
Abstract
Hemophilia A and B are X-linked monogenic disorders caused by deficiencies in coagulation factor VIII (FVIII) and factor IX (FIX), respectively. Current treatment for hemophilia involves intravenous infusion of clotting factor concentrates. However, this does not constitute a cure, and the development of gene-based therapies for hemophilia to achieve prolonged high level expression of clotting factors to correct the bleeding diathesis are warranted. Different types of viral and nonviral gene delivery systems and a wide range of different target cells, including hepatocytes, skeletal muscle cells, hematopoietic stem cells (HSCs), and endothelial cells, have been explored for hemophilia gene therapy. Adeno-associated virus (AAV)-based and lentiviral vectors are among the most promising vectors for hemophilia gene therapy. Stable correction of the bleeding phenotypes in hemophilia A and B was achieved in murine and canine models, and these promising preclinical studies prompted clinical trials in patients suffering from severe hemophilia. These studies recently resulted in the first demonstration that long-term expression of therapeutic FIX levels could be achieved in patients undergoing gene therapy. Despite this progress, there are still a number of hurdles that need to be overcome. In particular, the FIX levels obtained were insufficient to prevent bleeding induced by trauma or injury. Moreover, the gene-modified cells in these patients can become potential targets for immune destruction by effector T cells, specific for the AAV vector antigens. Consequently, more efficacious approaches are needed to achieve full hemostatic correction and to ultimately establish a cure for hemophilia A and B.
Introduction
As a monogenetic hereditary disorder, hemophilia has been considered to be a particularly significant target for gene therapy. Most aspects of the disease, including its genetics are quite well understood. Introduction of a functional FVIII or FIX gene copy into the target cells via gene therapy may provide a cure and eliminate the need for repeated clotting factor infusions. One of the hallmarks of hemophilia treatment is that there is no need to normalize circulating clotting factor levels to obtain a therapeutic effect since a slight increase in plasma clotting factor levels (above 1% threshold) suffices to reduce the risk of mortality and morbidity. Consequently a modest increase in clotting factor levels by gene therapy can profoundly improve the clinical symptoms. Moreover, therapeutic efficacy of gene therapy can be relatively easily ascertained based on robust clinical endpoints such as circulating clotting factor levels and bleeding frequency. Finally, there are several well-validated small and large hemophilic animal models available for preclinical testing.
Gene therapy for hemophilia aims at achieving prolonged high level expression of clotting factors to improve the clinical phenotype. Essentially, there are two distinct strategies to achieve this goal: (i) deliver the clotting factor genes into stem cells using integrating (viral or nonviral) vectors in order to achieve long-lasting expression through their progeny, and (ii) deliver the clotting factor genes into long-lived postmitotic cells, such as skeletal muscle cells or hepatocytes. In this case, the therapeutic genes should either persist as episomal DNA or integrated into the target cell genome. There are many comprehensive reviews on gene therapy for hemophilia (Pierce et al., 2007; High, 2001, 2011; Mátrai et al., 2010a; VandenDriessche and Chuah, 2012). The objective of the current review is to summarize and discuss the most salient and recent developments in this field. In particular, we will discuss current bottlenecks and outstanding questions that need to be addressed to ultimately achieve a cure.
Retroviral and Lentiviral Vectors
Liver-directed gene transfer
The liver is the main physiological site of FVIII and FIX synthesis; hence hepatocytes are well-suited target cells for hemophilia gene therapy. From this location, FVIII and FIX protein can easily enter into the circulation. The hepatic niche may favor the induction of immune tolerance towards the transgene product. The induction of immune tolerance may also, at least in part, depend on the induction of regulatory T cells. This antigen-specific immune tolerance to the transgene product could be a key factor in establishing long-term therapeutic clotting factor levels (Mingozzi et al., 2003). Another advantage of targeting liver cells is that the posttranslational modifications faithfully replicate the normal FVIII or FIX processing pathways.
Retroviral vectors derived from Moloney murine leukemia virus (MoMLV) can transduce a wide variety of target cells and integrate into the host genome, provided the target cells are actively dividing. MoMLV-derived vectors were among the first vectors to be considered for hemophilia gene therapy (Axelrod et al., 1990). The first proof-of-concept that hemophilia A mice could be cured by gene therapy had been established with γ-retroviral vectors after hepatic gene delivery in neonatal mice (VandenDriessche et al., 1999). Similarly, efficient γ-retroviral transduction could be obtained in the liver of neonatal hemophilia A and B dogs (Xu et al., 2003, 2005). Based on long-term expression data in adult rabbits following systemic administration of γ-retroviral vectors encoding FVIII, a phase I clinical trial was conducted in adults with severe hemophilia A. Though the procedure was well tolerated by the patients, only a transient low rise in FVIII activity could be detected in sporadic cases (Powell et al., 2003). The lack of efficacy in that trial could largely be ascribed to the inability of γ-retroviral vectors to transduce nondividing cells. However, it is unclear why the preclinical rabbit studies differed from the human, mouse, and dog studies.
