
Abstract
Adeno-associated viral (AAV) vectors 2 and 8 have been used in clinical trials for patients with hemophilia, and data suggest that the capsid-specific CD8+ T cell response has had a negative impact on therapeutic success. To date the pattern of capsid cross-presentation from AAV2 and AAV8 transduction in vivo has not been elucidated. Previously, we have demonstrated that an engineered AAV2 virus carrying the immune-dominant SIINFEKL peptide in the capsid backbone was indistinguishable from wild type with respect to titer, tropism, and the ability to induce capsid-specific CD8+ T cell responses in vivo. In this study, we used the same strategy to engineer an AAV8 vector and demonstrated that antigen from SIINFEKL peptide-integrated AAV8 capsid was effectively presented via either plasmid transfection or AAV8 transduction in vitro. The tissue tropism and transgene expression kinetics of the engineered AAV8 vector in vivo were identical to that of wild-type AAV8. Animal studies show that capsid antigen presentation from AAV transduction was dose dependent, and more importantly, the proliferation of capsid-specific CD8+ T cells had similar kinetics (detectable before 30 days and undetectable after 40 days) for both AAV2 and AAV8 vectors. Elucidation of the kinetics of capsid antigen presentation from AAV transduction by various serotypes provides new insight into the potential impact CD8+ T cells can have during clinical trials and may help with rational design of effective strategies to prevent capsid-specific CD8+ T cell-mediated elimination of AAV-transduced target cells.
Introduction
To mimic the findings from clinical trials, AAV2 vectors have been used to test the kinetics of antigen presentation in animal models. Such studies have indicated that capsid-specific T cell activation is observed at early time points after vector application (H. Li et al., 2011). Among 12 serotypes and more than 100 variants, AAV serotype 8 is considered to be the most efficient for liver transduction (Gao et al., 2006; Wu et al., 2008; C. Li et al., 2011; Wang et al., 2012). Furthermore, peak levels of transgene expression are obtained significantly sooner with AAV8 as compared with AAV2 packaged vectors (Wu et al., 2008). To determine whether the kinetics of a capsid-specific CTL response in vivo differ between AAV2 and AAV8, we adopted a previously used strategy of substituting the capsid HI loop with the chicken ovalbumin (OVA)-derived SIINFEKL peptide to generate an AAV8OVA cassette (Li et al., 2009b). Using the engineered AAV8OVA vector, we found that AAV8 capsid antigen was properly processed and presented on the cell surface with MHC class I molecules to activate OVA-specific lymphocytes either through endogenous production of capsid or by AAV8OVA vector transduction in vitro. In vivo administration of AAV8OVA induced tropism and kinetics of transgene expression similar to those of wild-type AAV8 vector. Prominently, similar in vivo kinetics of capsid antigen presentation were observed for AAV8OVA and AAV2OVA vectors.
Materials and Methods
Constructs
The SIINFEKL immunodominant epitope was cloned into the AAV8 capsid by substitution of the HI loop with the eight-amino acid sequence as described previously for generation of the pXR2/OVA cassette (Li et al., 2009). We first used the PCR primers F1R1-0 and F2-0R2 (Supplementary Fig. S1; supplementary data are available online at
Cells and virus
HEK293 and HepG2/H-2Kb were maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum, penicillin (100 U/ml), and streptomycin (100 g/ml). AAV virus production was done as previously described, using three-plasmid cotransfection in 293 cells (Xiao et al., 1998). The viral titer was determined by Southern dot blot.
Mice
C57BL/6 mice and BALB/c mice were purchased from Jackson Laboratory (Bar Harbor, ME). OT-1/Rag-1 mice express an H-2Kb -restricted T cell receptor (TCR) specific for the OVA-derived SIINFEKL peptide and were ordered from Taconic Farms (Germantown, NY). All mice were maintained in a specific pathogen-free facility at the University of North Carolina at Chapel Hill (Chapel Hill, NC), and the University of North Carolina Institutional Animal Care and Use Committee approved all procedures.
Antigen presentation assay in vitro after pAAV8/OVA transfection or AAV8OVA vector transduction
Activation of CD8+ OT-1 T cells, measured by upregulation of CD69 expression via flow cytometry, was used as a readout for the efficiency of antigen presentation of the SIINFEKL-containing capsids after transfection or transduction of AAVOVA plasmids and packaged vectors, respectively. For transfection studies, 293 cells at 70–80% confluency in a 6-well plate were transfected with 3 μg of each plasmid including pXR8/OVA, pXX6-80, and pCBA-H-2Kb, using polyethylenimine (PEI). After 24 hr, cells were incubated with OT-1 T cells overnight. In transduction studies, 2×105 HepG2/H-2Kb cells were incubated with 2×1011 particles of AAV vector for 24 hr, and then washed and incubated with OT-1 T cells overnight.
Flow cytometry
Phycoerythrin (PE)-labeled rabbit anti-mouse CD69 (clone H1.2F3; BD Biosciences, San Jose, CA) and a fluorescein isothiocyanate (FITC)-labeled anti-CD8 (clone 53-6.7; BD Biosciences) were used to stain OT-1 T cells for 1 hr at 4°C. Cells were then washed and analyzed by flow cytometry (UNC Chapel Hill Flow Cytometry Core Facility).
