
Abstract
Many DNA vaccine candidates have been developed for the treatment of human papillomavirus type 16 (HPV16)–induced malignancies. Most of these vaccines consist of a fusion of E7 with a “carrier-protein” that functions to increase the potency of the vaccine. The nature of these carrier-proteins varies widely, and the mechanisms proposed to explain the enhanced immunogenicity of such fusions are often linked to the biological function of the carrier-protein. However, the potentiating effect of these carrier-proteins might also be explained by more general mechanisms, such as the provision of CD4+ T-cell help, increased antigen stability, or altered subcellular localization of the antigen. To assess whether these more generic mechanisms could suffice to generate highly immunogenic DNA vaccines, we evaluated a series of modular HPV16 E7 DNA vaccines in which the presence of CD4+ T-cell help, the presence of an endogenous carrier-protein, and the subcellular localization of the antigen could be systematically altered. Using this approach, we demonstrate that the addition of an element that provides CD4+ T-cell help, elements that enforce endoplasmic reticulum (ER) localization/retention are both necessary and sufficient to create markedly effective HPV16 E7-directed DNA vaccines. Importantly, the resulting design rules also apply to an HPV16 E6–directed DNA vaccine. The developed “HELPER” HPV DNA vaccines encode only very limited additional sequences besides the antigen, thereby reducing the risk of antigenic competition and/or autoimmunity.
Introduction
As HPV type 16 is one of the most common subtypes associated with HPV-induced malignancies, a substantial interest has grown in the development of therapeutic vaccination strategies that aim to enhance HPV16 E6- and E7-specific T-cell responses (Brinkman et al., 2007; Hung et al., 2008). Among these strategies, DNA vaccination is attractive because of its relative simplicity, excellent safety record, and its ability to elicit strong cellular immunity (Schalk et al., 2006; Kutzler and Weiner, 2008). The number of DNA vaccine candidates that have been developed for the induction of HPV16 E7- or E6-specific T-cell responses is large (Lin et al., 2010). Most of these vaccine candidates consist of a genetic fusion of the E6 or E7 antigen and a so-called “carrier protein”. The nature of the carrier proteins used for this purpose varies widely and examples include heat shock proteins (HSPs) (Chen et al., 2000; Huang et al., 2007), calreticulin (CRT) (Cheng et al., 2001), E. coli beta glucoronidase (GUS) (Smahel et al., 2004), interferon gamma inducible protein 10 (IP-10) (Kang et al., 2011), herpes simplex viral protein 22 (HSV VP22) (Michel et al., 2002), and tetanus toxin fragment C (TTFC) (Oosterhuis et al., 2011) (see also Table 1). In most cases, the enhanced immunogenicity of these fusion proteins has been postulated to be directly related to the specific biological function of the carrier protein. To provide some examples, ER chaperones such as calreticulin have been suggested to deliver the antigen directly into the antigen presentation pathway, thereby increasing the efficiency of antigen presentation (Cheng et al., 2001; Lin et al., 2005); linkage to HSPs or Flt3 ligand is thought to result in an enhanced uptake of the antigen by antigen-presenting cells (Chen et al., 2000; Hung et al., 2001b); fusion with HSV VP22 is believed to result in enhanced immunogenicity as a consequence of transfer of the coupled antigen to neighboring cells, thereby increasing antigen cross presentation (Michel et al., 2002); fusion with IP-10 is thought to lead to enhanced immunogenicity by promoting the recruitment of T cells to the vaccination site through its chemoattractive function (Kang et al., 2011).
HPV16, human papillomavirus type 16; HSV VP22, herpes simplex viral protein 22; IP-10, interferon gamma inducible protein 10; TTFC, tetanus toxin fragment C; DC, dendritic cell; PADRE, Pan-Allelic DR Epitope.
Noting that such a diverse group of carriers can apparently enhance HPV vaccine immunogenicity, we considered the possibility that vaccine immunogenicity might primarily be determined by more general properties of the vaccine-encoded protein. For instance, in those cases in which the carrier is a foreign molecule (e.g., mycobacterial HSP-70, TTFC, HSV VP22, and Pseudomonas aeruginosa exotoxin A), provision of CD4+ T-cell help may at least partly explain the carrier effect (Chen et al., 2000; Wolkers et al., 2002; Stevenson et al., 2004; Holst et al., 2010;). Secondly, both HPV16 E6 and E7 proteins are known to have a short half-life (McLaughlin-Drubin and Munger, 2009; Tomaic et al., 2009), and for several of the carrier proteins that have been utilized, fusion has been shown to result in increased steady state levels of the antigen (Michel et al., 2002; Smahel et al., 2004; Oosterhuis et al., 2011). As it has previously been demonstrated that the extent of in vivo accumulation of DNA vaccine-encoded antigens correlates with the magnitude of the CD8+ T-cell response (Bins et al., 2007), such carrier-induced protein stabilization could contribute to vaccine immunogenicity.
Finally, many of the fusions that have been utilized result in an altered subcellular localization of the antigen (Cheng et al., 2001; Hung et al., 2001b; Peng et al., 2004; Smahel et al., 2008; Kang et al., 2011), and antigen localization—rather than the biological function of the carrier used—could conceivably alter immunogenicity. In line with this possibility, in some reports addition of only protein domains, or even signal sequences, was shown to already improve vaccine immunogenicity (Ji et al., 1999; Brulet et al., 2007). To directly determine the potential contribution and relative importance of these generic mechanisms, we have developed a modular DNA vaccine and utilized this to assess which elements are crucial for HPV E7 and also HPV E6 DNA vaccine immunogenicity.
Materials and Methods
Mice
C57BL/6 mice (6–10 weeks, male or female) were obtained from Jax® Mice (The Jackson Laboratory). All experiments were approved by the Experimental Animal Committee of The Netherlands Cancer Institute and in accordance with institutional and national guidelines. Within experiments all mice were age and sex matched.