To overcome the need for active cell division to achieve efficient transduction, lentiviral vectors were developed (Mátrai et al., 2010b). These vectors can transduce quiescent noncycling hepatocytes in vivo, leading to relatively efficient gene transfer of the adult liver after local or even after systemic gene delivery. A point of concern regarding liver-directed gene transfer would be to avoid unintentional transduction of antigen-presenting cells (APCs) present in the liver microenvironment, since ectopic FVIII or FIX expression in APCs increases the risk of inducing an immune response against the transduced cells and the resultant transgene product (VandenDriessche et al., 2002). Inadvertent transgene expression in APCs can increase the risk of immune responses against the clotting factors, inhibiting long-term expression. In particular, antibodies to FIX could be detected when ubiquitously expressed promoters were used, whereas the utilization of a hepatocyte-specific promoter reduced this immunological risk, consistent with long-term FIX expression (Follenzi et al., 2004; VandenDriessche et al., 2007). In addition, T-cell responses against the gene-modified cells may occur that could ultimately result in their elimination by the immune system. By including an additional layer of posttranscriptional control, mediated by the use of endogenous microRNA (miR), nonspecific expression in APCs could be further reduced, resulting in prolonged transgene expression and persistence of gene-modified cells (Brown et al., 2007; Mátrai et al., 2011) (Fig. 1). The mechanism of immune tolerance induction following hepatic FVIII or FIX delivery with lentiviral vectors may require induction of CD4+CD25+Foxp3+ regulatory T cells (Matsui et al., 2009; Annoni et al., 2009).

Gene therapy for hemophilia using micro-RNA (miR)-regulated lentiviral vectors. Ectopic transgene expression in APCs results in the induction of an antigen-specific T cell–dependent immune response that consequently eliminates the gene-engineered APCs and hepatocytes that express the transgene product (i.e., factor IX [FIX] or reporter protein such as GFP). To avoid ectopic transgene expression in APCs, miR target sequences can be incorporated in the lentiviral vectors that are complementary to an APC-specific miR (i.e., miR142-3p). The specific interaction of the APC-specific miR with its cognate miR target sequence embedded within the mRNA encoded by the vector results in its degradation via the cellular miR-processing machinery. This prevents inadvertent FIX (or GFP) expression in APC. Since miR142-3 is not expressed in hepatocytes, this specific degradation of the FIX mRNA does not occur in hepatocytes. Consequently, there is no induction of an antigen-specific T-cell response, and the lentivirally transduced APCs and hepatocytes are able to persist resulting in long-term expression of the gene of interest. APC, antigen-presenting cell; cPPT, central polypurine tract; GFP, green fluorescent protein; LV, lentiviral vector; WPRE, Woodchuck post-transcriptional regulatory element.
Nevertheless, miR regulation may not necessarily suffice to induce immune tolerance in all circumstances, and additional studies are required to address these outstanding issues. In particular, even if ectopic transgene expression in APCs is eliminated, it is still possible that immune responses to the transgene product can be induced (Matsui et al., 2011). Indeed, the efficiency of immune tolerance induction by miR regulation may vary depending on the transgene product, the target organ, and the underlying mutation of the defective endogenous gene. In particular, it is typically more challenging to induce immune tolerance in the context of a null mutation than when a missense mutation has occurred in the affected gene. In any case, though miR regulation may impact on the adaptive immune response against the transduced cells and/or the therapeutic proteins, it will not eliminate the innate immune response induced by the lentiviral particles themselves.
