4 demonstrate that EGFRvIII CAR 139-28BBZ engineered cells from both donors produced IFN-γ following coculture with the GSC lines equivalent to ERBB2 CAR-transduced T cells and showed specificity for the EGFRvIII glioma tumor antigen.
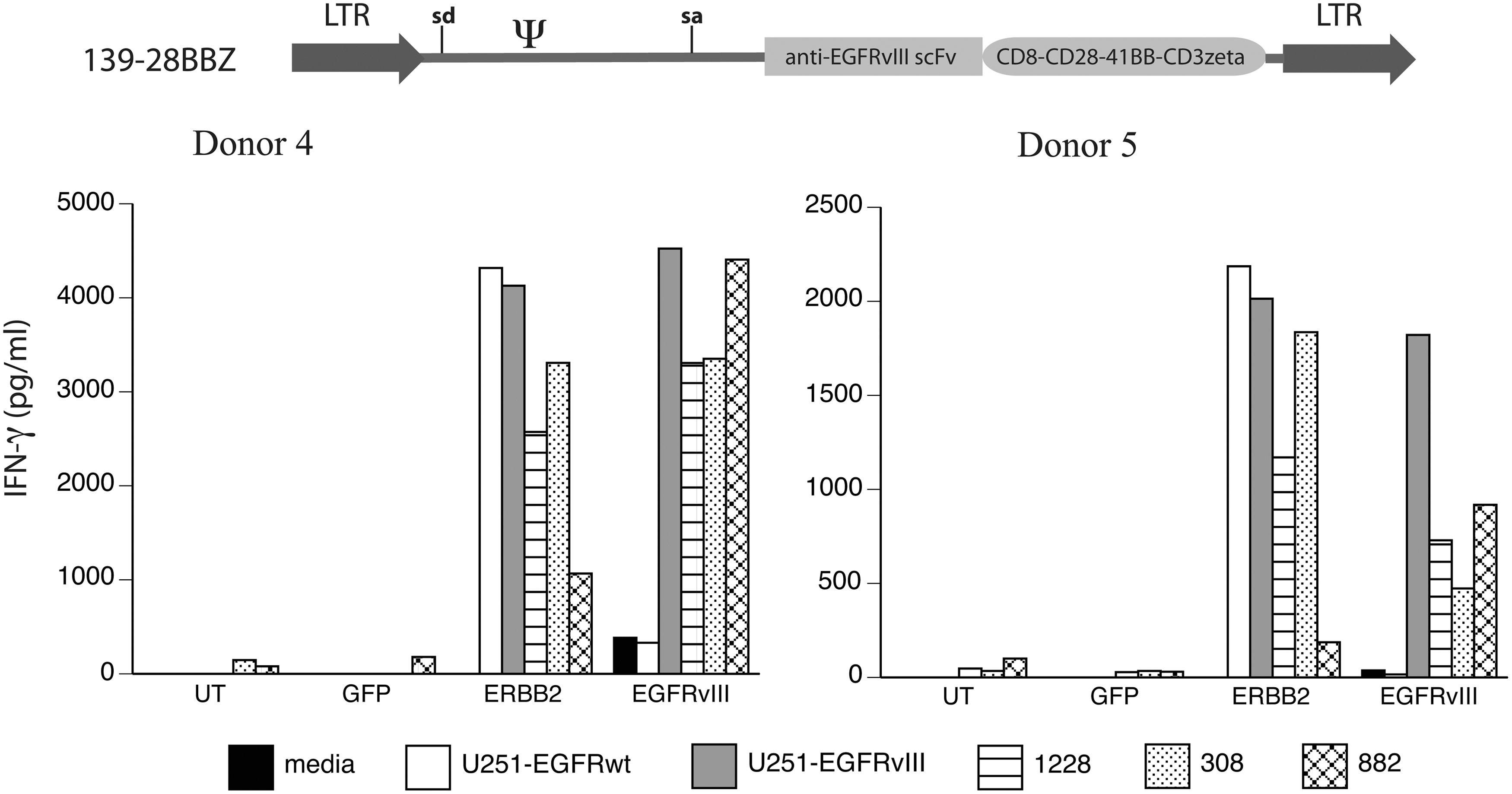
Recognition of GSCs by anti-EGFRvIII CAR-transduced T cells. The third-generation anti-EGFRvIII CAR vector (139-28BBZ) was used to transduce T cells from two donors (donors 4 and 5) along with a GFP-expressing vector (GFP) and a CAR vector targeting ERBB2. Transduced T cells were cocultured with wild-type EGFR-engineered U251 cells (U251-EGFRwt) or EGFR variant III engineered U251 cells (U251-EGFRvIII) and GSC lines 1228, 308, and 822. IFN-γ values were determined by ELISA (shown in pg/mL, mean of triplicate determinations). Data are representative of three independent experiments. Differences between recognition of EGFRvIII-transduced U251 versus EGFRwt-U251 were statistically significant (p<0.01, Student's t test).
