
Abstract
The loss of dystrophin or its associated proteins results in the development of muscle wasting frequently associated with cardiomyopathy. Contractile cardiac tissue is injured and replaced by fibrous tissue or fatty infiltrates, leading to a progressive decrease of the contractile force and finally to end-stage heart failure. At the time symptoms appear, restoration of a functional allele of the causative gene might not be sufficient to prevent disease progression. Alterations in Ca2+ transport and intracellular calcium levels have been implicated in many types of pathological processes, especially in heart disease. On the basis of a gene transfer strategy, we analyzed the therapeutic efficacy of primary gene correction in a δ-sarcoglycan (δ-SG)-deficient animal model versus gene transfer of the Ca2+ pump hSERCA2a (human sarco-endoplasmic reticulum calcium ATPase 2a), at a symptomatic stage of heart disease. Our results strongly suggest that restoration of δ-SG at this stage of disease will not lead to improved clinical outcome. However, restoration of proper Ca2+ handling by means of amplifying SERCA2a expression in the myocardium can lead to functional improvement. Abnormalities in Ca2+ handling play an important role in disease progression toward heart failure, and increased SERCA2a levels appear to significantly improve cardiac contraction and relaxation. Beneficial effects persist at least over a period of 6 months, and the evolution of cardiac functional parameters paralleled those of normal controls. Furthermore, we demonstrate that a plasmid formulation based on amphiphilic block copolymers can provide a safe and efficient platform for myocardial gene therapies. The use of synthetic formulations for myocardial gene transfer might thus overcome one of the major hurdles linked to viral vectors, that is, repeat administrations.
Introduction
H
As such, mutations in the dystrophin gene and in many of its associated proteins such as the sarcoglycans have been described to cause cardiomyopathies (Morita et al., 2005). The sarcoglycan complex constitutes a transmembrane component of the dystrophin–glycoprotein complex (DGC), which links the cytoskeleton to the extracellular matrix (Wheeler and McNally, 2003). δ-Sarcoglycan (δ-SG) mutations may lead most frequently to a complete loss or a strong reduction of the whole SG complex (Duggan et al., 1997) and are associated with a form of autosomal recessive limb-girdle muscular dystrophy 2F (LGMD2F). Patients with LGMD2F present progressive weakness, with respiratory failure and dilated cardiomyopathy (Nigro et al., 1996). The precise pathophysiology of the cardiomyopathy remains largely unknown, but disruption of the DGC, due to a δ-SG mutation, is the leading molecular event, which triggers the disease as it induces weakening of the mechanical connection between the contractile apparatus and the cell membrane. However, the membrane instability also allows increased entry of calcium via nonspecific channels and mitochondrial disorganization (Fraysse et al., 2010). Ample evidence suggests that abnormal elevation of cytosolic calcium may play a central role in the pathogenesis of heart disease (Dunn and Radda, 1991; Alloatti et al., 1995; Williams and Allen, 2007; Fauconnier et al., 2010; Sarma et al., 2010).
The sarcoplasmic reticulum (SR) contains large stocks of calcium, which it sets apart and then releases when the cardiac cell is stimulated; SR Ca2+ uptake from the cytosol is regulated mainly by the SERCA2a (sarco-endoplasmic reticulum calcium ATPase 2a) Ca2+ pump, which plays a major role in cardiac contraction and relaxation (Toyoshima et al., 2000). Key defects in Ca2+ cycling occur at the level of the sarcoplasmic reticulum during the development of heart failure (Ribadeau et al., 1997; Netticadan et al., 2000; Frank et al., 2003).
Therapeutic approaches aiming at correcting the molecular defects and based on gene transfer are gaining increased interest as transfer efficacy improves. However, this raises questions about the proper drug to administer. Is correction of the primary genetic defect enough the correct the phenotype? Is the therapeutic effect mainly a problem of quantitative gene transfer? It might have seemed obvious, at least in autosomal recessive diseases, that addition of a normal allele of the disease causing gene should lead to cure. Some experimental results sustain such options at early stages of disease in animal models.
Our hypothesis is that the initial hereditary genetic defects trigger a heart failure program that will be responsible for the symptoms at various stages of the disease. Over time these secondary molecular changes might become prevailing and independent, and therefore should be considered the primary therapeutic targets.
To evaluate this working hypothesis we used the CHF147 hamsters described by Hunter and colleagues (1984). This is a representative model of DCM, which results from a mutation in the δ-SG gene leading to a complete deficiency in the corresponding protein (Nigro et al., 1997; Sakamoto et al., 1997). As shown previously, CHF147 hamsters display a clear cardiac phenotype revealing progressive left ventricle dysfunction at about 155 days of age (Blain and Straub, 2011). On the basis of adeno-associated virus (AAV), gene transfer of δ-SG early in the life of deficient hamsters provides beneficial results regarding cardiac disease (Hot et al., 1998; Li et al., 2003). Taking into account these studies in young hamsters, we considered AAV-based treatment for the reexpression of δ-SG in older hamsters with symptomatic cardiac disease. As shown previously, intracardiac injections of AAV-based vectors allow the achievement of significant reporter gene expression in adult hamsters. Furthermore, in speculating on secondary molecular changes, we evaluated expression of an exogenous human cDNA encoding SERCA2a in this same setting. Considering that such a strategy might need repeat administration of gene products and that the use of AAV might limit the efficacy of repeated doses because of immune responses, we opted for a nonviral formulation of human SERCA2a (hSERCA2a). On the basis of former work, we used a Pluronic block copolymer formulation. Some studies showed their efficacy in cardiac and skeletal muscle gene transfer and an optimized formulation for dystrophic muscle (Kabanov et al., 2002; Roques et al., 2009a).