The most important safety concern related to γ-retroviral and lentiviral vectors is that of insertional mutagenesis and oncogene activation (or tumor-suppressor gene inactivation) resulting from genomic integration (Mátrai et al., 2010b). The vector design or the presence of transcriptionally active long terminal repeat (LTR) can influence this genotoxic risk. Removal of approximately 400 bp in the LTR region to abolish its transcriptional activity (i.e., self-inactivating vector or SIN design) coupled with the use of a moderately active promoter in an internal position may lower the risk of insertional oncogenesis. By contrast, the potential oncogenic risk of lentiviral vectors may be reduced compared to γ-retroviral vectors because they integrate more distantly from transcriptional start sites. An alternative approach to minimize the risk of insertional mutagenesis is the use of integration defective lentiviral vectors (IDLVs) that harbor an inactivating mutation in the integrase. Hepatocyte-targeted expression using IDLVs resulted in the sustained and robust induction of immune tolerance to both intracellular and secreted proteins, like FIX, despite the reduced transgene expression levels in comparison with their integrase-competent vector counterparts (Mátrai et al., 2011). IDLV-mediated and hepatocyte-targeted FIX expression prevented the induction of neutralizing antibodies to FIX even after antigen rechallenge in hemophilia B mice and accounted for relatively prolonged therapeutic FIX expression levels, at about 2% of normal levels. Upon the delivery of intracellular model antigens, hepatocyte-targeted IDLVs induced transgene-specific regulatory T cells that contributed to the observed immune tolerance. Deep sequencing of IDLV-transduced livers showed only rare genomic integrations that occurred mostly by a mechanism inconsistent with residual integrase activity. However, since hepatic transgene expression is typically reduced with IDLVs compared to their integration-competent counterparts, we engineered the FIX transgene itself to boost the levels of active FIX protein. By incorporating a hyper-activating mutation in a codon-optimized FIX, we obtained sustained levels 20% of normal cFIX activity, which is 20-fold higher than the therapeutic threshold of 1% active FIX (VandenDriessche et al., unpublished observations). This indicates that IDLVs provide an attractive platform for the tolerogenic expression of intracellular or secreted proteins in the liver with a substantially reduced risk of insertional mutagenesis.
Stem cell–based gene transfer
Alternatively, γ-retroviral and lentiviral vectors could be used for ex vivo gene transfer into stem cells, particularly hematopoietic stem cells (HSCs) and other stem/progenitor cells populations (reviewed in Mátrai et al., 2010a, 2010b). HSCs are an attractive source of target cells for hemophilia gene therapy because they can self-renew and differentiate into all blood lineages during hematopoiesis. Another advantage is their accessibility for genetic modification and ease of transplantation. Furthermore, there is a possibility of inducing immune hyporesponsiveness to the transgene product. HSC-derived erythrocytes, megakaryocytes, and their platelet pogeny could then serve as a delivery platform to secrete the clotting factors in the circulation following HSC-targeted gene transfer.
Targeting hematopoietic cells by ex vivo transduction, followed by their transplantation, allows for direct access of the clotting factors to the circulation. Initially, transplantations of bone marrow cells transduced ex vivo with FVIII-encoding γ-retroviral vectors failed to show clinically relevant FVIII plasma levels (Hoeben et al., 1992). Nonetheless, these levels were sufficient to induce immunological tolerance to the transgene product (Evans et al., 1998). Recently, however, advances in methodology could result in sustained phenotypic correction of hemophilia A mice, after γ-retrovirus–mediated FVIII expression in transplanted bone marrow cells (Moayeri et al., 2004; Ramezani et al., 2009). Moreover, no FVIII antibodies developed after transplantation of transduced bone marrow cells, not even after immunological challenge (Moayeri et al., 2005). Similarly, sustained curative FVIII activity could be demonstrated in hemophilia A mice after transplantation of transduced HSCs encoding a (B-domain deleted) porcine FVIII transgene, with evidence for immunological nonresponsiveness to FVIII (Ide et al., 2007, 2010).
Long-term therapeutic FVIII expression could also be demonstrated in hemophilia A mice after transplantation of lentivirally transduced HSCs encoding a hybrid human–porcine FVIII transgene (Spencer et al., 2010). Remarkably, this hybrid transgene could significantly increase FVIII expression levels compared to the (human) FVIII transgene. Using lentiviral transduction of HSCs, FVIII and FIX expression can be specifically directed to platelets, resulting in phenotypic correction of the bleeding diathesis in hemophilia A or B mice (Shi et al., 2007; Montgomery and Shi, 2012). Remarkably, phenotypic correction of hemophilia A mice could be achieved in the presence of high-titer inhibitory antibodies after lentiviral platelet-directed (human) FVIII expression (Kuether et al., 2012; Chuah and VandenDriessche, 2012). These strategies could be especially beneficial to treat hemophilia patients with inhibitors. To facilitate HSC engraftment and create a “niche” in the bone marrow, it is necessary to condition the stem cell recipients using busulfan or other conditioning regimens. However, since conditioning is not without side effects, the risk/benefit ratio would need to be carefully assessed in the context of HSC-based hemophilia gene therapy. In addition to HSC, other stem/progenitor cells, and in particular endothelial progenitors, are attractive targets for hemophilia gene therapy by virtue of their expression of von Willebrand factor that helps to stabilize FVIII (Matsui et al., 2007). However, efficient and persistent engraftment of these endothelial progenitors remains challenging.