DNA vaccines
DNA vaccines based on HPV16 E6 and E7 genes were generated by the introduction of target genes or gene fragments into pVAX 1 (Invitrogen). The generation of E7SH, TTFC-E7SH, E6SH, and TTFC-E6SH has been described previously (Oosterhuis et al., 2011). FM4 consists of four moieties of a mutated human FK506-binding protein (97% homology to the mouse protein) with the signal peptide of human growth hormone fused to the N-terminus (Rivera et al., 2000). FM4-HELP-E7SH (see Fig. 1A for a schematic representation) was ordered from GeneArt with codon-optimization for expression in human cells and was cloned between the HindIII and XbaI sites of pVAX. FM4-E7SH and HELP-E7SH were generated by removal of either the BamHI flanked helper cassette or the SpeI-flanked FM4 moiety. Histone 2B, endoplasmic reticulum protein 29, keratin 14, and cluster of differentiation antigen 8a (all mouse origin) were ordered from GeneArt with codon optimization for expression in human cells and flanked by SpeI sites. FM4(minus sig)-HELP-E7SH was generated by PCR using FM4-HELP-E7SH as a template. sig-HELP-E7SH-KDEL was constructed by replacing the complete FM4 sequence with only the signal peptide. The four amino acid KDEL sequence (ER retention signal) was fused to E7SH and E6SH by PCR. The different E6SH-encoding DNA vaccines were constructed by replacing E7SH with E6SH or E7SH-KDEL by E6SH-KDEL. Identity of all fusion genes was confirmed by sequence analysis. Plasmids were expressed in E. coli DH5α and were purified using an endotoxin-free DNA purification kit (Qiagen). DNA vaccines for intradermal tattoo application were dissolved at a concentration of 2 mg/ml in water for injections (Aqua B. Braun).
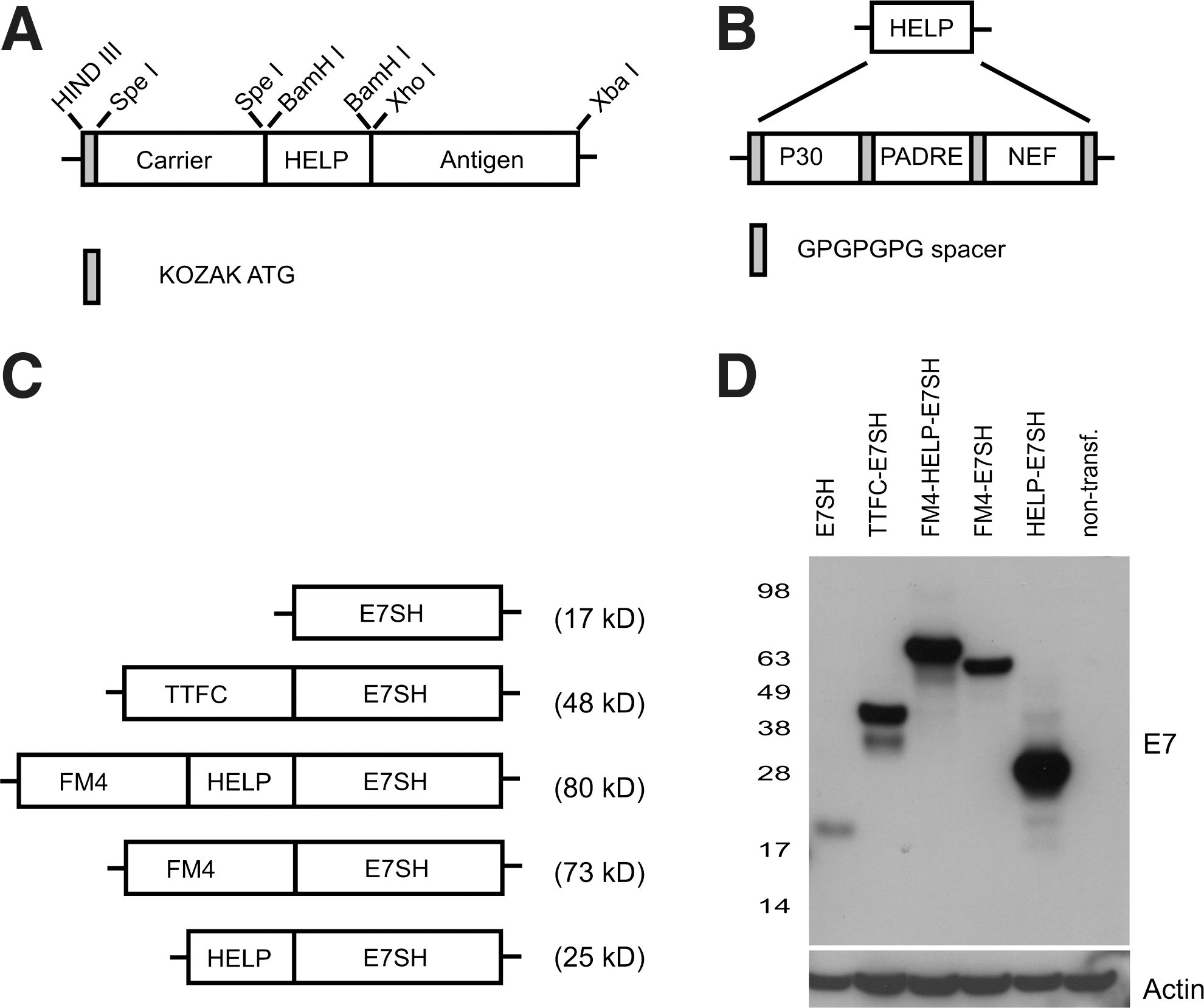
Design and validation of a modular DNA vaccine construct.
Transfection and immunoblotting
HEK 293 cells were transfected with 10 μg of a mixture of a green fluorescent protein (GFP)-encoding plasmid and the indicated constructs at a ratio of 3:7 by use of FuGENE 6 (Roche), according to the manufacturer's instructions. Cells were harvested 24 hr after transfection, and equal transfection efficiency was confirmed by analyzing the percentage of GFP-positive cells by flow cytometry. Subsequently, the remainder of the samples was lysed on ice in RIPA buffer (Sigma-Aldrich) supplemented with protease inhibitor cocktail (Roche) and 0.1 mM PMSF (Thermo Fisher Scientific). Cell lysates were subsequently cleared by centrifugation at 4°C. Total cellular protein was determined using a Bradford assay (Bio-Rad Corporation), and proteins were separated at 30 ug per lane on 4–12% NuPage Bis-Tris gradient gels (Invitrogen) in MES buffer, according to the manufacturer's instructions. Following immunoblotting, E7 expression was detected using a monoclonal mouse anti-HPV16 E7 antibody (Invitrogen, clone 8C9, 1:100 dilution), and actin expression was detected using a mouse anti-human actin antibody (Millipore, clone C4, 1:10000 dilution). In both cases, HRP-rabbit anti-mouse antibody (DAKO, P 0161) was used as secondary antibody at a 1:7500 dilution and detection was performed by enhanced chemiluminescence (Pierce Biotechnology).