Materials and Methods
Plasmid pCMV-XL5-hSERCA2a expressing Homo sapiens cardiac sarco-endoplasmic reticulum calcium ATPase 2a (SC326316; OriGene, Rockville, MD), and plasmid pCMV-δ-SG expressing the Mesocricetus auratus δ-sarcoglycan (GenBank accession no. AB001508), were used in the study. The plasmids were amplified in One Shot TOP10 chemically competent Escherichia coli (Life Technologies/Thermo Fisher Scientific, Lyon, France) and purified with an EndoFree plasmid giga kit (Qiagen, Courtaboeuf, France). Pluronic P85 was obtained from BASF (Parsippany, NJ). Ringer lactate solution (Sigma-Aldrich, Lyon, France) and all solvents were of analytical grade. P85 solution was prepared in Ringer lactate at 10% (w/v), and incubated overnight at 4°C. Transfection solutions were prepared by mixing an equal volume containing 750 or 1500 μg of pCMV-XL5-hSERCA2a with 0.6% P85; both were preincubated for 30 min at 37°C. Final solutions were incubated at 37°C for 30 min before injection. AAV2/8 CMV-δ-SG (lot #3373) and AAV2/9 CMV-δ-SG (lot #33713372) were provided by the Gene Vector Production Center of Nantes.
Animal studies
Male dystrophic hamsters (strain CHF147; Institute of Myology, Paris, France) and wild-type Syrian hamsters (strain RjHan:AURA; Centre d'Elevage R. Janvier, Le-Genest-St-Isle, France) aged 5 months were used for all experiments. Animals were housed in local facilities, they were maintained at 20°C with 10 hr:14 hr light/dark cycles, and had free access to food and water. The investigation conformed to the Guide for the Care and Use of Laboratory Animals ( National Institutes of Health Publication No. 85-23, revised 1996). Our protocol was approved by the local committee, and gained formal authorization (A-75-11-13). Animals were randomized into six groups: normal group (wild-type control, n=9), saline group (CHF147 not treated, n=9), AAV2/8 CMV-δ-SG-treated group (CHF147 δ-AAV8, n=3), AAV2/9 CMV-δ-SG-treated group (CHF147 δ-AAV9, n=3), hSERCA2a (750 μg)-treated group (CHF147 SERCA2a dose 1, n=3), and hSERCA2a (1500 μg)-treated group (CHF147 SERCA2a dose 2, n=7).
In vivo transfection
CHF147 hamsters were anesthetized by intraperitoneal injection of a mixture containing ketamine (75 mg/kg), xylazine (15 mg/kg), and midazolam (0.75 mg/kg) for induction and eventually by inhalation of isoflurane (0.25–0.5%) in oxygen for maintenance of anesthesia. Animals were placed in the dorsal position; heart rate and body temperature were monitored. Injections were performed with a 27-gauge needle, using an integrated rail system and image-guided needle injection system echograph (Vevo 770; VisualSonics/FUJIFILM SonoSite, Toronto, ON, Canada). One hundred microliters of AAV2/8 CMV-δ-SG or AAV2/9 CMV-δ-SG containing 1×1012 viral particles was delivered directly into the left ventricle. pCMV-XL5-hSERCA2a/P85 formulations (3×100 μl) were injected into three different sites of the left ventricle.
In vivo evaluations
Clinical evaluation based on daily observation of all animals was performed during the study period. Body weight was measured every 2 weeks and weight variations between death and inclusion date were further analyzed.
Echocardiography
All in vivo studies were performed with a high-resolution Vevo 770 system (VisualSonics/FUJIFILM SonoSite), including a high-frequency (40-MHz) transducer. Echographic studies were performed in parasternal long- and short-axis views. All images were recorded and measurements were made offline, using analytic software provided with the system. Two-dimensional and M-mode measurements were performed at the level of papillary muscles in order to obtain left ventricle (LV) dimensions: LV inner and outer diameters during systole and diastole (LVID,s and LVID,d; LVOD,s and LVOD,d). Left ventricular systolic function was assessed by ejection fraction (EF), which was calculated using M-mode, according the usual formula: EF%=100×(LV Vol,d – LV, Vol,s)/LV Vol,d. The average of at least three representative cardiac cycles was considered. All values are expressed as means±SEM. Echocardiograms were performed twice before vector administration (baseline), and then every 2 weeks after the injections.
Electrocardiogram
ECG recordings were made with a restraining system (emka Technologies, Paris, France) similar to the method previously described (Mongue-Din et al., 2007). Briefly, six ECG leads were recorded noninvasively in conscious hamsters for 20 min and repeated every 2 weeks. Analyses were made with dedicated software (ECG-auto; emka Technologies). Rhythm analysis was based on form recognition and allowed identification and quantification of premature extra beats. The duration of cardiac cycles as described by R–R interval duration was analyzed quantitatively according to standard criteria of heart rate variability (HRV). The standard deviation of normal R–R intervals (SDNN) was calculated for each period of 20 min.
Left ventricular pressure–volume loops
Anesthesia was induced with a single intraperitoneal injection of ketamine, xylazine, and midazolam, as described previously. The animals were maintained under stable sedation by additional gas anesthesia (Sevoflurane; Abbott Laboratories, Abbott Park, IL) in O2 under spontaneous ventilation. Briefly, an incision in the neck was made and the right carotid artery and right jugular vein were exposed. A 1.4-Fr high-fidelity pressure-conductance catheter (Millar Instruments, Houston, TX) was introduced through the right carotid artery into the left ventricle. After hemodynamic stabilization, baseline pressure–volume (PV) loops were recorded. A 27-gauge catheter was introduced into the right jugular vein, and parallel volume was determined by a bolus injection of 100 μl of NaCl solution. The inferior caval vein was compressed percutaneously at the level of the liver, while PV loops were recorded (occlusion loops). These maneuvers were performed three times. Only technically acceptable loops (no arrhythmia, stable baseline, etc.) were included in the analysis for each experiment. These analyses were performed blinded for the group to which the animal belonged. Only after all data sheets were established was the group code of the animal unblinded for statistical analysis.
Sacrifice
After 6 months of follow-up, animals were killed with an overdose of anesthetics. Heart muscle was quickly harvested and weighed. The ratio of heart weight to body weight was calculated. Lung, kidney, spleen, and liver were also collected to evaluate the toxicity.
In vitro analysis
Histological analysis
Heart, lung, kidney, spleen, and liver were snap frozen in isopentane cooled in liquid nitrogen and stored at −80°C. Tissues samples were cryosectioned (8–10 μm) and fixed in a 4% paraformaldehyde and 0.2% glutaraldehyde solution. Hematoxylin–eosin (H&E) staining was done and infiltrations of fibrotic tissue and calcifications in heart were observed, respectively, by sirius red (SR) and von Kossa (VK) staining. Fibrosis on SR slides and calcifications on VK slides were quantified with NIS-Elements software (Nikon Instruments, Melville, NY). The ratio of collagen (and calcification) area and total area (%) were determined and compared among the groups.