Luciferase imaging
BALB/c mice were retro-orbitally injected with 1×1011 particles of AAV/luc, and 1 week later, imaging was performed with a Xenogen IVIS Lumina imaging system (Caliper Life Sciences/PerkinElmer, Hopkinton, MA) after intraperitoneal injection of
ELISA for human AAT
The AAT concentration in blood was determined by enzyme-linked immunosorbent assay (ELISA) as described previously (C. Li et al., 2011).
In vivo CTL killing assay
As described previously (Li et al., 2009), 107 spleen cells from C57BL mice were incubated with either a high dose (5 M) or low dose (0.5 M) of carboxyfluorescein succinimidyl ester (CFSE) in phosphate-buffered saline (PBS) at room temperature for 12 min. Fetal bovine serum (FBS) was then added to stop CFSE labeling. After washing, CFSEhigh cells were incubated with OVA SIINFEKL (10 μg/ml). After peptide pulsing, two populations (CFSEhigh and CFSElow) of target cells were washed and mixed together, and then 107 cells of each population were injected into AAV8OVA-immunized mice via the tail vein. Spleen cells were collected 24 hr later and analyzed by flow cytometry. The percent specific lysis was determined by the following formulas: the ratio of recovery of non-peptide-treated control spleen cells to peptide-sensitized spleen cells=(percentage of CFSElow cells)/(percentage of CFSEhigh cells). The percent specific lysis (%)=100×[1−(ratio of cells recovered from naive mice/ratio of cells recovered from infected mice)].
In vivo T cell proliferation
C57BL/6 mice received AAVOVA vector via retro-orbital administration. At various time points after AAV injection, CFSE-labeled OT-1 T cells were transferred and T cell proliferation was measured. OT-1 T cells were isolated from spleen and incubated for 10 min at room temperature in PBS with 5 μM CFSE at 2×107 cells/ml. The reaction was stopped by the addition of FBS, cells were washed with PBS, and 5×106 CFSE-labeled OT-1 T cells were retro-orbitally injected into C57BL/6 mice. On day 10 after OT-1 T cell transfer, spleen cells were harvested and stained with antibodies specific for TCR α-chain Vα2 and CD8, or with PE-conjugated H-2Kb/SIINFEKL tetramer. The frequency of proliferating T cells was determined by the percentage of CFSE dilution (Krishnan et al., 2007). The proliferation index (PI) was calculated as the ratio of the total number of cells to the number of cells initially residing in the parent generation.
Statistical analysis
The Student t test was used to carry out statistical analysis. p<0.05 was considered statistically significant.
Results
AAV8OVA capsid stimulates CD8+ OT-1 T cell reactivity
The AAV8 capsid protein is associated with increased transduction efficiency and broader tissue tropism compared with the AAV2 capsid, based on animal studies (Wu et al., 2008). To study capsid-specific CTL reactivity, we previously established a system in which the AAV2 virus was engineered with an HI loop substitution in the AAV2 capsid, using the chicken ovalbumin immunodominant SIINFEKL peptide sequence (AAV2OVA) (Li et al., 2009). Using this engineered AAV2OVA virus, we demonstrated that incubation of AAV2OVA-transduced H-2Kb-expressing cells with spleen cells from OT-1 mice induced the activation of spleen cells in vitro, and administration of AAV2OVA virus elicited a capsid-specific CTL immune response in mice (Li et al., 2009, 2013). To investigate AAV8 capsid antigen presentation and immunogenicity, an AAV8OVA capsid, prepared similarly to AAV2OVA, was engineered by replacing the AAV8 HI loop with the OVA SIINFEKL peptide. First we confirmed that the SIINFEKL epitope was processed and presented, leading to OT-1 T cell stimulation. 293 cells were cotransfected with AAV8OVA plasmid, an H-2Kb expression plasmid, and pXX6-80, which encodes adenoviral proteins to induce capsid expression. The stimulatory capacity of the 293 transfectants was then tested by coculture of the cells with OT-1 T cells and by measuring upregulation of CD69 expression. 293 cells transfected with AAV8OVA plasmid activated OT-1 T cells to a similar extent as AAV2OVA transfectants, whereas cells transfected with a plasmid encoding wild-type AAV8 capsid or mock transfected did not (Fig. 1). Next, we assessed the T cell-stimulatory capacity of HepG2/H-2Kb cells, an H-2Kb-expressing hepatocyte cell line (Li et al., 2013) that were transduced with AAV8OVA packaged vector. Whereas wild-type AAV8 vector-transduced HepG2/H-2Kb cells did not activate OT-1 T cells relative to the no-vector control (0.20 and 0.12% CD8+CD69+ cells, respectively), AAV8OVA transduction resulted in an increased percentage of CD8+CD69+ T cells (1.29%). However, this frequency was significantly lower than that detected in cultures of AAV2OVA-transduced HepG2/H-2Kb cells (10.89%; Fig. 1). These results indicate that the SIINFEKL peptide in the context of the AAV8 capsid is processed and presented, and stimulates CD8+ T cells albeit at a reduced level compared with that resulting from the AAV2OVA capsid.