Tattoo vaccination
Intradermal DNA tattoo vaccination was performed as described previously (Oosterhuis et al., 2011). In brief, on day −1 the hair on the hind leg was removed using depilating cream (Veet®, Reckitt Benckiser). On day 0, mice were anesthetized and 10 μl of a 2 mg/ml DNA solution in water was applied to the hairless skin of the hind leg. The DNA vaccine was then applied with a permanent make up (PMU) tattoo machine (kindly provided by MT Derm), using a sterile disposable 9-needle bar with a needle depth of 1 mm, oscillating at a frequency of 100 Hz for 30 seconds. Unless indicated otherwise, vaccination was repeated on days 3 and 6. If the mice received a secondary challenge/boost, the hair removal step was repeated and the mice received a single tattoo at 3 months (between day 80 and 90), using 15 μl of the DNA vaccine used for priming. If the mice received only a single tattoo for priming, 15μl of the DNA solution was used.
Detection of HPV-specific CD8+ T cells
Peripheral blood cells were obtained by tail bleeding, and erythrocytes were removed by incubation in erythrocyte lysis buffer (155 mM NH4Cl, 10mM KHCO3, 0.1mM EDTA [pH 7.4]) on ice. Cells were subsequently stained in FACS-buffer (1× PBS, 0.5% BSA, and 0.02% sodium azide) with allophycocyanin (APC)-conjugated anti-CD8a mAb (BD Pharmingen) plus phycoerythrin (PE)-conjugated H-2Db E749–57 or H-2Kb E648-57 tetramers for 15 min at 20°C. Subsequently, cells were washed two times in FACS-buffer before analysis. Live cells were selected based on PI exclusion. MHC tetramers were produced by UV-induced peptide exchange, as described previously (Toebes et al., 2006).
Detection of P30 and PADRE-specific CD4+ T cells
Intracellular IFN-γ staining was performed using the BD Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's protocol. Peripheral blood cells were stimulated for 16 hr with either the PADRE (AKFVAAWTLKAAA) or P30 (FNNFTVSFWLRVPKVSASHLE) peptide at a 1 μg/ml concentration. Cells were subsequently stained using PE-conjugated anti-IFN-γ mAb (BD Pharmingen), and APC-conjugated anti-CD8a mAb (BD Pharmingen). All samples were analyzed on a FACScalibur (BD Biosciences), using Flow-Jo® software for data analysis.
Area under the curve calculation
The total area under the curve (AUC) value is used as a measure for total vaccine potency and was calculated using the trapezoid rule, using the following formula: AUC0-tlast=((C0+C1)/2) x (Time1-0)+((C1+C2)/2) x (Time2-1) etc., where C is the % of tetramer+ CD8+ cells at a certain time point.
TC-1 tumor challenge
C57BL/6 mice were injected subcutaneously with 1×105 TC-1 tumor cells that express both HPV16 E6 and E7 (Lin et al., 1996). DNA tattoo vaccination was subsequently performed on either day 4, or on day 4, 7, and 10 after tumor challenge, as indicated. Tumor growth was monitored by caliper measurements in two dimensions, tumor volume was calculated as (width2 x length)/2. Mice were sacrificed when tumor length reached 15 mm or when the tumor volume exceeded 1,000 mm3.
Statistical analysis
Statistical analysis was performed using the student's t-test. A p-value <0.05 was considered to be significant (two-tailed). For evaluation of survival data a log-rank test was used.
Results
Design of a modular DNA vaccine
In order to be able to separate the contribution of factors such as antigen localization, antigen stabilization, and the presence of T-helper cell epitopes on vaccine immunogenicity, we designed a modular HPV E7-directed DNA vaccine that contains an endogenous carrier-protein, and a separate minimal domain for the provision of CD4+ T-cell help (Fig. 1A). The use of this design minimizes the risk of antigenic competition at the level of CD8+ T-cell response induction as only minimal foreign sequences are incorporated besides the antigen. As a first carrier protein, we selected an engineered human protein called FM4. This protein contains four repeats of a point-mutated version of the FK506 binding protein, FKBP12, and is targeted to the ER by the inclusion of a signal peptide. As a result of these modifications, FM4 forms stable aggregates inside the ER (Rivera et al., 2000). To generate a minimal domain that would provide CD4+ T-cell epitopes that can be presented by a large series of MHC class II alleles, we created a “helper-cassette” (Fig. 1B), consisting of the TTFC P30 pan DP epitope (Panina-Bordignon et al., 1989), the PADRE pan DR epitope (Alexander et al., 1994), and the HIV NEF pan DQ epitope (Pancre et al., 2002). Both P30 and PADRE are also recognized in the context of mouse MHC class II, allowing evaluation of this strategy in mice (Alexander et al., 1994; Rice et al., 2001). In order to avoid the potential formation of CD8+ T-cell epitopes at the junctions of these three sequences, all epitopes were linked by GPGPGPG spacers (Livingston et al., 2002).
To obtain insight into the value of the individual components used, we also generated HPV E7-encoding vaccines that only contained the FM4 carrier-protein or only the helper-cassette (Fig. 1C). In all cases, a gene-shuffled version of E7 (E7SH) was used as antigen, in order to avoid the risk of cellular transformation at the vaccination site (Ohlschlager et al., 2006; Oosterhuis et al., 2011). All fusion constructs displayed the expected molecular weight when expressed in HEK 293 cells. Furthermore, the expression level of all E7 fusion vaccines was strongly increased compared to that of a nonfused E7SH vaccine (Fig. 1D).
The combined inclusion of a carrier protein and helper-cassette results in superior vaccine immunogenicity
In order to reveal possible differences in immunogenicity between the above-described vaccines, C57BL/6 mice (n=5 per group) were immunized by short interval DNA tattooing (Bins et al., 2005), and E7-specific CD8+ T-cell responses in peripheral blood samples were analyzed over time. As shown in Figure 2A and B, a clear pattern in vaccine immunogenicity is noted. First, HPV16 E7-specific CD8+ T-cell responses are not above background in mice that received a vaccine that solely encodes E7SH, but clear CD8+ T-cell responses are detected in groups that were treated with the fusion vaccines, thereby underlying the value of such genetic fusions. Second, when comparing the magnitude of the CD8+ T-cell responses induced by the different fusion vaccines at the peak of the primary response (Fig. 2C), it is apparent that both the addition of the FM4 carrier (FM4-E7SH) and, in particular, the helper-cassette (HELP-E7SH), results in a substantial improvement in immunogenicity of E7SH, but that the combination of the carrier and the helper-cassette (FM4-HELP-E7SH) is clearly superior (mean peak response of 19.05±5.02% E749-57-specific CD8+ T cells directly ex vivo, p<0.05 relative to HELP-E7SH the second-best vaccine). FM4-HELP-E7SH also significantly (p<0.05) outperformed a vaccine encoding a fusion of tetanus toxin fragment C and E7SH (TTFC-E7SH) that was previously developed in our lab (Oosterhuis et al., 2011). Differences in T-cell response magnitude were not only observed at the peak of the primary CD8+ T-cell response but remained visible over time and were maintained after a secondary challenge (indicated by the arrow in Fig. 2B). As a measure of total construct potency the AUC values for each group were calculated (Fig. 2D).