Biodistribution
Total DNA was extracted with a DNeasy blood & tissue kit (Qiagen) at apical and basal ventricular levels. PCR was carried out for 40 cycles with a GoTaq kit (Promega, Charbonnieres, France) with specific primers to the expression cassette CMV-δ-SG and CMV-hSERCA2a (see Supplementary Table S1; supplementary data are available online at
Messenger expression
Total RNA was extracted from the middle levels of the ventricle, using an RNeasy fibrous tissue mini kit (Qiagen). Reverse transcription was performed with an Omniscript RT kit (Qiagen). For each gene, the qPCR conditions were optimized to obtain one specific product. Each sample was measured in triplicate. The housekeeping gene RPL18 was used as an endogenous control (Zivcec et al., 2011). Efficiency was calculated from dilution curves and used for quantification according to the Pfaffl method (Pfaffl, 2001) (see Supplementary Table S1).
Immunohistochemistry
δ-SG and hSERCA2a protein expression was revealed by immunofluorescence staining. Frozen heart sections (8 μm) were blocked with 10% goat serum at room temperature for 30 min. A rabbit monoclonal anti-δ-SG antibody (diluted 1:100; CovalAb, Villeurbanne, France) and a mouse monoclonal anti-hSERCA2a antibody were used (diluted 1:200; CliniSciences, Nanterre, France) for 2 hr at room temperature. Slides were mounted with VECTASHIELD mounting medium with 4′,6-diamidino-2-diphenylindole (DAPI) (Vector Laboratories). Microscopy observations were performed with a Leica microscope and image acquisition was carried out with TRIBVN ICS software (TRIBVN, Chatillon, France).
Statistical analysis
Significance was tested using nonparametric statistical tests. The Dunn test was used as a post-hoc test in analysis of variance. The Kruskal–Wallis test was used to compare groups and the Mann–Whitney test was used for nonparametric comparisons. The Dunnett test was performed for post-hoc analysis for each treatment group compared with a single control group. Data are presented as means±SEM. p<0.05 was considered significant.
Results
Our therapeutic strategy was based on myocardial gene transfer via direct injections. One animal died prematurely in the sham group, and one died in the experimental group hSERCA2a (dose 2). No drug-related cause of death could be highlighted for these animals; not having completed the full study period they were not included in the final statistical evaluations. At autopsy of the various cohorts, no significant lesions of vital organs or lesions related to the injection technique could be detected and hearts were excised carefully for further studies. Variations of body weight and heart weight/body weight ratio are reported in Supplementary Table S2. Overall ratios of heart weight to body weight did not reveal significant changes. However, it is noteworthy that CHF147 hamsters treated with δ-AAV9 had a significant degree of cardiac hypertrophy.
δ-SG gene restoration
Assuming that δ-SG is the main triggering event in the development and progression of cardiomyopathy, we started our study by injecting AAV2/8 CMV-δ-SG or AAV2/9 CMV-δ-SG into the left ventricle of symptomatic CHF147 hamsters with the aim of reversing or halting the cardiac disease. Careful in vivo follow-up did not reveal episodes of arrhythmia. Heart rate variability parameters demonstrated stable evolution over the study period (see Supplementary Table S3).
Repeated echocardiographic evaluation did not demonstrate improved cardiac function (Fig. 1A). In fact, the ejection fraction continuously decreased over the treatment period in CHF147 hamsters treated with AAV8 or AAV9, similarly to untreated animals.

δ-Sarcoglycan (δ-SG) gene transfer in hamster heart.
Biodistribution and expression of the δ-SG transgene were assessed in various organs. CMV-δ-SG was clearly detected in total heart DNA extracts by PCR assay and analysis by agarose gel electrophoresis (Fig. 1B). Unlike other organs, CMV-δ-SG appeared at a high copy number in liver after the administration of AAV2/8 CMV-δ-SG (data not shown).
Relative quantification of δ-SG gene expression confirmed a background expression level of only 2.6% compared with the normalized wild-type control (p<0.001). Groups treated with δ-SG did not show a significant increase in δ-SG with reference to the nontreated group (Fig. 1C).
Protein expression was assessed by immunohistology and no significant cardiac expression of δ-SG could be found in CHF147 hamsters treated with AAV8 or AAV9.
The δ-SG deficiency triggers a progressive cardiomyopathy, which induces over time secondary molecular changes in the myocardium. mRNA expression of endogenous SERCA2a and phospholamban (PLN) was analyzed: SERCA2a and PLN expression was decreased to 47 and 26%, respectively, compared with wild-type control (Fig. 2).
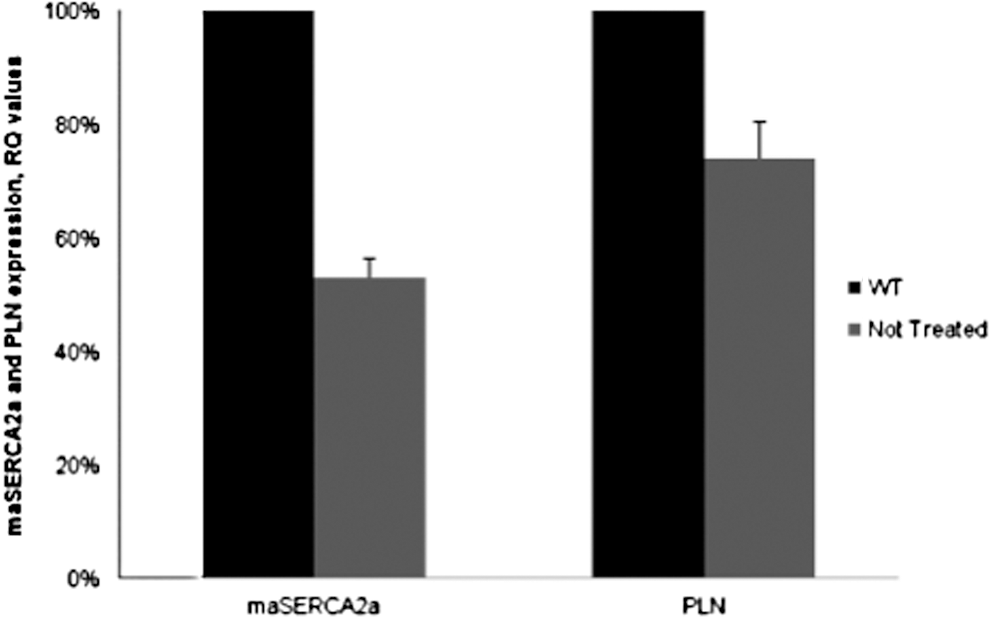
Relative quantification of endogenous hamster SERCA2a (mesocricetus auratus, maSERCA2a) and phospholamban (PLN) expression in hamster heart. RQ values correspond to fold changes in ratios of nontreated CHF147 hamsters normalized to wild-type controls. Values are shown as means±SEM.