Preclinical evaluation
To continue the preclinical evaluation of the EGFRvIII CAR vector, we used the 139-28BBZ vector to transduce PBLs from two glioblastoma patients, as well as a healthy donor, and tested for expression and reactivity. Transduced cells were cocultured with EGFRvIII-engineered U87 cells and then assayed by intracellular cytokine staining. Engineered T cells from both patients and the healthy donor demonstrated specific IFN-γ production in both CD8+ and CD8− (presumably CD4+) CD3+ T cells (7.8–16.2% IFN-γ versus <0.36% against the control U87 line) (Fig. 5A). Transduction efficiency was also similar between the glioblastoma patient T cells and the healthy donor (Fig. 5A, right-side panels). If large numbers of T cells (>1×109) are required for future clinical applications, these can be obtained via a 14-day REP (Riddell and Greenberg, 1990). To verify that 139-28BBZ CAR-transduced T cells could be expanded to numbers sufficient for patient treatment and still maintain reactivity, these T cells were subject to REP and retested. As demonstrated in Fig. 5B (and Supplementary Fig. S2), the 139-CAR-transduced T cells retained their ability to specifically produce IFN-γ as shown here in an Elispot assay.
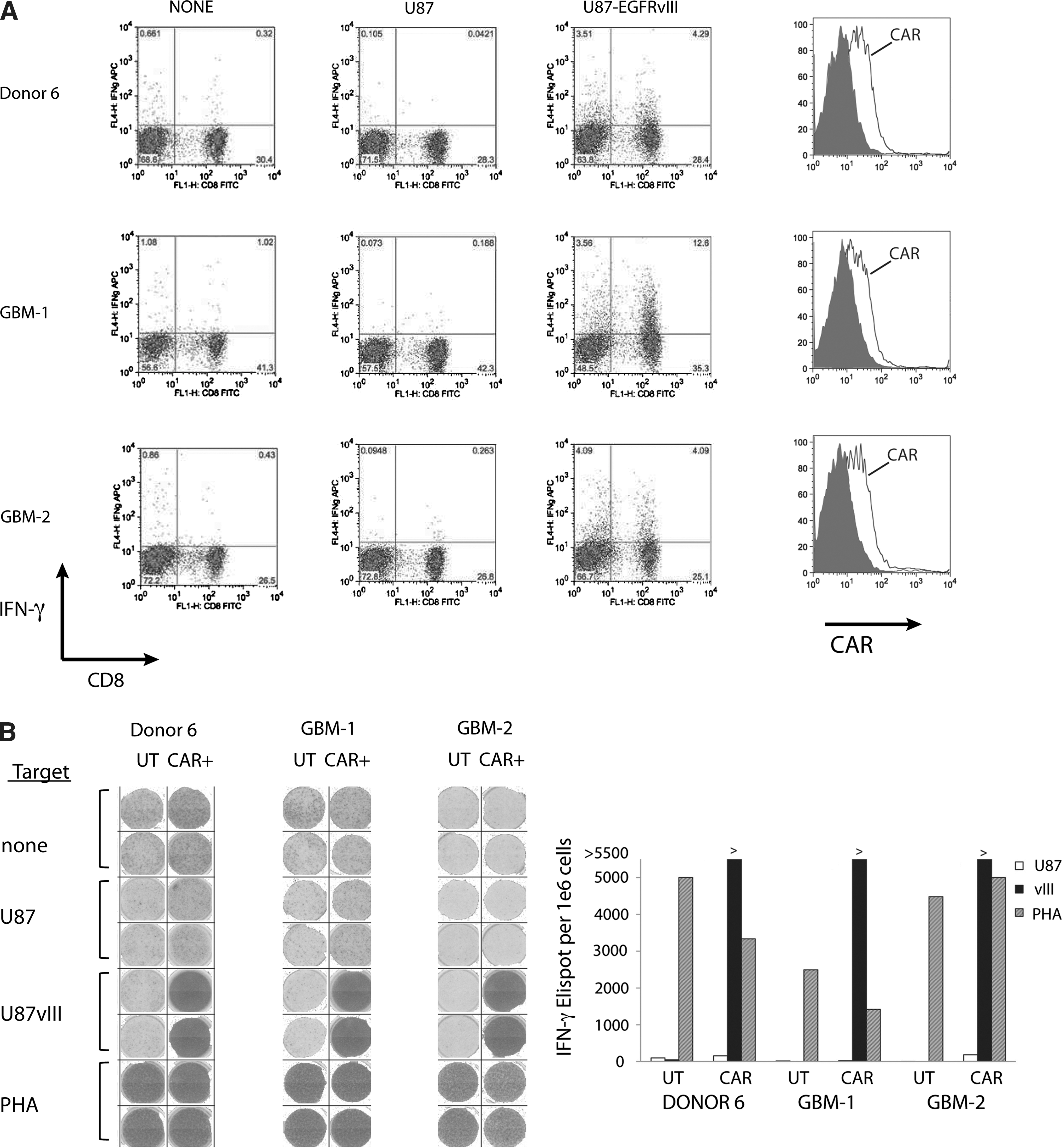
EGFRvIII CAR vector-transduced glioblastoma patient T cells recognize EGFRvIII-expressing tumors.
Using the 139-28BBZ EGFRvIII CAR construct, we prepared a PG13 γ-retroviral vector producer cell clone under conditions that meet FDA guidelines for human gene therapy clinical trials. One cell clone (clone F10) was use to produce 18 L of viral vector supernatant in six harvests collected over 4 days. Each harvest was used to transduce donor PBL, and the gene transfer efficiency and biologic activity were determined (Fig. 6A). All harvests produced biologically active supernatant based on the ability of transduced T cells to express the CAR and to specifically recognize EGFRvIII-expressing cell lines. Harvest 1 was slightly less reactive than harvests 2–6 in this assay. To test for possible toxicity against normal human tissues, a pool of harvests 3 and 4 was used to transduce a different donor, and these transduced T cells were cocultured with seven different primary human adult and neonatal cell cultures of epithelial, endothelial, and fibroblast origin. As determined by IFN-γ production, there was no reactivity of the EGFRvIII CAR-transduced T cells with any primary human cell culture tested (Fig. 6B).