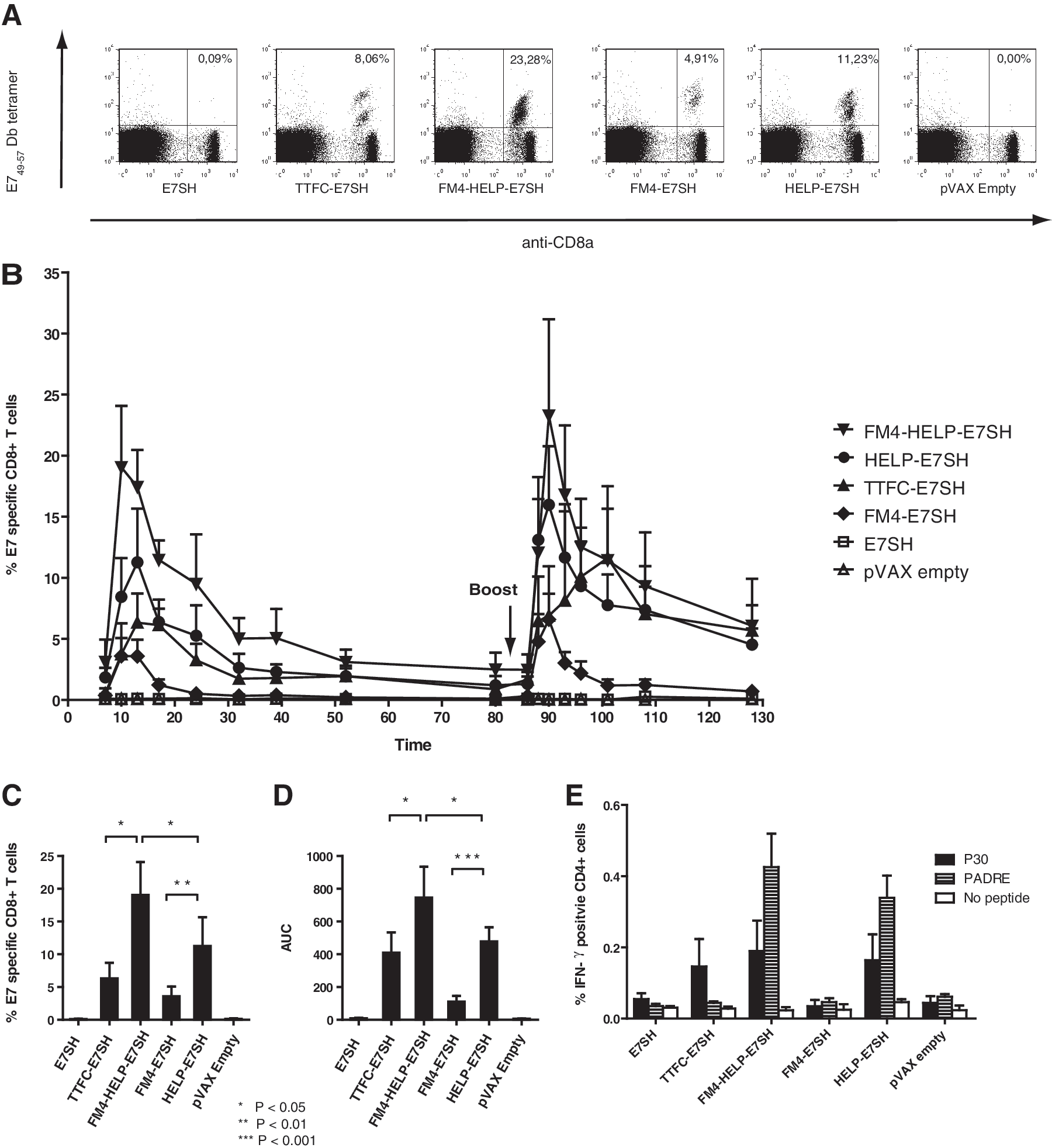
Synergistic effect of an endogenous carrier protein and a helper-cassette on vaccine immunogenicity. C57BL/6 mice (n=5 per group) were immunized by DNA tattoo vaccination on day 0, 3, and 6 with the indicated vaccines (primary challenge) and homologously boosted at 3 months. Peripheral blood was analyzed over time for antigen-specific CD8+ T cells by MHC tetramer staining.
In order to evaluate whether the inclusion of the helper-cassette resulted in the expected CD4+ T-cell responses toward the encoded epitopes, CD4+ T-cell reactivity against P30 and PADRE epitopes was evaluated by intracellular IFN-γ staining. In mice that received DNA vaccines encoding the helper-cassette, P30 and PADRE-specific CD4+ T-cell responses could readily be demonstrated. In mice vaccinated with TTFC-E7SH, which contains the P30 but not the PADRE epitope, P30-specific CD4+ T-cell responses could also be observed (Fig. 2E).
Only ER-localized carriers enhance vaccine immunogenicity
Having demonstrated that the combination of a carrier molecule and the helper-cassette results in superior immunogenicity, we wished to explore which aspects of the carrier molecule determined its effect on vaccine potency. To this purpose, we selected four endogenous proteins—histone 2B (H2B, nuclear localization), endoplasmic reticulum protein 29 (ERP-29, ER localized), keratin 14 (KRT 14, cytosolic localization), cluster of differentiation antigen 8a (CD8a, plasma membrane localized)—for which there are no prior data that would suggest a specific role in antigen presentation. As shown in Fig. 3B, transfection of HEK 293 cells with the different constructs leads to expression of proteins with the expected size (see Fig. 3A). Furthermore, while expression levels vary widely, in all cases the expression level of the fusion vaccine is strongly enhanced relative to that of unmodified E7SH.