hSERCA2a gene therapy
Impact of transfer of an additional SERCA2a gene copy into CHF147 animals was evaluated after injection of a P85 formulation of pCMV-XL5-hSERCA2a. Animals were assessed prospectively for a period of 6 months.
Exogenous SERCA2a did not induce arrhythmogenicity. Time-domain analysis of cardiac cycle length variability confirmed the reduced HRV in CHF147 hamsters with reference to wild-type hamsters. HRV in CHF147 hamsters remained stable during the study period (see Supplementary Table S3).
Repeated echocardiographic studies revealed that animals treated with hSERCA2a experienced beneficial effects on cardiac structure and function (Fig. 3). Basically, left ventricular systolic function, as measured by EF, decreased in all CHF147 hamsters. However, starting with comparable EF values in all CHF147 groups, over time the parameter decreased by 15% in the nontreated group compared with 4% in the treated groups (p<0.001) (Fig. 3A).
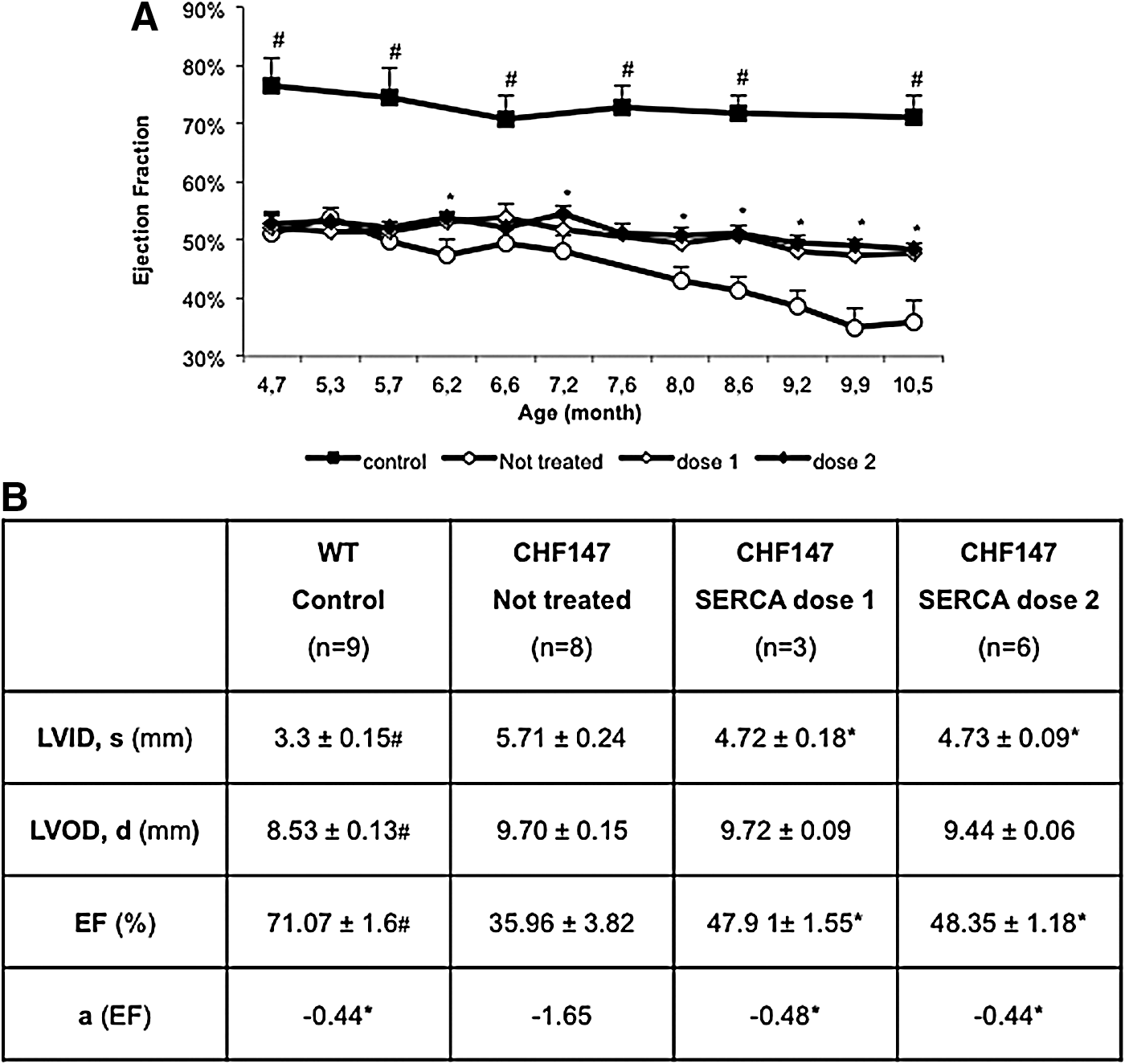
Cardiac function and size parameters assessed by echocardiography.
More interestingly, we observed that the slope of evolution of the EF of the treated groups paralleled that of wild-type animals (−0.44 vs. −0.48 at dose 1, and −0.44 vs. −0.44 at dose 2) (Fig. 3B).
Cardiomyopathy evolved mostly toward dilation by the end of the study period. LVID had increased by 28% in the nontreated group (p<0.001) whereas dilation was limited to 12 and 15% in groups hSERCA2a dose 1 and dose 2, respectively, with reference to data obtained at inclusion (Fig. 3B). Interestingly, hSERCA2a treatment allowed a significant degree of protection (p<0.001) against LV dilation, but no clear dose effect could be observed. LVOD values were not significantly different between CHF147 groups at any time point, suggesting greater wall thinning in untreated animals.