Adeno-Associated Viral Vectors
Adeno-associated viruses (AAVs) are naturally occurring replication-defective nonpathogenic viruses with a single-stranded DNA genome. AAV vectors have a favorable safety profile and are capable of achieving persistent transgene expression. Long-term expression is predominantly mediated by episomally retained AAV genomes. More than 90% of the stably transduced vector genomes are extrachromosomal, mostly organized as high molecular weight concatemers. Therefore, the risk of insertional oncogenesis is minimal, especially in the context of hemophilia gene therapy in which no selective expansion of transduced cells is expected to occur. Nevertheless, oncogenic events have been reported following AAV-based gene transfer (Donsante et al., 2007), but it has been difficult to reproduce these findings in other model systems (Li et al., 2011b). The major limitation of AAV vectors is the limited packaging capacity of the vector particles (i.e., approximately 4.7 kb), constraining the size of the transgene expression cassette to obtain functional vectors. Several immunologically distinct AAV serotypes have been isolated from human and nonhuman primates (Gao et al., 2004), although most vectors for hemophilia gene therapy were initially derived from the most prevalent AAV serotype 2. The first clinical success of AAV-based gene therapy for congenital blindness underscores the potential of this gene transfer technology (Bainbridge et al., 2008).
Muscle-directed gene transfer
The muscle was one of the first targets for AAV-mediated gene therapy for hemophilia B (High et al., 2011). Muscle does not normally express FVIII or FIX. It is an easily accessible site with a robust secretory capacity. Muscle cells are also equipped with the cellular machineries for posttranslational modifications to generate functional transgene product. Nevertheless, the proteolytic cleavage and glycosylation are slightly impaired compared to hepatocytes. Muscle-directed gene transfer in hemophilia B dogs with a missense mutation resulted in stable FIX expression (Herzog et al., 1999). By contrast, dogs with a FIX null mutation formed high-titer inhibitors even at low doses of the same vector (Herzog et al., 2001). These results clearly indicate that the underlying FIX mutation impacts on the risk of inhibitor formation. Furthermore, the likelihood of inhibitor development appeared to rise with increasing AAV vector doses and correlated most strongly with dose per site of intramuscular injection (Herzog et al., 2002). It was demonstrated that a local immune response was largely responsible for the induced antibody response, with local FIX antigen doses in the transduced muscle being a critical parameter (Wang et al., 2005). Transient immune suppression (Fields et al., 2001; Herzog et al., 2001) or limitation of vector dose per site (Herzog et al., 2002) sufficed to prevent the development of anti-FIX antibodies.
Translational studies in hemophilic animal models (Herzog et al., 1997, 1999; High et al., 2011) revealed stable FIX expression levels and phenotypic correction of the bleeding diathesis. This justified a phase I clinical trial in patients suffering from severe hemophilia B. Subjects were injected repeatedly into the skeletal muscle using AAV vectors that expressed FIX from a ubiquitous cytomegalovirus promoter. Although FIX expression could be detected at the site of injection in all patients, circulating FIX levels were mostly below the therapeutic range. Nevertheless, expression was sustained even 10 years posttreatment in at least one of the subjects treated (Buchlis et al., 2012). It is encouraging that no antibodies to the FIX transgene product were ever observed, though the subjects that were enrolled in this trail did not have a history of inhibitors to begin with. This clinical trial supports the feasibility of long-term muscle-directed gene transfer, if patients are carefully selected and only those with a missense mutation are enrolled. However, the vector dose per site was limiting, which contributed to the low systemic FIX expression levels. An intravascular delivery system of AAV-FIX vectors to skeletal muscle was developed, to obviate concerns associated with multiple localized intramuscular injections (Arruda et al., 2010). This procedure resulted in widespread muscular transduction and sustained dose-dependent therapeutic levels of canine FIX transgene in the hemophilia B dog model, yielding circulating FIX levels that were up to 10-fold higher than those obtained by localized intramuscular delivery. Transient immunosuppression prevented inhibitory antibody development, whereas a transient inhibitor was detected following vector delivery with no immunosuppression. Increased FIX levels and reduced bleeding time correlated with a significant reduction of spontaneous bleeding episodes. This preclinical study demonstrates the feasibility of this intravascular approach for the treatment of hemophilia B and highlights the importance of transient immune suppression to prevent FIX-specific inhibitor development.