Only endoplasmic reticulum (ER) localized self-carriers provide an advantage over the addition of the helper-cassette alone.
In order to study possible differences in vaccine immunogenicity, mice were vaccinated and immune responses were monitored as described above. Of the five fusion vaccines tested, both FM4-HELP-E7SH and ERP29-HELP-E7SH were significantly (p<0.05) more immunogenic than HELP-E7SH, whereas the immunogenicity of H2B-HELP-E7SH and CD8a-HELP-E7SH was comparable to that of HELP-E7SH (Fig. 3C and D). Finally, KRT14-HELP-E7SH was significantly (p<0.01 based on AUC) less immunogenic than HELP-E7SH (Fig. 3E).
These differences in vaccine immunogenicity did not directly correlate with differences in expression levels as displayed in Figure 3B. For example, the expression level of CD8a-HELP-E7SH was extremely high but immunogenicity was mediocre. Vice versa, the expression level of FM4-HELP-E7SH was intermediate, but immunogenicity was high. Thus, when a series of different endogenous carriers is utilized (for which the presence of CD4 T-cell epitopes is therefore not a confounding factor), the enhancement of vaccine immunogenicity can only partially be ascribed to increased accumulation of the antigen. An interesting observation in this regard is that the two carrier proteins that result in a significant enhancement of E7 vaccine immunogenicity are both ER localized.
The carrier effect can be fully explained by ER targeting of the antigen
In order to determine whether the observed correlation between ER localization of the carrier and vaccine immunogenicity was due to chance or reflected a true effect of ER localization, we generated vaccine variants of FM4-HELP-E7SH, in which either the signal peptide of FM4 was removed (FM4(minus-sig)-HELP-E7SH), or in which only the signal peptide of the FM4 moiety was retained. In the latter case (sig-HELP-E7SH-KDEL), a C-terminal KDEL sequence was included to achieve ER retention. As a control, a variant that encoded the antigen with the signal peptide and ER retention signal but that lacked the helper-cassette (sig-E7SH-KDEL) was also generated (see Fig. 4A for a schematic representation of the constructs). Expression of protein products of the expected size was demonstrated by western blotting (Fig. 4B) as described above. As shown in Figure 4C and E, removal of the signal peptide from FM4 resulted in a strong decrease in immunogenicity of FM4-HELP-E7SH. Furthermore, the same detrimental effect of signal peptide removal was observed for ERP29, the second ER localized carrier protein (data not shown).
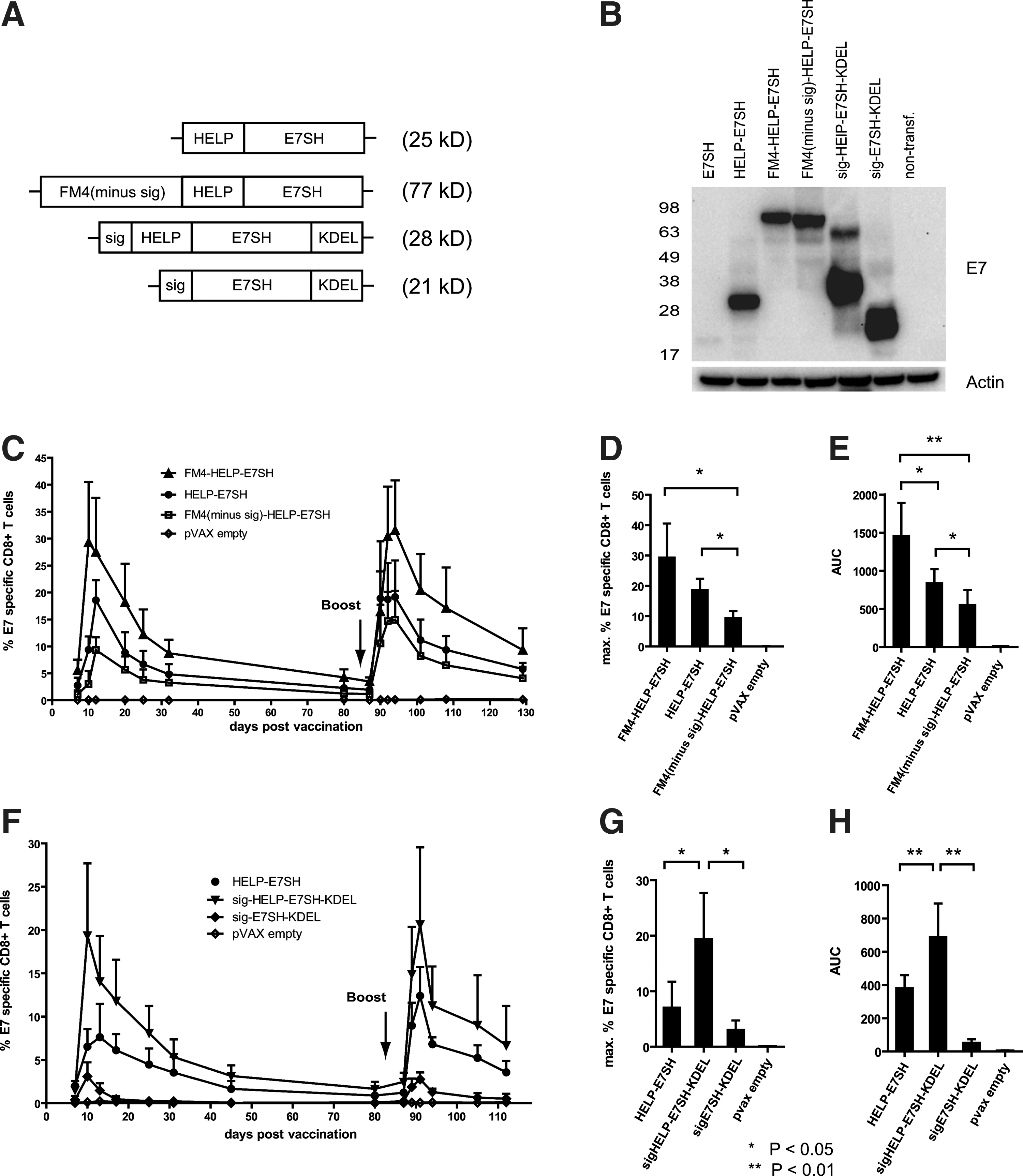
The “carrier effect” can be solely explained by ER targeting of the antigen.