Further functional studies were based on in vivo cardiac catheterization and allowed us to obtain more detailed insight at the end of the observation period (Fig. 4B). Developed pressure (DP) decreased significantly in sham animals, and hSERCA2a treatment allowed this parameter to be restored to normal. End-diastolic pressure (P ed) increased in sham animals, and hSERCA2a treatment induced a significant decrease in P ed. Under our experimental conditions treated animals had slightly lower P ed than did wild-type control animals. The maximal rate of increase in pressure during contraction (dP/dt max) was sharply decreased in sham hamsters with reference to wild-type controls (p<0.01). SERCA2a treatment allowed significant improvement of the contractile function (p=0.0437) and treated animals barely differed from controls (p=0.0529). Changes in heart rate were limited as investigations were obtained under anesthesia.
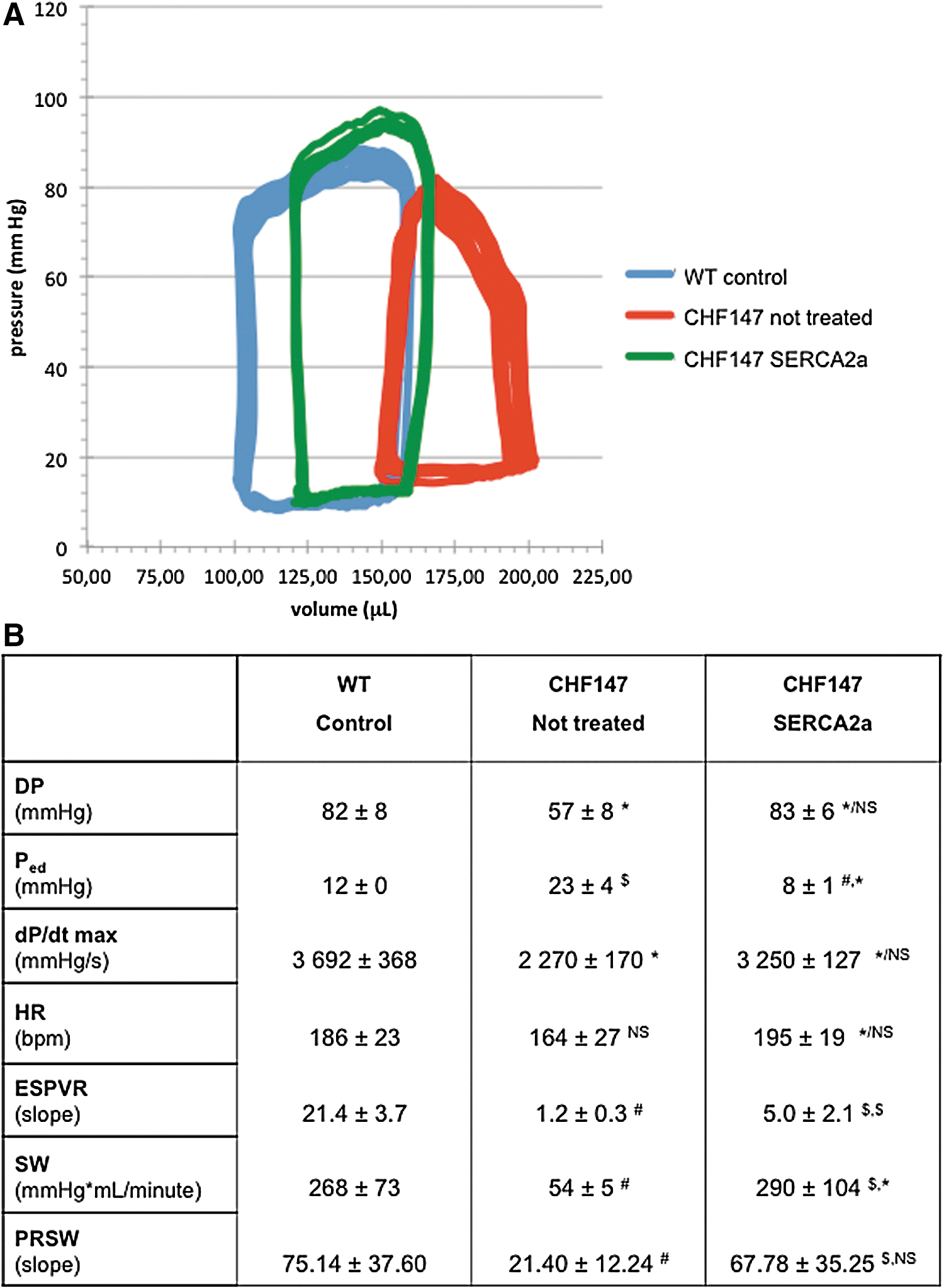
Intracardiac hemodynamics.
Focusing further on intrinsic contractile parameters, the end-systolic pressure–volume relationship was investigated by conduction catheterization. Even if in vivo data might be considered less robust than those observed in isolated heart preparations, our experimental data indicate a clear trend toward improved myocardial contractility after hSERCA2a gene transfer. Figure 4A displays a typical PV loop at steady state in control versus sham and hSERCA2a-treated animals. A clear increase in preload (right shift of the PV curves), increased P ed, and decreased DP can be seen. Statistical analysis of the three groups of animals allowed us to further refine the evaluation (Fig. 4B). On the basis of a linear regression model, the slope of the end-systolic pressure–volume relationship (ESPVR) demonstrated a sharp decline in sham hamsters, as a hallmark of the decrease in contractile function in this cardiomyopathy. hSERCA2a treatment allowed significant restoration (p<0.001), although even so, only partly normal contractility.
Stroke work (SW), as analyzed by ventricular pressure–volume curves, demonstrated a significant drop (268±73 vs. 54±5 mmHg×μl/min; p<0.001) in sham animals, whereas hSERCA2a restored normal SW (Fig. 4B). Furthermore, the relationship between stroke work and end-diastolic volume was studied by its slope, assuming this relationship is linear in hamsters. We observed that this preload-recruitable stroke work was strongly decreased in sham animals (75.1±37.6 vs. 21.4±12.2; p<0.001) and that hSERCA2a treatment also allowed normalized values for this parameter to be obtained with reference to wild-type animals (75.1±37.6 vs. 67.8±35.2; p=NS).