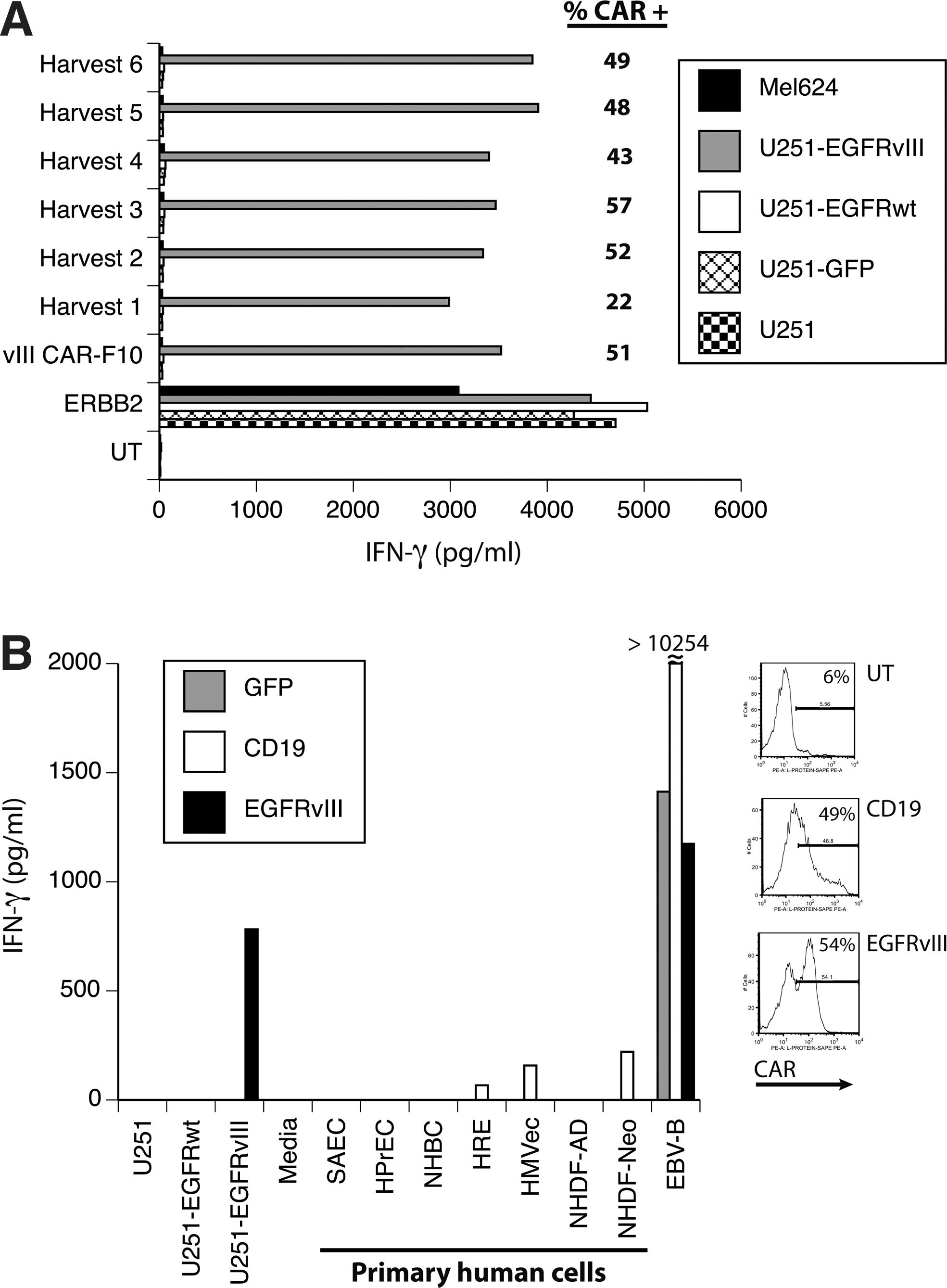
Development of an anti-EGFRvIII CAR vector for clinical applications.
Discussion
Immunotherapy for glioma has been investigated by a number of groups (Johnson and Sampson, 2010), but one of the underlining assumptions in these studies was that the target cells for immunological assays (glioma cell lines) were representative of primary tumors, but this may not be the case. By detailed molecular analysis of many different glioma cell lines, it has now been demonstrated that established glioma cell lines do not mirror the molecular characteristics of primary human brain tumors (Venere et al., 2011). An alternative to the use of established glioma cell lines is the analysis of GSC lines. We have previously demonstrated that in situ glioma cells share properties not found in glioma cell lines and harbor features consistent with TSCs (Lee et al., 2006). We further demonstrated marked phenotypic and genotypic differences between primary human tumor–derived GSCs and their matched glioma cell lines. GSCs derived directly from primary glioblastomas harbor extensive similarities to normal neural stem cells and recapitulate the genotype, gene expression patterns, and in vivo biology of human glioblastomas (Lee et al., 2006; Liu et al., 2006; Venere et al., 2011). These findings suggest that GSCs may be a more reliable model than commonly utilized glioma cell lines for the development of therapies for these primary human brain tumors. In our hunt for immunological targets in GSC lines, we discovered that they express EGFRvIII.