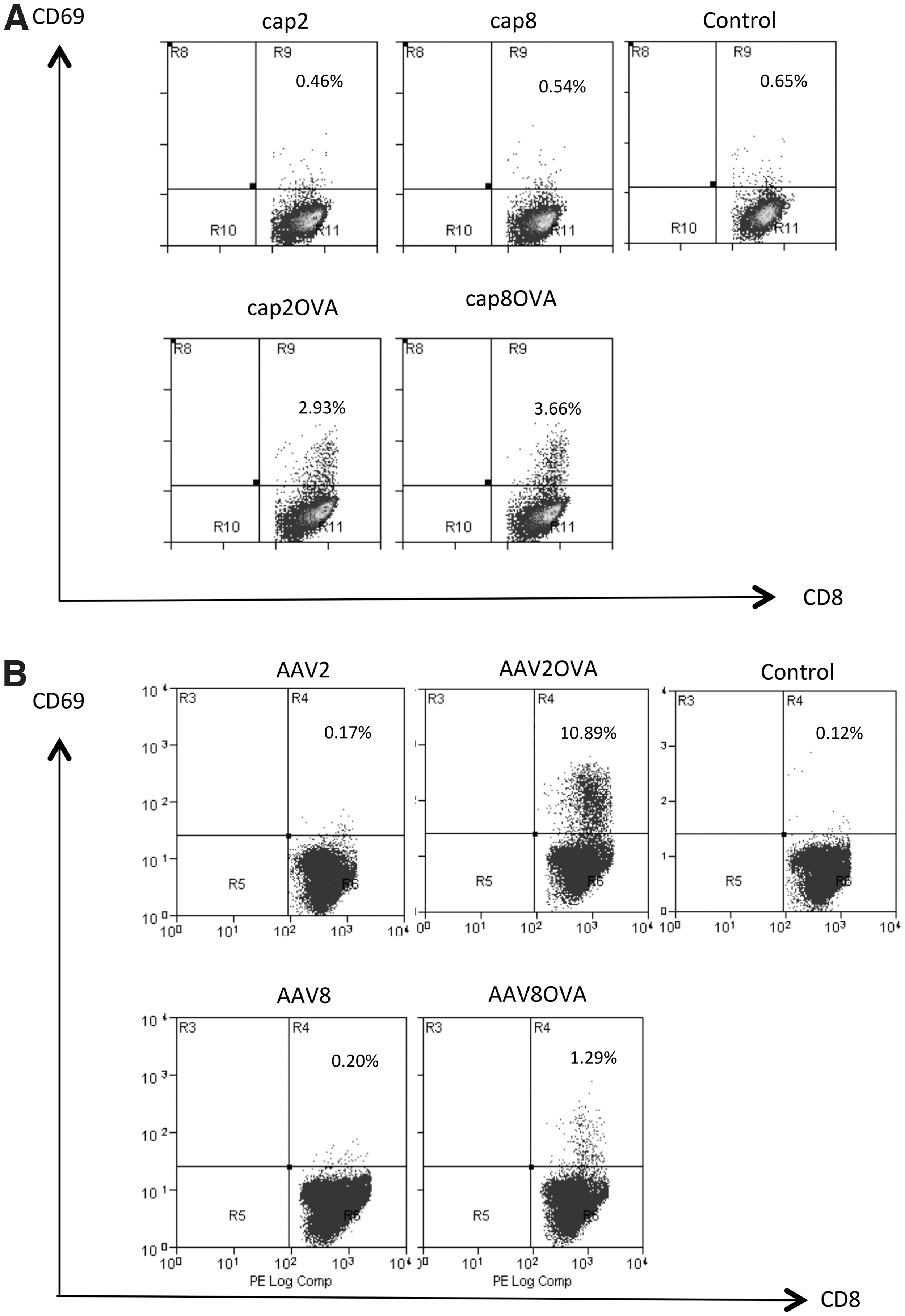
AAV8OVA capsid protein stimulates OT-1 T cells in vitro. Activation of OT-1 T cells, based on upregulation of CD69 expression, was measured via flow cytometry on coculture with
Similar transduction efficiency and tropism in vivo between AAV8OVA and wild-type AAV8 vectors
To study antigen presentation of the AAV8 capsid, we initially determined whether insertion of the SIINFEKL epitope into the HI loop of the AAV8 capsid altered tissue tropism in vivo by monitoring luciferase expression in vivo in mice injected with AAV8OVA/luc vector. As shown in Fig. 2A, the pattern of luciferase expression was similar for animals injected with AAV8OVA/luc and wild-type AAV8/luc. Notably, for both AAV8 vectors the strongest signal was localized to the liver. Next, we injected AAV8OVA/AAT vector intravenously into mice, and measured blood levels of AAT. Similar levels and kinetics of AAT expression over 3 months were detected in mice treated with the AAV8/AAT and AAV8OVA/AAT vectors. Transgene levels peaked within 2 weeks postinjection, remained stable over 10 weeks, and decreased slightly at week 14 in both groups (Fig. 2B).