In stark contrast, the sole inclusion of an ER localization signal (i.e., signal peptide and KDEL) sufficed to significantly enhance the immunogenicity of HELP-E7SH (p<0.05). Together these results demonstrate that retention in the ER suffices to explain the potentiating effect of endogenous carriers on E7 vaccine-specific T-cell responses. Consistent with the notion that such a “minimal carrier” does not provide CD4+ T-cell epitopes, inclusion of the helper-cassette was required for maximal immunogenicity as illustrated by the very moderate immunogenicity of sig-E7SH-KDEL (Fig. 4F and H).
Application of design rules to HPV E6SH–encoding vaccines
The above data indicate that the combination of a helper-cassette and ER targeting/ retention signal suffices to create a highly immunogenic E7 DNA vaccine. In order to further validate this minimal “HELPER” design for HPV-directed vaccines, we generated a set of HPV16 E6SH DNA vaccines (Fig 5A) in which the value of the helper cassette or the combination of this cassette with ER localization were compared. Mice were tattoo-vaccinated with these vaccines, and E6-specific CD8+ T-cell responses were monitored directly ex-vivo by MHC tetramer staining. Similar to what was observed for E7SH-encoding vaccines, the addition of the helper cassette by itself strongly improved the immunogenicity of E6SH. In addition, the joint inclusion of the minimal ER localization/retention signal led to a further increase in immunogenicity, although the latter difference was not statistically significant (Fig. 5C and E). Importantly, the CD8+ T-cell responses induced by sig-HELP-E6SH-KDEL significantly outperformed those elicited by a previously developed TTFC-E6SH fusion vaccine (Oosterhuis et al., 2011) (p<0.01 based on AUC). Superiority of the “HELPER” DNA vaccine design was also apparent when immunogenicity was assessed in HLA-A2 transgenic mice (Pascolo et al., 1997), demonstrating that these design rules also apply in the context of human MHC class I (Supplementary Fig. 1; Supplementary Material available online at
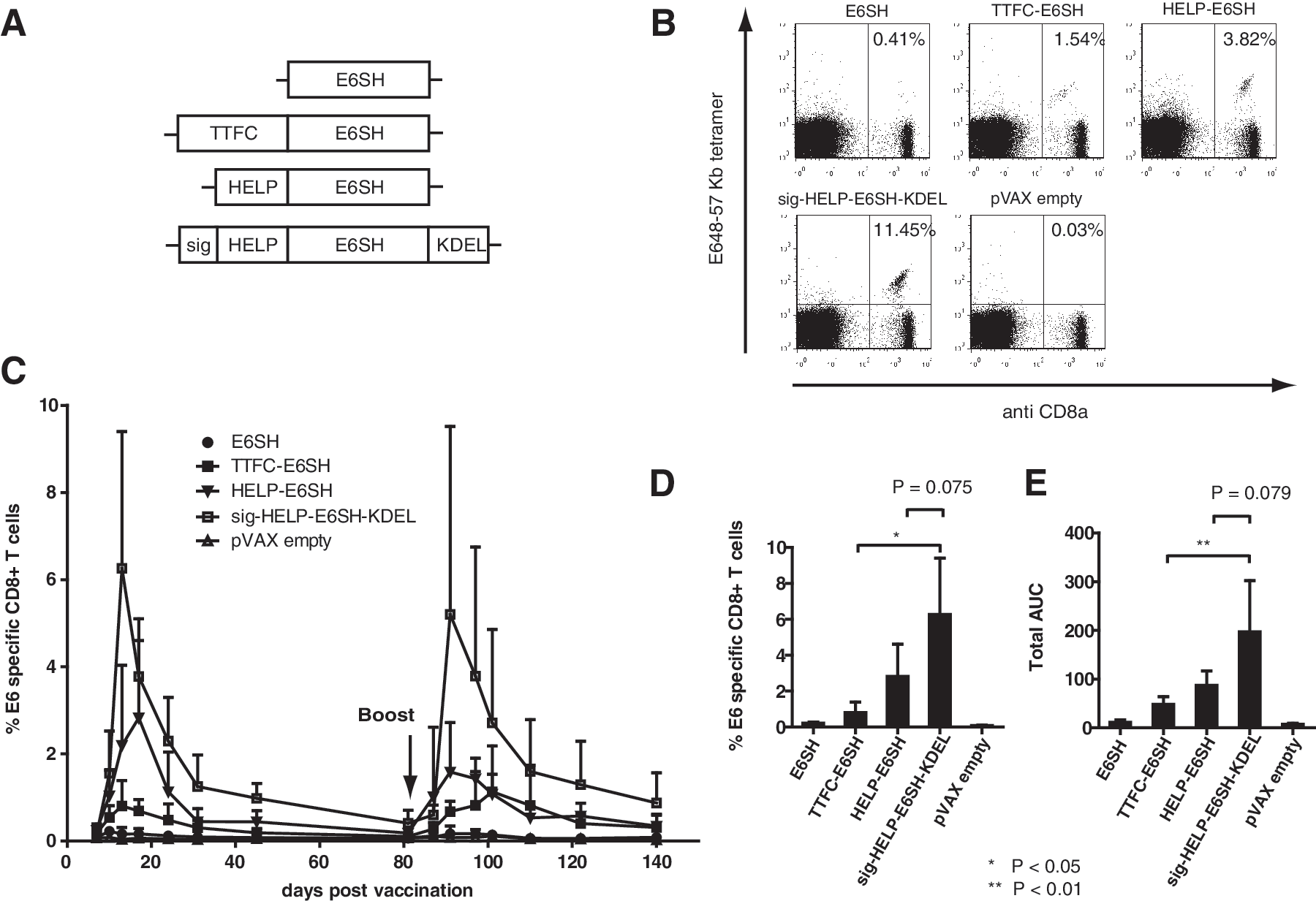
The combined addition of the helper-cassette and ER entry/retention signal also improves the immunogenicity of E6SH. C57BL/6 mice (n=5) DNA were tattoo-vaccinated on day 0, 3, and 6 with the indicated vaccines (primary challenge) and homologously boosted at 3 months.