Liver-directed gene transfer
AAV-mediated hepatic gene transfer is an attractive alternative for gene therapy of hemophilia. Preclinical studies with the AAV vectors in murine and canine models of hemophilia or nonhuman primates have demonstrated persistent therapeutic expression, leading to partial or complete correction of the bleeding phenotype in the hemophilic models (Snyder et al., 1997, 1999; Wang et al., 1999, 2000; Mount et al., 2002; Nathwani et al., 2002). Particularly, hepatic transduction conveniently induces immune tolerance to FIX that required induction of regulatory T cells (Tregs) (Mingozzi et al., 2003; Dobrzynski et al., 2006). Long-term correction of the hemophilia phenotype without inhibitor development was achieved in inhibitor-prone null mutation hemophilia B dogs treated with liver-directed AAV2-FIX gene therapy (Mount et al., 2002). In order to further reduce the vector dose, more potent FIX expression cassettes have been developed. This was accomplished by using stronger promoter/enhancer elements, codon-optimized FIX, or self-complementary, double-stranded AAV vectors (scAAV) that overcome one of the limiting steps in AAV transduction (i.e., single-stranded to double-stranded AAV conversion) (McCarty et al., 2001, 2003; Nathwani et al., 2006; Wu et al., 2008). Alternative AAV serotypes could be used (e.g., AAV8) that result in increased transduction into hepatocytes, improve intranuclear vector import and reduce the risk of T-cell activation (Gao et al., 2002; Vandenberghe et al., 2006). Liver-directed gene therapy for hemophilia B with AAV8 or AAV9 is more efficient than when lentiviral vectors are used, at least in mice, and resulted in less inflammation (VandenDriessche et al., 2006). Furthermore, recent studies indicate that mutations of the surface-exposed tyrosine residues allow the vector particles to evade phosphorylation and subsequent ubiquitination and thus prevent proteasome-mediated degradation, which resulted in a 10-fold increase in hepatic expression of FIX in mice (Zhong et al., 2008). As far as FVIII is concerned, the packaging constraints of AAV initially hampered the production of AAV vectors for hemophilia A gene therapy due to the large size of the FVIII transgene. However, this limitation was eventually overcome by using small regulatory elements to drive expression of a B-domain–deleted form of FVIII (Jiang et al., 2006). AAV vector–mediated gene transfer resulted in sustained FVIII expression in mice and dog, though high vector doses were required.
These liver-directed preclinical studies paved the way toward the use of AAV vectors for clinical gene therapy in patients suffering from severe hemophilia B. Hepatic delivery of AAV-FIX vectors resulted in transient therapeutic FIX levels (maximum 12% of normal levels) in subjects receiving AAV-FIX by hepatic artery catheterization (Kay et al., 2000). However, the transduced hepatocytes were able to present AAV capsid-derived antigens in association with MHC class I to T cells (Manno et al., 2006, Mingozzi et al., 2007). Although antigen presentation was modest, it was sufficient to flag the transduced hepatocytes for T cell–mediated destruction. Recently, gene therapy for hemophilia made an important step forward (Nathwani et al., 2011; commentary by VandenDriessche and Chuah, 2012). Subjects suffering from severe hemophilia B (<1% FIX) were injected intravenously with scAAV8 vectors expressing codon-optimized FIX from a liver-specific promoter. This AAV8 serotype exhibits reduced cross-reactivity with pre-existing anti-AAV2 antibodies. Interestingly, its uptake by dendritic cells may be reduced compared to conventional AAV2 vectors, resulting in reduced T-cell activation (Vandenberghe et al., 2006). In mice, AAV8 allows for a substantial increase in hepatic transduction compared to AAV2, though this advantage may be lost in higher species, like dog, rhesus monkeys, and man. Subjects received escalating doses of the scAAV8-FIX vector, with two participants per dose. All of the treated subjects expressed FIX above the therapeutic 1% threshold for several months after vector administration, yielding sustained variable expression levels (i.e., 2% to 11% of normal levels). The main difference with the previous liver-directed AAV trial is that for the first time sustained therapeutic FIX levels could be achieved after gene therapy. Despite this progress, T cell–mediated clearance of AAV-transduced hepatocytes remains a concern consistent with elevated liver enzyme levels in some of the patients. Transient immune suppression using a short course of glucocorticoids was used in an attempt to limit this vector-specific immune response (Fig. 2).