Lusitropy of the CHF147 myocardium was severely altered over time, probably related to the increase in interstitial collagen. Our results showed that hSERCA2a gene transfer partly but significantly improved myocardial relaxation (Supplementary Table S4).
Cardiac muscle structure was analyzed by histology. No significant differences were observed by H&E and VK staining for each CHF147 group (data not shown). In contrast, SR staining (Fig. 5A) revealed that untreated animals had important myocardial fibrosis with increased interstitial collagen (Fig. 5B) (11.2%) compared with wild-type animals (3.8%; p<0.001) and those treated with SERCA2a dose 1 (7.9%; p<0.05) and dose 2 (6.2%; p<0.001). On the other hand, no significant difference was observed between δ-SG-injected and nontreated animals, with 10–12% interstitial collagen content in each group (data not shown).
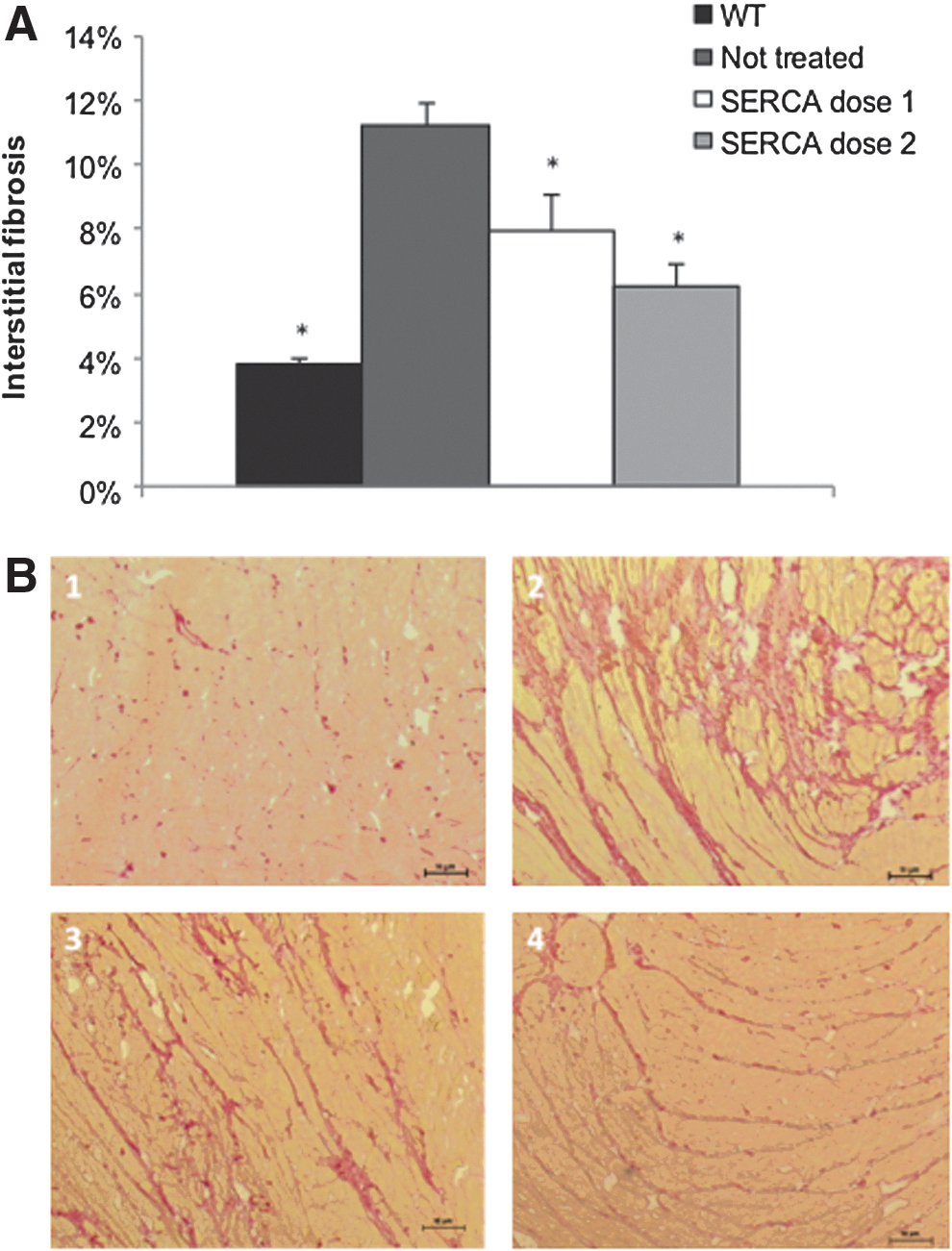
Interstitial fibrosis of the myocardium.
Biodistribution and expression of the hSERCA2a gene were assessed by a PCR assay testing total heart DNA extracts. pCMV-XL5-hSERCA2a was detected in the heart at different levels (Fig. 6A), but was not detected in other organs (data not shown). To compare the expression of hSERCA2a in the injected groups, we performed RT-qPCR. RQ values of hSERCA2a gene expression were higher in the group treated with dose 2 (2.6) compared with dose 1 (2.11) (p<0.001) (Fig. 6B).