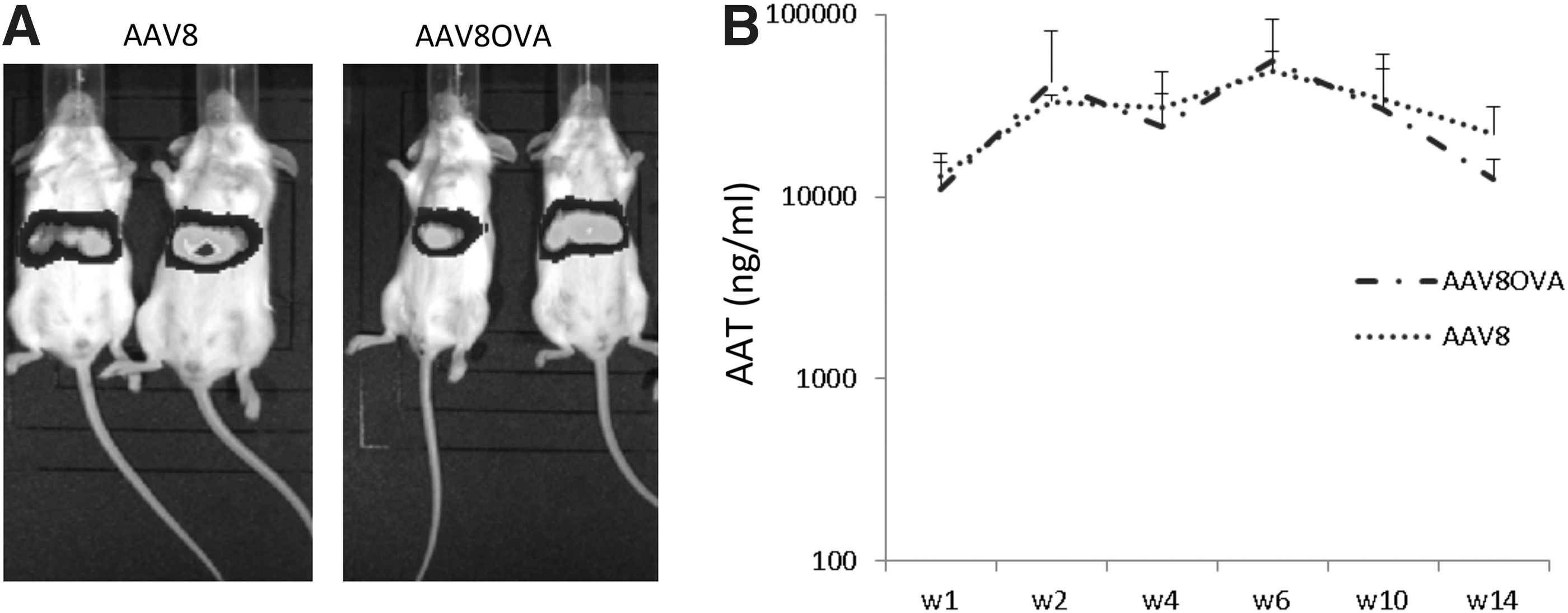
AAV8OVA- and wild-type AAV8-packaged vectors exhibit similar transduction profiles.
T cell response to AAV8OVA capsid in mice
In previous studies, we demonstrated that muscular administration of AAV2OVA vector mounts an OVA peptide-specific CTL response (Li, Hirsch et al., 2009). To determine whether administration of the AAV8OVA vector elicited a capsid CTL response in vivo, we administered AAV8OVA/AAT vector at a dose of 1×1011 particles via muscular injection into C57BL6 mice. Thirty days after AAV8OVA injection, CFSE-labeled naive syngeneic mouse spleen cells pulsed with OVA SIINFEKP peptide were infused into mice. After 24 hr, the mice were killed and the amount of cells pulsed with OVA peptide was calculated by flow cytometry. As shown in Fig. 3, compared with control mice, no obvious killing of OVA peptide-pulsed cells was observed in mice receiving AAV8 or AAV8OVA vector.