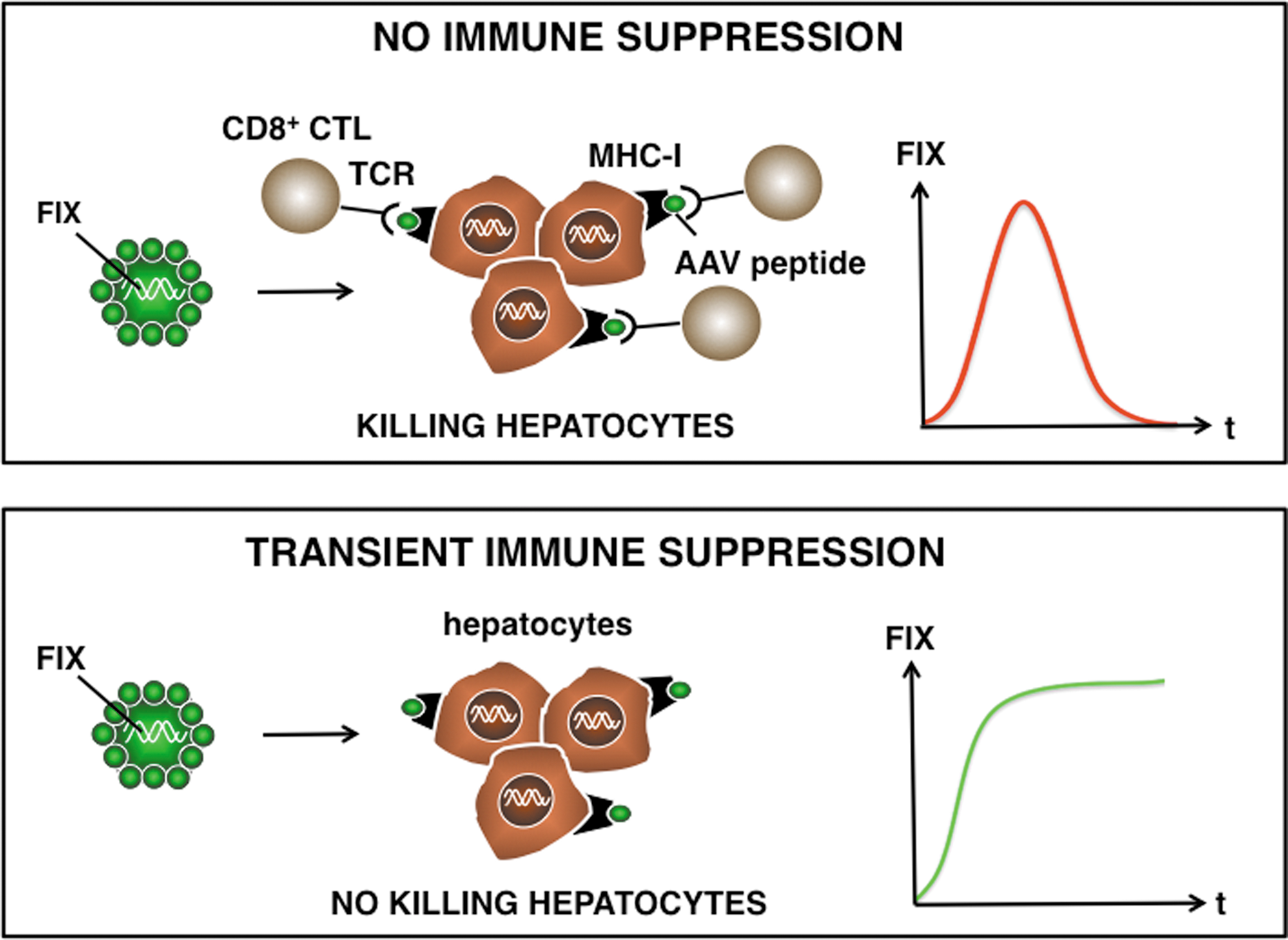
Hypothetical model: AAV-based gene therapy for hemophilia. AAV-based gene therapy may evoke AAV capsid-specific T-cell responses that eliminate the gene-modified cells, such as hepatocytes. These AAV capsid antigenic peptides are derived from the incoming vector particles and presented in association with MHC-I to the TCR of the AAV-specific T cells. Consequently, this would result in a transient production of FIX and short-term correction of the bleeding diathesis. In contrast, transient immune suppression may prevent AAV capsid-specific T cell–mediated immune rejection of AAV-FIX (or FVIII) transduced hepatocytes, resulting in sustained FIX expression. AAV, adeno-associated virus; CTL, cytotoxic T cell; TCR, T-cell receptor; MHC-I, major histocompatibility complex class I antigen.