“HELPER” vaccine formats allow for dose reduction and show superior anti-tumor effects
As the CD8+ T-cell responses observed after vaccination with sig-HELP-E7SH-KDEL were highly potent, we evaluated whether substantial CD8+ T-cell responses could still be induced in case either the number of vaccinations or the vaccine dose was reduced. As shown in Figure 6A, vaccination with a single dose of the control vaccines HELP-E7SH and TTFC-E7SH led to the induction of only very modest CD8+ T-cell responses. In contrast, vaccination with a single dose of the sig-HELP-E7SH-KDEL vaccine still induced CD8+ T-cell responses of around 15% ex vivo (Fig. 6A, compared with Fig. 4F and G). Likewise, marked (>20% ex vivo) CD8+ T-cell responses were still induced by sig-HELP-E7SH-KDEL when mice were vaccinated with a five-fold reduced DNA dose (0.4 mg/ml instead of 2 mg/ml) (Fig. 6B). The latter finding is of particular relevance for clinical translation as the inability to scale DNA doses used in mice to humans is considered one of the main explanations for the lower efficacy of DNA vaccination in humans as compared to small animals (Johnston et al., 2002).
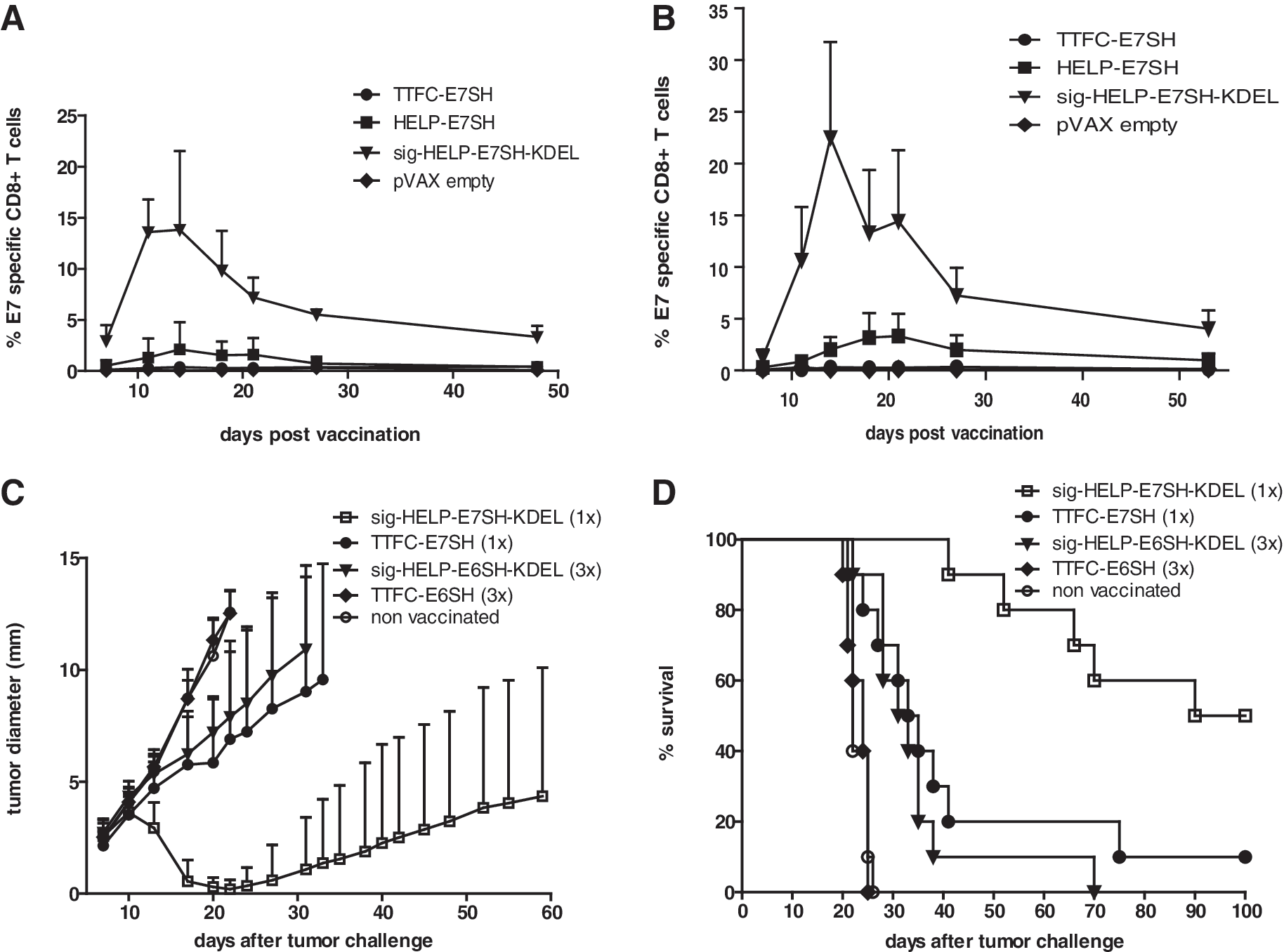
The novel design allows for dose-sparing and shows superior functionality.
In order to evaluate whether the enhanced immunogenicity of the “HELPER” E6 and E7 DNA vaccines also translated into a superior anti-tumor effect, mice (n=10) were challenged with HPV E6/E7-expressing TC-1 cells, and vaccination was started at day 4. Notably, a single vaccination with sig-HELP-E7SH-KDEL induced initial regression of tumors in 10 out of 10 mice, whereas a single vaccination with TTFC-E7SH only induced regression in 2 out of 10 mice (Fig 6C). This difference also translated into a significant effect on survival (P=0.0019) (Fig. 6D). A similar pattern was observed in mice vaccinated with the E6SH encoding variants: sig-HELP-E6SH-KDEL significantly (p=0.0002) outperformed TTFC-E6SH in terms of tumor control and survival. These results show that both E6 and E7 “HELPER” DNA vaccines are superior with respect to CD8+ T-cell induction and with respect to in-vivo tumor clearance.
Discussion
In this study we aimed to rationally design DNA vaccines that induce immune reactivity against the HPV16 E6 and E7 oncoproteins. The resulting vaccine designs, sig-HELP-E6SH-KDEL and sig-HELP-E7SH-KDEL, contain only minimal additional sequences apart from the antigen of interest and induce extremely potent E6- and E7-specific CD8+ T-cell responses. Our results underscore the importance of the addition of CD4+ T-cell help in DNA vaccination and indicate that the enhanced immunogenicity that can be observed after addition of a carrier molecule can be achieved by the mere ER localization of the fused antigen.