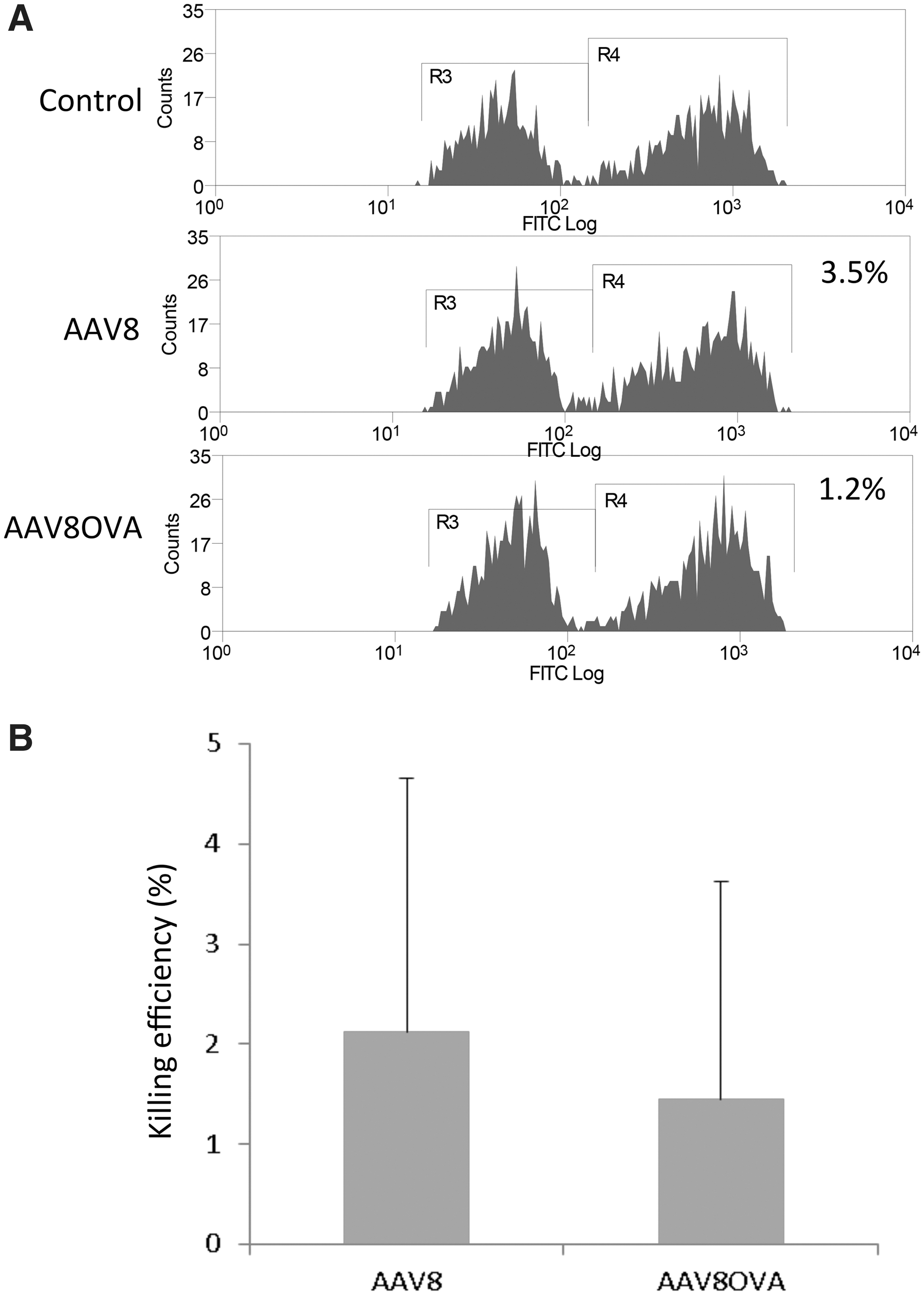
No OVA-specific CTL-induced cytotoxic activity was elicited by muscular injection of AAV8OVA vector. AAV8OVA/AAT vector (1×1011 particles) was injected into the muscle of C57BL/6 mice, and 4 weeks later the mice received via tail vein injection a mixture of C57BL/6 mouse spleen cells labeled with either a high concentration of CFSE (CFSEH) and pulsed with SIINFEKL peptide, or a low concentration of CFSE (CFSEL). After 24 hr, single cells were isolated from the spleens and analyzed by flow cytometry to determine in vivo CTL-mediated killing.
Cross-presentation of AAV capsid antigen is dose dependent in vivo
The efficiency of cross-presentation is weak compared with endogenous antigen presentation. Usually high exogenous antigen is required to elicit a strong cellular immune response. In this study, we determined the efficiency of in vivo SIINFEKL presentation by measuring the proliferation of transferred OT-1 T cells. To investigate the effects of the dose of AAVOVA capsid on antigen presentation and OT-1 T cell stimulation, various particle numbers of AAV2OVA vector were injected into C56BL/6 mice. When compared with control mice without AAV2OVA treatment, a dose of 5×1010 particles and higher induced OT-1 T-cell proliferation (Fig. 4). However, no proliferation of OT-1 T cells was detected in groups with doses of AAV2OVA less than or equal to 1×1010 particles. Similar dose-dependent antigen presentation was observed when using the AAV8OVA vector in vivo (data not shown). These data suggest that efficient T cell stimulation occurs only with high doses of AAV vector.