This original study raises several outstanding questions. Intriguingly, several participants that received the intermediate vector dose had no elevated liver enzyme levels despite an increased AAV8 capsid-specific T-cell response. This suggests that the detection of a vector-specific T-cell response in the peripheral blood may not necessarily result in the elimination of transduced hepatocytes and concomitant liver toxicity. This may perhaps be due to differences in the liver micro-environment and cytokine milieu that could impact on the local T-cell response. Alternatively, since MHC class I presentation of AAV capsid-derived peptides may be proportionate to the vector dose, the intermediate dose may not have sufficed to flag the hepatocytes for T cell–mediated destruction, in contrast to the higher, cytotoxic vector doses. Finally, it cannot be formally excluded that the liver enzyme elevation in some of the subjects was unrelated to AAV-specific T-cell responses. Future clinical trials are needed to address some of these unresolved issues.
Other Vector Systems
Nonviral vectors typically rely on a plasmid-based gene delivery system, where only the naked DNA is delivered, potentially in conjunction with physicochemical methods that facilitate transfection. Consequently, the nonviral approach may be less immunogenic and potentially safer than viral vectors, though innate immune response may still occur. The nonviral gene transfer method is simple, but the efficiency is generally low compared to most viral vector-mediated gene transfer approaches. Moreover, nonviral transfection is typically short-lived and gives transient expression of the transgene, unless selection is applied on ex vivo transfected cells (Lin et al., 2002). A phase I clinical trial for hemophilia A has been conducted with stably transfected autologous fibroblasts electroporated with FVIII-expressing plasmids. Transfected cells were selected, expanded, and subsequently implanted into the patient. Nevertheless, this approach resulted in only minimal and transient improvement in FVIII activity (Roth et al., 2001).
Efficient in vivo gene delivery of nonviral vectors remains a bottleneck. Typically, for hepatic gene delivery, plasmids are administered by hydrodynamic injection. In this case, a hydrodynamic pressure is generated by rapid injection of a large volume of DNA solution into the circulation, in order to deliver the gene of interest in the liver (Miao et al., 2000). Efforts are being made to adapt hydrodynamic injection towards a clinically relevant modality by reducing the volume of injection along with maintaining localized hydrodynamic pressure for gene transfer. Alternative approaches based on targetable nanoparticles are being explored to achieve target-specific delivery of FVIII or FIX into liver sinusoidal endothelial cells (LSECs) and hepatocytes. Expression could be prolonged by removing bacterial backbone sequences that interfere with long-term expression (i.e., mini-circle DNA) Finally, to increase the stability of FVIII or FIX expression after nonviral transfection, transposons could be used that result in stable genomic transgene integration. We and others have shown that transposons could be used to obtain stable clotting factor expression following ex vivo or in vivo gene therapy (Yant et al., 2000; Ohlfest et al., 2004; Mátés et al., 2009; VandenDriessche et al., 2009; Kren et al., 2009).
Adenoviral vectors have been intensively studied for hemophilia gene therapy. The latest version high-capacity adenoviral vectors are devoid of any viral genes resulting in reduced adaptive immune responses and improved stability of transgene expression. There is an added advantage of minimized risk of oncogene activation because they are essentially nonintegrating. One disadvantage of adenoviral vectors is their robust interaction with APCs. This interaction triggers innate immune responses which is a safety concern. High-capacity adenoviral vectors encoding FVIII or FIX from a liver-specific promoter gave rise to robust supraphysiologic clotting factor expression levels with limited toxicity in hemophilic mice (Ehrhardt and Kay, 2002; Chuah et al., 2003; Brown et al., 2004). These results were later confirmed in large animal models, in particular hemophilic dogs. A phase I clinical trial was conducted, involving systemic administration of high-capacity adenoviral vectors encoding FVIII from a liver-specific promoter to a severe hemophilia A patient. Although very low FVIII levels (approximately 1%) may have been obtained, the trial was stopped upon detection of a transient inflammatory response with hematologic and liver abnormalities. Therefore, the interaction of (high-capacity) adenoviral vectors with the innate immune system needs to be minimized to enable further clinical use. For instance, localized hepatic delivery results in efficient, long-term transgene expression in nonhuman primates, while minimizing such side effects (Brunetti-Pierri et al., 2009).