EGFRvIII is one of the few tumor-specific antigens and thus, potentially, an excellent candidate for the development of immunotherapy for glioblastoma patients. Preclinical studies using mAbs and small-molecule inhibitors against EGFRvIII have provided modest results; however, only peptide vaccines against EGFRvIII are currently offered in experimental clinical trials. A phase I clinical trial in patients with newly diagnosed glioblastoma demonstrated the safety of vaccination in this population using dendritic cells loaded with an EGFRvIII peptide vaccine (Sampson et al., 2008). The safety of EGFRvIII peptide vaccines in conjunction with standard care using temozolomide chemotherapy has also been established. These various trials have demonstrated tolerability of peptide vaccines and documented their ability to elicit antigen-specific cellular and humoral responses; however, the effectiveness of any of these approaches has yet to be proven, and, most recently, the induction of low-avidity T-cell responses by peptide vaccination was suggested to be associated with EGFRvIII antigen loss and disease progression in some patients (Sampson et al., 2010).
There has also been preclinical reports on adoptive immunotherapy using EGFRvIII-targeted T cells. Bullian and colleagues created an EGFRvIII-specific CAR, MR1-ζ, and demonstrated that human T cells genetically modified with MR1-ζ specifically recognized EGFRvIII-expressing tumor cell lines and delayed tumor growth in vivo in mouse models (Bullain et al., 2009). Similarly, Ohno and colleagues engineered T cells expressing a CAR against EGFRvIII based on a mouse mAb, 3C10; they demonstrated in vivo inhibition of intracranial tumor growth in mice (Ohno et al., 2010). Although suggestive of potential clinical efficacy, immunodeficient-mouse models based on the treatment of artificially engineered human tumor xenografts are unlikely to replicate the multitude of factors to be encountered in human applications (Morgan, 2012). In the case of EGFRvIII-expressing glioma cell lines, we feel that murine models add minimal value to the development of immunotherapy protocols aimed at treating patients with recurrent glioblastoma. Based on the data reported here and the investigations of others, we have proposed a human gene therapy clinical trial to treat recurrent glioblastoma using infusions of autologous T cells engineered with the anti-EGFRvIII CAR based on human mAb 139. Although we recognize the dangers of giving a nonmyleoablative chemotherapy and IL-2 to patients with recurrent brain tumors (e.g., capillary leak syndrome and thrombocytopenia), we are encouraged by our experience treating metastatic tumors to the brain in patients with melanoma using TILs along with IL-2 following the same preparative regimen to be used in our proposed trial (Hong et al., 2010). Seven of 17 melanoma patients (41%) achieved a complete response in the brain following TIL therapy. In addition, two of nine patients treated using TCR gene-transduced cells (anti-MART-1 or anti-gp100) achieved a complete response. One of the 26 patients experienced a tumor-associated subarachnoid hemorrhage during the thrombocytopenic phase of therapy and had an uneventful metastasectomy.
The targeting of GSCs by gene-engineered T cells may have potential benefits in addition to the T cell–mediated destruction of mature glioma cells. GSCs have been reported to promote tumor angiogenesis and to be resistant to chemotherapy and radiotherapy (Singh et al., 2004; Bao et al., 2006a,b; Liu et al., 2006). These properties may be associated with the near 100% recurrence rate of glioblastoma patients following standard-of-care treatment with surgery, radiation, and temozolomide chemotherapy. Our experience with TILs and TCR/CAR gene therapy in patients with melanoma has demonstrated that T cells can mediate an immune response that results in the elimination of the last cancer-initiating cell, resulting in long-term complete responses. Thus, a clinical trial using adoptive cell therapy with EGFRvIII CAR gene-engineered T cells in patients with recurrent glioblastoma was reviewed and approved by the appropriate U.S. regulatory agencies and is currently accruing patients (
Footnotes
Acknowledgments
This work is supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. L.A.J. was supported by Voices Against Brain Cancer (VABC). We thank Pamela Norberg for technical assistance. Experiments were performed by R.A.M., L.A.J., J.L.D., Z.Z., S.A.F., and N.C. Reagents and other experimental support was provided by K.D.W., C.T.K., H.S., and W.Z. The manuscript was written by R.A.M., L.A.J., K.D.W., H.A.F., and S.A.R.
Author Disclosure Statement
The authors declare that they have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.