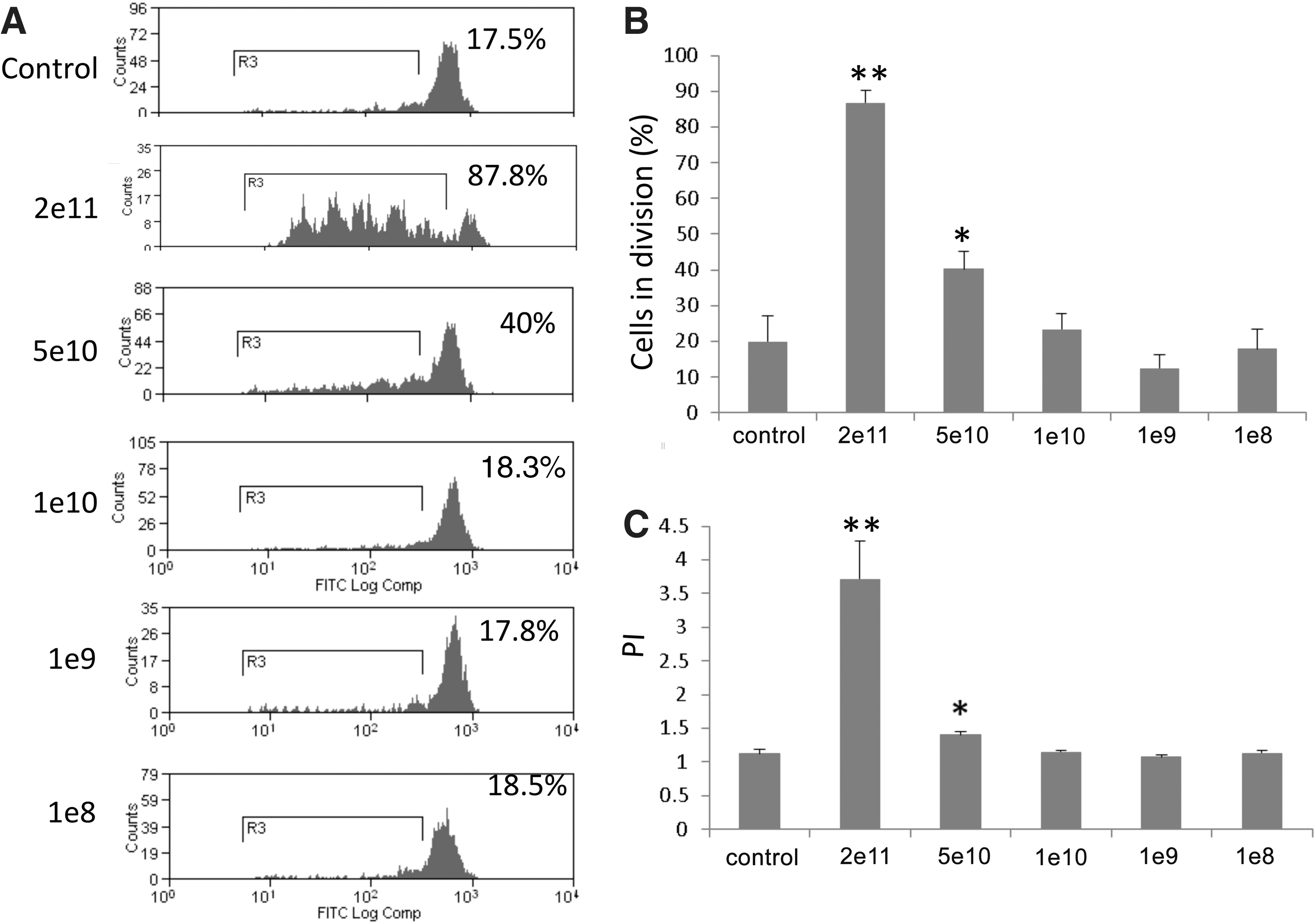
Capsid antigen presentation after AAVOVA transduction is dose responsive in vivo. Various doses of AAV2OVA/AAT vector were injected intravenously into C57BL/6 mice and 3 days later, CFSE-labeled OT-1 T cells were transferred. On day 10 after transfer OT-1 T cell proliferation in the spleen was assessed via flow cytometry.
The extent and kinetics of AAV8 capsid antigen presentation in vivo are similar to that of the AAV2 capsid
It has been shown that transgene expression gradually increases after AAV2 administration in mice and peaks at week 6, remaining stable long-term after vector injection. Although the epitopes from the AAV2 capsid are cross-presented by antigen-presenting cells (APCs) to elicit a capsid-specific CTL response, it is unclear whether the kinetics of antigen presentation of capsid epitopes in vivo after AAV vector administration correspond with the dynamics of transgene expression. To address this question, AAV2OVA/AAT vector (2×1011 particles) was injected intravenously, and at various time points splenic OT-1 T cells labeled with CFSE were transferred into mice. Ten days posttransfer OT-1 T cell division was measured by flow cytometry. As shown in Supplementary Fig. S2, over days 3–12 and days 21–30, OT-1 T cell division was significantly increased in AAV2OVA/AAT vector-treated (86.73 and 55.63%, respectively) versus control recipients (30 and 24.7%, respectively; p<0.01 and p<0.05, respectively). However, no difference was observed for OT-1 cell proliferation between AAV2OVA and control groups over days 41–50 and days 61–70 (p>0.05). This result indicates that antigen cross-presentation from the AAV2 capsid occurs during the earlier time period after AAV2 systemic administration.
To elucidate the kinetics of antigen presentation from AAV8 transduction in vivo, we injected 1×1011 particles of AAV8OVA/AAT or AAV2OVA vector into C57BL/6 mice, and assessed proliferation of CFSE-labeled OT-1 T cells transferred at various times after vector treatment. As shown in Fig. 5, the frequency of proliferating OT-1 T cells was significantly increased in mice injected with AAV8OVA between days 3–12 and days 21–30, relative to the control group (p<0.05). Although slightly more cells in division from AAV8OVA application were observed compared with that from control at later time points (days 41–50 and days 61–70), these findings were not statistically significant (Fig. 5B). Furthermore, no marked difference was observed in the level and kinetics of OT-1 T cell proliferation between animals treated with AAV8OVA and AAV2OVA. These results demonstrate that presentation of the SIINFEKL epitope is similar for both AAV8 and AAV2 capsids.