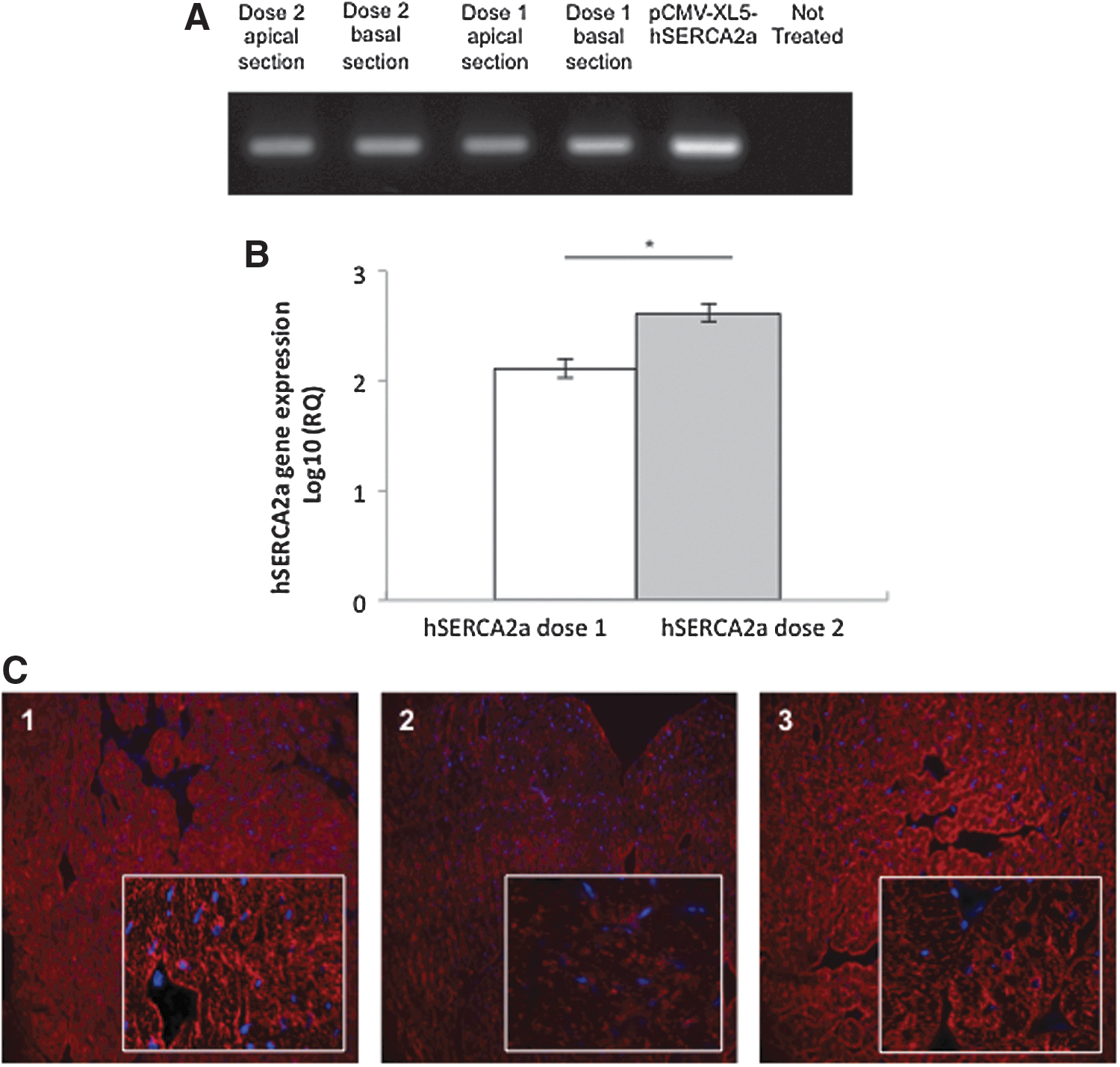
hSERCA2a biodistribution, gene expression, and immunostaining.
Immunostaining with a primary antibody raised against human SERCA2a demonstrated low-intensity staining of untreated hamster heart, revealing interspecies cross-reactivity. However, in treated animal heart, the immunoreactivity clearly increased above this background expression of endogenous hamster SERCA2a (mesocricetus auratus, maSERCA2a), strongly suggesting expression of exogenous hSERCA2a. A similar expression level was observed in the two SERCA2a-treated groups (data not shown). Furthermore, immunostaining showed significant disorganization of the intracellular architecture in the nontreated group compared with SERCA2a-injected animals. SERCA2a treatment seemed to induce improved preservation of intracellular organization of the sarcolemma (Fig. 6C).
Regarding drug development, detection of potential side effects was performed. No macroscopic lesions were observed. Tissue architecture of the spleen, liver, lung, and kidney was observed after H&E staining. No cellular disorganization or infiltrates were found in any group (data not shown).
Discussion
The goal of the current study was to determine the more effective therapeutic approach to treat symptomatic cardiac disease in an animal model for progressive cardiomyopathies.
Our initial approach was to inject a functional allele of the causative genetic defect affecting the δ-SG gene in the CHF147 hamster model, assuming that secondary molecular changes determining the cardiac disease might vanish later on. A second hypothesis based on the assumption that secondary modifications are autonomous and therefore need specific correction was tested by administration of an exogenous gene encoding hSERCA2a.
Many studies have been done concerning gene delivery using AAV in young animals. Holt and colleagues demonstrated the functional rescue of the sarcoglycan complex, using gene transfer in BIO14.6 hamsters (Holt et al., 1998). Later, Zhu and colleagues presented the reconstitution of δ-SG expression in young TO-2 hamsters after an intravenous injection of AAV8 vectors (Zhu et al., 2005). Only a few studies have been done in the late stage, when cardiac disease is established. Hoshijima and colleagues showed that intravenous injection of AAV9/δ-SG in BIO14.6 hamsters in late-stage disease might stabilize the cardiac phenotype and prevent the later development of heart failure (Hoshijima et al., 2010). Intramuscular injections using AAV vector have also proven their efficacy in gene transfection of the heart in mice (Straub et al., 1998). Considering these and other data it appeared that AAV8 and AAV9 vectors have a high tropism to muscular tissues, and that AAV9 might even have a particularly high tropism to the myocardium (Bish et al., 2008; Lipskaia et al., 2010). Therefore, we based our initial tests on intracardiac injections using AAV8 and AAV9 vectors in order to transfer the δ-SG gene.
Similar to other authors, our gene transfer was efficient; the δ-SG transgene was detected in the LV of all treated CHF147 hamsters. However, our results showed a great variability of δ-SG gene expression in groups treated with AAV8 and AAV9. AAVs did not diffuse widely after direct injection into the myocardium, and transgene expression was found only adjacent to the needle track; similar results had been described by Mitsuhashi and colleagues (2003). Significant δ-SG protein expression could not be found in the LV of treated animals and no difference was observed with nontreated hamsters concerning the tissue remodeling, that is, interstitial fibrosis. Moreover, functional results have shown that the EF of injected animals decreases over the treatment period and did not differ from that of nontreated animals.
Thus the attempt to restore a functional δ-SG allele in CHF 147 hamsters with symptomatic heart disease did not stop or slow down the progression of the cardiac disease. Therefore, alternative or combined approaches might be necessary.