Future Considerations
In recent years substantial progress has been made in the field of gene therapy. Various preclinical studies using different vectors and target cells demonstrates unequivocally that gene therapy can result in a sustained therapeutic effect in animal models of hemophilia A or B. AAV and lentiviral vectors are among the most promising vectors to date for hemophilia gene therapy based on the levels and duration of expression and the immune considerations. Hepatocytes, muscle cells, or HSCs are particularly attractive target cells and have all been explored with regard to their ability to stably express clotting factors. Convincing evidence continues to emerge from clinical trials that gene therapy is effective in patients suffering from hemophilia B and results in sustained therapeutic FIX levels.
However, there are still a number of hurdles that need to be overcome. Though sustained therapeutic FIX levels were obtained in an AAV-based trial for hemophilia B, these levels are not sufficient to prevent bleeding induced by trauma or injury. Consequently, improved vector designs are warranted to achieve full hemostatic correction in patients with severe hemophilia B. Moreover, the proportion of functional infectious vector particles versus empty particles could be further improved. Last but not least, the AAV capsids themselves could be engineered further to minimize the risk of inducing dose-limiting T-cell responses and liver toxicity. It is tempting to speculate that the same approach could ultimately also be used for gene therapy of the more common hemophilia A form, but more efficient vector designs are needed to help accomplish this goal. The challenge here lies in identifying small regulatory elements that are sufficiently potent to drive expression of the relatively large FVIII transgene and then can be accommodated by the small AAV vector particles. The use of a codon-optimized FVIII that enhances expression nearly 50-fold may at least in part overcome some of the limitation with conventional B-domain deleted FVIII cDNAs (Ward et al., 2011).
If the T-cell responses observed in the AAV-based trial reflect memory responses from prior natural exposure with wild-type AAV, then this may be less of a concern when lentiviral vectors are used, given the relatively low prevalence of HIV compared to AAV. Moreover, HIV can more readily incorporate larger transgenes like FVIII. The ability to achieve hemostatic correction following platelet-directed FVIII expression in the face of high-titer inhibitory antibodies is unprecedented and may pave the way towards treatment of patients with inhibitors that are resistant to conventional immune tolerization approaches (Shi et al., 2007). Since the behavior of gene delivery vectors does not always translate well from mice to humans, it is important to also validate lentivirus-based approaches in large animal models. Preliminary experiments suggest that lentiviral FIX vectors, equipped with a miR-regulated hepatocyte-specific expression cassette, can give rise to sustained therapeutic effects in the canine hemophilia model without inducing inhibitory antibodies (L. Naldini, personal communication). Nevertheless, this requires manufacturing relatively large vector doses, which remains challenging and warrants the development of improved and more efficient lentiviral vector production systems.
It will be particularly important to also assess the risk of inhibitor development following gene therapy using these different vector approaches. It will be challenging to achieve hemostatic correction following gene therapy in patients with pre-existing inhibitory antibodies to FVIII. Nevertheless, it is encouraging that even in this case, gene therapy may provide a potential solution by coaxing FVIII expression in platelets (Shi et al., 2006) and that liver-directed approaches by result in Treg induction and clotting factor–specific immune tolerance (Mingozzi et al., 2003). Though other emerging vector systems received less attention than AAV or lentiviral vectors for hemophilia gene therapy, they could potentially overcome some of their limitations. In particular, the use of transposons or engineered nucleases, are particularly attractive, provided more efficient gene delivery can be achieved (Mátés et al., 2009; Li et al., 2011a; Daboussi et al., 2012). It is important therefore to keep an open mind and explore all possible options in the hope to ultimately develop a safe and efficacious gene therapy approach for these serious congenital bleeding disorders that could lead to a bona fide cure.
Footnotes
Acknowledgments
We thank the members of the Department of Gene Therapy & Regenerative Medicine and our collaborators for their various contributions to some of the work presented in this review. We also wish to thank EU FP7 PERSIST, FWO, Bayer-Schering Pharma, IWT, VUB-GOA (EPIGEN) for providing financial support.
Author Disclosure Statement
No competing financial interests exist.