The importance of CD4+ T-cell help for the generation of effective CD8+ T-cell responses is well established (Bevan, 2004). In the field of DNA vaccination, multiple strategies have been developed to provide such CD4+ T-cell help, and the most commonly used method is to fuse the antigen of interest to an exogenous (e.g., bacterial) protein (Wolkers et al., 2002; Stevenson et al., 2004). An extensively studied example of such as carrier-protein is domain 1 of tetanus toxin fragment C that contains several CD4+ helper epitopes, which can be presented by multiple MHC class II alleles (; (Stevenson et al., 2004; Oosterhuis et al., 2011).
A potential drawback to the use of foreign proteins as carrier molecules is that such proteins are likely to contain competing CD8+ T-cell epitopes as well. This could result in skewing of the CD8+ T-cell response toward the carrier molecule by the principle of immunodominance (Yewdell and Bennink, 1999). A more elegant strategy is therefore to fuse the antigen of interest to one or multiple minimal CD4 T-cell epitopes. Following this line of thought, prior work has shown the immunogenicity of DNA vaccines in which a modified version of the MHC class II invariant chain was used, in which the class II-associated invariant chain peptide (CLIP) peptide was replaced by CD4+ T-cell epitopes such as P30 or PADRE (Stevenson et al., 2004; Wu et al., 2011), although clinical application of this strategy may potentially be limited by the concern that vaccination could result in the induction of autoimmune reactivity toward the invariant chain. In the current study we show that robust CD4+ T-cell help can simply be provided by fusing a set of promiscuous CD4+ helper epitopes that can be presented by a variety of common DP, DR, and DQ alleles (see Fig. 2 and 5) to HPV E6 and E7 antigens. Because of the very broad MHC class II coverage of those epitopes (Panina-Bordignon et al., 1989; Alexander et al., 1994; Pancre et al., 2002), there may be little advantage in the inclusion of further CD4+ T-cell epitopes, but if necessary, this set can obviously be expanded. Importantly, as only minimal foreign sequences are added to the antigen of interest in this approach, the risk of antigenic competition should be small.
The observation that the immunogenicity of DNA vaccines can also be improved by the genetic fusion to carrier molecules that are of self-origin indicates that mechanisms other than the provision of CD4+ T-cell help must also play a role. Examples of endogenous carrier-proteins that have been shown to improve the immunogenicity of HPV16 E6 and/or E7 DNA vaccines include calreticulin, HSP 60, and IP-10 (Cheng et al., 2001; Peng et al., 2004; Huang et al., 2007; Kang et al., 2011). As discussed within the introduction, the enhanced immunogenicity of these self carriers is often attributed to the specific biological function of the carrier molecule (see also Table 1). However, noting that vaccine immunogenicity can be enhanced by such a variety of approaches, we speculated that more generic mechanisms may be involved, and such mechanisms could, for instance, involve the effect of fusion on antigen accumulation or subcellular localization (Bins et al., 2007).
Comparison of the immunogenicity of E7 DNA vaccines utilizing five different endogenous carrier molecules indicated that immunogenicity was strongly enhanced by fusion to two different ER localized carrier-proteins for which there is no prior data suggesting a specific role in antigen presentation. Direct support for the notion that antigen localization is the key parameter in these vaccines was provided by the demonstration that the mere addition of ER targeting and ER retention signals is sufficient to enhance immunogenicity, and this effect applied to E7SH, HELP-E7SH, and HELP-E6SH. In this regard, it is noteworthy that many of the strategies previously shown to improve the immunogenicity of E7 and/or E6 also do result in ER localization (Cheng et al., 2001; Hung et al., 2001b; Peng et al., 2004; Smahel et al., 2008). A conceptual advantage of the use of a minimal ER localization/retention system as developed here is the reduced risk of antigenic competition in case a foreign sequence is used, and the reduced risk of induction of autoimmune reactivity in case an endogenous protein is utilized.
At present it is unclear why ER localization of E6 and E7 benefits the induction of CD8+ T-cell responses. It may be speculated that ER localization could increase the half-life of the (likely unfolded) vaccine-encoded antigens and thereby increase the size of the protein pool available for cross presentation (Bins et al., 2007). However, the observation that the correlation between the extent of antigen accumulation and immunogenicity is at best partial indicates that this cannot form the sole explanation. Freigang et al. (2007) previously observed that modifications that resulted in the ER localization of the lymphocytic choriomeningitis virus (LCMV) glycoprotein also strongly enhanced immunogenicity in a mouse model system that was dependent on antigen cross-presentation, and several mechanisms have been proposed to explain such enhanced cross presentation.
Firstly, upon cell death, ER localized antigens may be enclosed within ER-derived membranes/vesicles that could protect them from degradation in the extracellular milieu. Secondly, defects in the folding of ER-localized proteins could lead to association with ER chaperones that could conceivably be involved in antigen delivery to APCs (Binder and Srivastava, 2005). Thirdly, the destruction of (misfolded) proteins in the ER via the endoplasmic reticulum–associated protein degradation (ERAD) pathway could potentially lead in higher numbers or qualitatively different (Golovina et al., 2002) antigenic peptides. Finally, the accumulation of (misfolded) proteins in the ER may induce an ER-stress response, thereby triggering the cells that express the vaccine-encoded antigen to undergo apoptosis or by some other mechanism become “visible” for APCs (Martins et al., 2011).
To distinguish the first three from the latter hypothesis is of interest, as in the latter case the induction of ER stress could suffice to enhance immunogenicity even for cytosolic antigens. To evaluate the importance of ER presence of the antigen versus the induction of ER stress, we inserted a 2A linker in between FM4 and HELP-E7SH. As a result, the FM4 moiety remains ER-resident, whereas the genetically fused HELP-E7SH (the antigen) will now remain cytosolic. Interestingly, the immunogenicity of this vaccine was not higher than that of a HELP-E7SH vaccine that lacks the ER-located FM4 moiety (Supplementary Fig. 2), thereby indicating that ER localization of the antigen itself is required. Future studies should reveal whether—and if so by which mechanis—the ER forms a dedicated compartment for antigen cross presentation.
In conclusion, here we have developed highly effective DNA vaccines for the treatment of HPV16 positive malignancies. The resulting candidate vaccines, “HELPER” E6 and “HELPER” E7, contain only minimal additional sequences apart from the antigen, thereby limiting potential safety risks and the risk of antigenic competition. The fact that these novel vaccine candidates strongly outperform two previously developed candidate vaccines both in terms of CD8+ T-cell induction and tumor control warrants their clinical evaluation.
Footnotes
Author Disclosure Statement
Koen Oosterhuis, Ton N. Schumacher, and John B. Haanen declare a potential conflict of interest as they are inventors on a patent application that has been submitted based on the findings described in this manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.