Disruption of membrane stability by the loss of δ-SG may lead to increased intracellular Ca2+ levels that seem to be responsible for cell death (Fraysse et al., 2010). Previously, we have demonstrated that δ-SG deficiency triggers a cascade of molecular modification in the myocardium. Indeed, proteins PLN and SERCA2a, both implicated in Ca2+ cycling, were reduced. In the CHF147 hamster model we showed that the decrease in SERCA2a was more significant than that of PLN. Although gene transfer of a pseudo-phosphorylated mutant of PLN had already been studied with no promising results (Hoshijima et al., 2010), many studies revealed a benefit of SERCA2a gene transfer in heart failure disease (Lipskaia et al., 2007, 2010). More recently, the Calcium Up-regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) phase 2 clinical trial showed the safety and efficacy of such an approach in patients with class III/IV heart failure (Jessup et al., 2011).
In our hamster model, we could confirm that the SERCA2a–PLN complex contributes to heart failure. Thus, we evaluated the efficacy of injecting an exogenous gene copy by intracardiac injection. In the context of multiple or combined gene transfers, we based the formulation of the plasmid encoding hSERCA2a on a synthetic vector, that is, with P85 Pluronics. The SERCA2a transgene presented stable expression in the various levels of the studied sample heart. Unlike results with δ-SG, hSERCA2a messenger expression was detected in the entire LV of injected animals. Moreover, the percentage of interstitial fibrosis was significantly decreased compared with untreated animals. Gene transfer of hSERCA2a, using P85, led to improved preservation of cellular and intracellular organization.
Quantification of cardiac function in small animals remains difficult, and no single measure appears to reflect the complexity of the pathophysiology of heart failure. Within the classical Frank–Starling framework, we tried to assess the modifications induced by hSERCA2a gene transfer in the CHF147 hamster model. Echocardiographic follow-up of the animal cohorts has shown partial preservation of the contractile function of the LV and a stable evolution of this parameter over the observation period. Using conduction catheterization, we observed several beneficial modifications of the pressure–volume relationship. Stroke work was restored close to normal in treated animals. Furthermore, by analyzing the relationship of stroke work and the end-diastolic volume, defined as preload-recruitable stroke work, we were able to confirm improvement of the intrinsic myocardial performance independent of loading and geometry (Glower et al., 1985). Heart rate did not show large differences, but all measurements were obtained under stable anesthesia, which might have lowered the influence of heart rate regulation. The concept of the end-systolic ventricular pressure–volume relationship as developed by Suga and Sagawa might be controversial for in vivo evaluation in small animals (Suga and Sagawa, 1974). However, conduction catheterization gives some insight on this relationship. Our data provide a coherent argument in favor of improved myocardial contractility after hSERCA2a gene transfer into the LV. Considering that a linear function can describe the ESPVR, quantification is possible. The slope of the ESPVR appears to be clearly increased although not normalized. Our study did not demonstrate a significant dose effect, but the dose range might have been too restrictive. Our data support the hypothesis that improved LV function relies more on improved Ca2+ handling and restoration of proper excitation–contraction than on correction of the triggering genetic mutation in the δ-SG gene.
At the stage of symptomatic heart disease, decreased amounts of SERCA2a Ca2+ pump lead to lower recapture of SR Ca2+ during diastole and low SR Ca2+ content in failing cardiomyocytes (Kawada et al., 1999). Decreased SERCA2a in CHF147 hamster heart thus appears to be a major hallmark of the contractile dysfunction and a new therapeutic target.
Gene transfer of hSERCA2a allowed animals to maintain stable cardiac function, as evolution of EF paralleled that of the normal control group. Our results suggest that correction of specific secondary molecular changes in the myocardium prevails over correction of the triggering hereditary mutation. However, in the long-term evaluation, combined therapies might be useful, raising the question of adequate formulation for repeat administrations. Rather high cardiac transgene expression seems necessary for efficient clinical benefits. Direct intramyocardial delivery by echocardiography image-guided injections appeared to be a simple and realistic approach in small animals. Proper formulation of genetic cargo for therapeutic options remains a subject of controversy (Lipskaia et al., 2010; Blain and Straub, 2011). Viral vectors have demonstrated their potentialities in several instances, but carry some intrinsic limitations. Unlike intravenous injection, intramyocardial transfer can limit the propagation of AAV in the body (Kawada et al., 2002; Toyo-oka et al., 2002). However, high levels of CMV-δ-SG were found in the liver with AAV8 transfection. This serotype has a strong tropism for hepatocytes (Lipskaia et al., 2010). The large amount found compared with the heart generates an issue of safety due to body-wide diffusion. Indeed, immunity related to AAV has become a major problem in biotherapy and dissemination of the virus throughout the organism tends to increase immune reactions (Glower et al., 1985). Here, we have shown that Pluronic block copolymers might enhance transgene expression in vivo. The transfection mechanism of Pluronics is not yet completely understood (Netticadan et al., 2000; Yang et al., 2008), but the fact that they do not condense DNA, and their flexibility and efficiency in releasing plasmid DNA, might explain their wide range of transfection (Kabanov et al., 2005; Roques et al., 2009b). Pluronics have been shown to be safe and repeatable synthetic formulations to deliver nucleic acids without adverse immune reactions (Sriadibhatla et al., 2006; Roques et al., 2009a). Furthermore, we did not detect transgene dissemination in the liver, lungs, and kidneys. On the other hand, transgene was found in the spleen, similar to previous biodistribution studies with Pluronics (Gaymalov et al., 2009). No adverse effect could be linked to this specific distribution.
In our study, P85 Pluronics demonstrated better gene transfer to the myocardium with reference to AAV8 or AAV9. This combination gives rise to an efficient platform for the treatment of cardiomyopathies. We have shown that, in the context of hereditary cardiomyopathies, gene transfer aiming at correction of the causative genetic defect does not modify the deleterious progression of the disease. On the other hand, correction of some prominent secondary molecular changes, such as the decrease in SERCA2a, can be achieved by gene transfer and leads to improved functional outcomes.
Footnotes
Acknowledgments
The authors gratefully acknowledge the technical aid of Martine DiPietro. The authors thank the team of the Gene Vector Production Center in Nantes for providing all AAV stock solutions.
Author Disclosure Statement
No relevant financial or nonfinancial relationship in the products, techniques, or companies exists for any of the authors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.