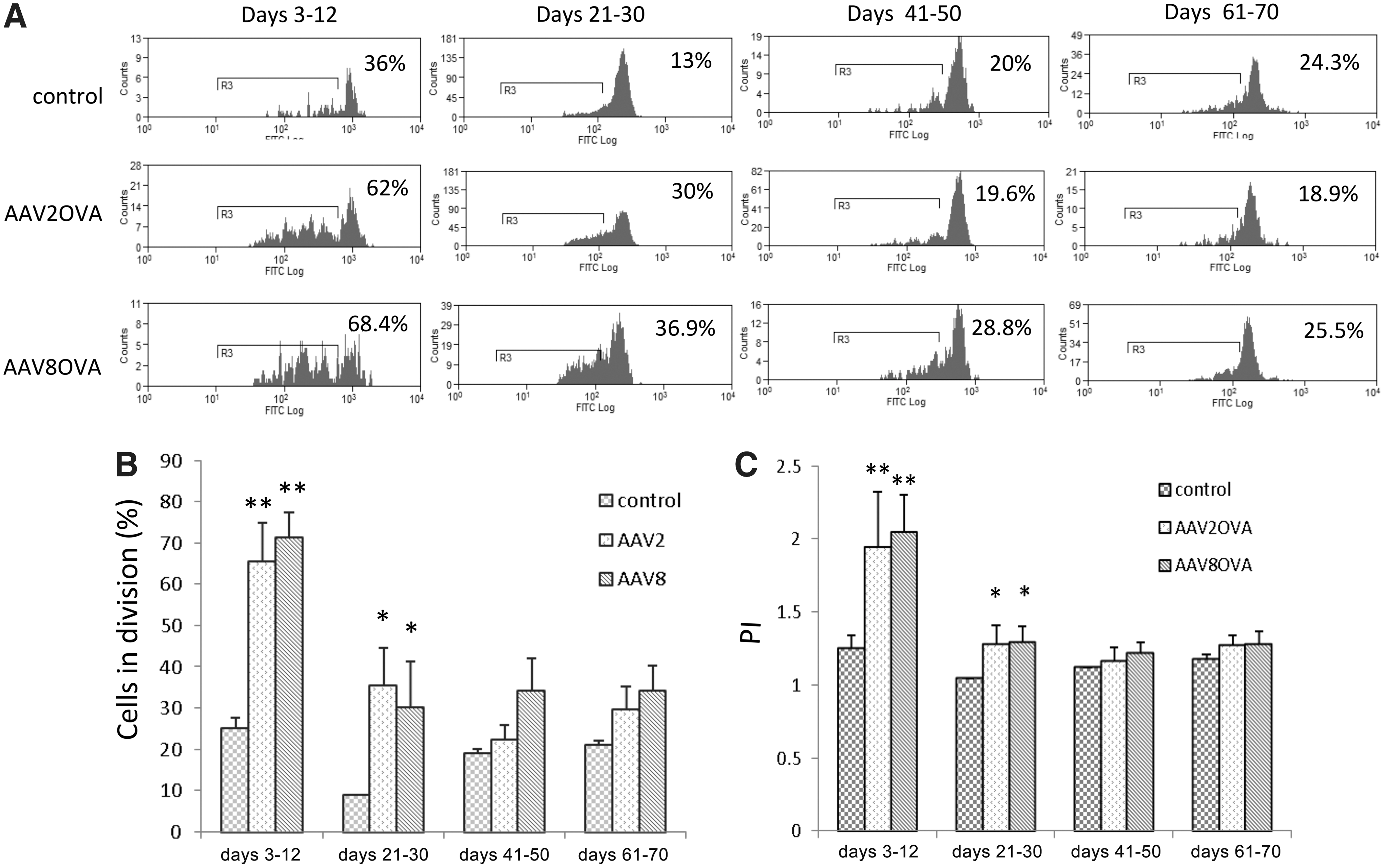
The kinetics of capsid antigen presentation after AAV8OVA transduction in mice. Particles of AAVOVA/AAT virus (1×1011) were injected intravenously into C57BL/6 mice, and at the indicated time points 5×106 CFSE-labeled OT-1 T cells were transferred. Ten days after transfer proliferation of CD8+ OT-1 T cells was measured by flow cytometry.
Discussion
In this study we demonstrated through plasmid transfection and in vitro virus infection that integration of the OVA immunodominant peptide SIINFEKL into the AAV8 capsid induces antigen processing and presentation. Transgene expression pattern and kinetics subsequent to AAV8OVA vector administration were similar to those of wild-type AAV8 in vivo. A functional T cell response against OVA peptide was not observed in vivo after AAV8OVA administration. Antigen cross-presentation from AAV2 transduction in vivo is dose dependent. Most significantly, similar to the AAV2OVA vector, efficient antigen presentation on AAV8OVA vector transduction in vivo occurred within an early time period.
It has been thought that AAV vectors induce a low cellular immune response against the capsid because AAV vector transduction does not produce any endogenous capsid protein. However, clinical trials have suggested that capsid antigen can be cross-presented on AAV transduction and that AAV-transduced target cells can be eliminated by capsid-specific CTLs (Manno et al., 2006; Nathwani et al., 2011). In one clinical trial, an AAV2 vector was used to deliver F9 into the liver via the arterial route. High F9 expression was observed at week 4 after AAV2 injection and remained stable for 2 weeks before returning to the pre–gene therapy baseline. It was suggested that memory CTLs against the vector capsid were activated to clear AAV2-transduced hepatocytes (Manno et al., 2006). Although capsid-specific CTL clones were isolated from the patient, and numerous experiments confirmed that capsid cross-presentation can also be induced on AAV vector transduction in animal models, the results from this study and others about the kinetics of AAV capsid cross-presentation, in which efficient antigen presentation occurs in earlier periods after AAV transduction, do not support the conclusion from the clinical trial (H. Li et al., 2011). Alternative hypotheses have been proposed, such as cryptic epitopes derived from the F9 cDNA alternative reading frame (Li et al., 2009a), which may provide an alternative rationale for the time course of F9 decline observed in patients.
AAV8 is one of the most efficient liver-transducing serotypes in animal models (Gao et al., 2006; Wu et al., 2008; C. Li et al., 2011; Wang et al., 2012). AAV8 has been employed to treat patients with severe hemophilia B in a phase 1 clinical trial (Nathwani et al., 2011). Eight patients were enrolled for treatment with self-complementary AAV8 vectors encoding a codon-optimized human F9 transgene via peripheral vein administration. At initial report, four of six patients have discontinued F9 prophylaxis without spontaneous bleeding after a follow-up of 6–16 months. One of two patients receiving a high dose of 2×1012 AAV8 particles per kilogram had elevated liver transaminases with a drop in F9 level on day 62 after gene transfer. Enzyme-linked immunospot analysis detected a capsid-specific T cell response at 9 weeks, suggesting that the decrease in F9 was related to capsid-specific CTL reactivity (Nathwani et al., 2011). In this study we have demonstrated that the efficiency of antigen cross-presentation on AAV8 transduction in vivo was confined to the earlier time points, similar to that with the AAV2 vector. So, similarly, these results do not support the current interpretation from AAV8/F9 clinical trials. To completely validate in vivo mouse studies further experiments are needed to test whether the kinetics with human hepatocytes is different from that with mouse cells. In addition, alternative interpretations of AAV liver transgene expression and CTL response correlation remain open-ended and may require further clinical studies specifically designed to answer these concerns.
Several studies have demonstrated that muscular injection of AAV2 elicits a CTL response against capsid (Chen et al., 2006; Li et al., 2007; Wang et al., 2007); however, the application of AAV8 vector did not induce a capsid-specific CTL response in mice (Wang et al., 2007). Consistent with this observation, activation of capsid-specific T cells was not induced after muscular injection of AAV8OVA vector in this study. One possibility is that AAV8 binding on the surface of APCs is not mediated by heparan sulfate proteoglycan, the primary receptor for AAV2 (Summerford and Samulski, 1998; Vandenberghe et al., 2006). It has been demonstrated that heparan binding directs activation of T cells against AAV 2 capsid (Vandenberghe et al., 2006). The lack of heparan binding by AAV8 may therefore account for the lack of T cell activation by this vector.
Results from the aforementioned two clinical trials in patients with hemophilia B suggest that capsid-specific CTL responses eliminate AAV-transduced cells at increasing vector doses (Manno et al., 2006; Nathwani et al., 2011). The data from our study are consistent with the finding that higher doses of vector result in elevated antigen presentation on the surface of target cells. To achieve optimal transgene expression and decrease antigen load from input AAV vectors, several approaches have been exploited: (1) generation of double-stranded AAV vectors that can induce more rapid and increased transgene expression compared with conventional single-stranded cassettes (McCarty et al., 2001, 2003; Wu et al., 2008); (2) modification of transgene cassette to enhance transgene expression through optimization of the promoter, poly(A), and/or transgene cDNA codon (Wu et al., 2008); and (3) removal of empty particles in AAV preparations to decrease virus antigen load and increase transduction by decreasing competition for AAV binding and intracellular trafficking with full virions (Urabe et al., 2006).
In conclusion, our study has demonstrated that AAV8 engineered with an OVA peptide can induce proliferation of OVA-specific T cells through transfection of plasmid and infection with virus, thus allowing for the monitoring of in vivo capsid antigen presentation kinetics. There is a dose dependency for AAV capsid antigen presentation in vivo. Last, the kinetics of capsid antigen cross-presentation from AAV8 is similar to that of AAV2 after in vivo administration—antigen presentation for both serotypes occurs within an early time-frame. Continued studies on the kinetics of capsid antigen presentation from AAV transduction in vivo will help in the design of effective approaches to prevent and block capsid-specific CTLs against AAV-transduced target cells in future clinical trials.
Footnotes
Acknowledgments
This work was supported by National Institutes of Health grants 1R01AI080726 and 5R01DK084033 (to C.L. and R.J.S.), 5U54AR056953 and 5R01AI072176 (to R.J.S.), F32NS070356 (to M.S.W), and 5R01DK081585 (to R.T.). The UNC Flow Cytometry Core Facility is supported in part by an NCI Center Core Support grant (P30CA06086) to the UNC Lineberger Comprehensive Cancer Center. The authors thank Xiaojing Chen, Karen Hogan, and H. Sophia Shih for excellent technical assistance.
Author Disclosure Statement
No competing financial interest exists.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.