
Abstract
Here we describe a series of replication-defective adenovirus vectors designed to express transgene products from two expression cassettes placed into the deleted E1 and E3 domains. Vectors that contained an E1 cassette with a cytomegalovirus promoter in the forward orientation and an E3 cassette with the chicken β-actin promoter in the reverse orientation grew to acceptable yields and expressed both transgenes. Additionally, they elicited immune responses to both transgene products. Levels of expression and the vectors' immunogenicity were influenced by the presence of regulatory elements shared between the two expression cassettes. Specifically, vectors that carried the same intron and enhancer in both expression cassettes could be rescued and expanded, but they were poorly immunogenic. Deletion of the enhancer or both the enhancer and the intron from the E3 cassette increased T- and B-cell responses to both transgene products.
Introduction
E1-
Our results showed that inserting expression cassettes containing either the promoter from respiratory syncytial virus (RSV) or chicken β-actin (CB) placed in the clockwise orientation (5′–3′) in relation to the Ad genome resulted in molecular viral clones that commonly failed to rescue in HEK 293 cells, which contain the Ad human serotype 5 sequences for transcomplementation of the deleted E1 in the AdC7 molecular clones. The same promoters carried in an expression cassette inserted in the counterclockwise orientation (3′–5′), on the other hand, resulted in virus that could be expanded on HEK 293 cells. Vectors containing the RSV promoter within the E3 expression cassette grew poorly. We further focused on viruses that carried an expression cassette with the cytomegalovirus (CMV) promoter in the forward orientation in E1 and an expression cassette with the CB promoter in the reverse orientation in E3. Here we show that the presence of shared sequences, for example, the CMV enhancer and/or an intron, between the E1 and E3 expression cassette affects levels of transcription of the transgenes and thereby protein expression and immunogenicity of the transgene products.
Materials and Methods
Construction of recombinant Ad vectors
The construction of shuttle plasmids for insertion of transgenes into the Ad E1 and E3 loci and of adenoviral vectors has been described previously (Zhou et al., 2010). Briefly, expression of transgenes in shuttle plasmids (pShuttle) used for insertion into the E1 locus was driven by the human CMV promoter. Transgenes inserted into the E3 locus were either under the control of the RSV or the CB promoter. E1 pShuttle vectors had an intron and an enhancer, or only an intron. To construct E1 pShuttle vectors lacking the CMV enhancer, restriction enzymes NdeI and SpeI were used to remove the CMV enhancer followed by a Klenow fragment fill-in reaction and blunt-end ligation. E3 pShuttle vectors were designed to place the cassette into either a forward (5′–3′) or a reverse (3′–5′) orientation. Three types of vectors were constructed: one that contained only the promoter, one that contained the promoter with intron, and one that contained the promoter, the intron, and the CMV enhancer. For construction of viral molecular clones, all E1 expression cassettes were inserted into the E1 locus in a forward orientation using the rare-cutter restriction enzymes I-CeuI and PI-SceI. To insert the expression cassettes into the E3 locus, the E3 pShuttle vectors, regardless of orientation, and the viral molecular clone were digested with I-SceI followed by ligation. All adenoviral vectors were rescued and propagated in HEK 293 cells, followed by CsCl purification, titration, and quality control as described (Zhou et al., 2010). Briefly, virus particle (vp) contents were determined by spectrophotometry at 260 and 280 nm, with the latter indicating the purity of the preparation. IU contents were determined by nested reverse transcription polymerase chain reaction (RT-PCR) with hexon-specific primers (outer forward primer, 5′-AGGTACAGATGACAGTAGCTC-3′; outer reverse primer, 5′-CATGTAATCGTAGGTGTTGG-3′; inner forward primer, 5′-ACAGACCCAACTACATTGGC-3′; inner reverse primer, 5′-GATTCCACATACTGAAATACC-3′) on RNA isolated from HEK 293 cells infected for 5–7 days with serial dilutions of vector. Eight replicas are used for each Ad vector dilution. As part of quality control, vector stability was measured by serially passaging the rescued virus 10 times in HEK 293 cells. Viral DNA was isolated for the original rescued virus (P1) and for the viral isolate from the 10th passage (P10). They were compared by restriction digestion for 1 hr with BglII or EcoRI and SpeI. Digests were run on a 1% (vol/wt) agarose gel.
RT-PCR analysis
Total RNA of HEK 293 cells transduced with AdC7 vectors 48 hr earlier was isolated using RNeasy Mini prep kit (Qiagen, Valencia, CA). The quantity of RNA was determined by spectrometry. RNA samples were stored at −80°C till use. For each sample, 2 μg of RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA) following the manufacturer's protocol. The real-time PCR was subsequently carried out using Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA) at a final volume of 25 μl. For amplification of nucleoprotein (NP), the following primers were used at 5 pmol/μl: forward, AGCAGGTACTGGGCCATAAGG; reverse, ACTGAGAACGTAGGTTGTATGCTG. For amplification of simian immunodeficiency virus (SIV) gag, the following primers were used at 5 pmol/μl: forward, AAGCCCATCAAGTGCTGGAAC; reverse, TCTTGCCGCACTTCCAC. The housekeeping gene GAPDH was used as an internal control and was amplified using 5 pmol/μl of the following primers: forward, TGCCCCCATGTTGTGATGG; reverse, AATGCCAAAGTTGTCATGGATGACC. About 1 μl of cDNA was used as template. Quantitative RT-PCR amplification of each gene was performed as follows: initial amplification of primers was performed at 95°C for 20 sec, followed by 40 cycles of denaturation at 95°C for 3 sec and amplification at 60°C for 30 sec. All real-time PCRs were carried out on ABI Prism 7500 Fast Sequence Detection System (Life Technologies, Grand Island, NY). Reactions were run in triplicate in two separate experiments. Standard curves used to calculate the amount of NP and SIV gag RNA in each sample were determined by serially diluting plasmids encoding NP and SIV gag from 5 to 0.31625 ng/well. Expression data for NP and SIV gag were normalized to GAPDH to control for variability in samples.
Western blots
HEK 293 cells plated on six-well plates were infected with two different doses of viral vectors (103 and 104 vp/cell). About 24 hr postinfection, cells were harvested and treated with RIPA buffer containing HALT protease inhibitors (Thermo Fisher Scientific, Pittsburgh, PA). Protein samples were diluted in a reducing sample buffer and electrophoresed on 4–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels (Biorad, Hercules, CA) with Tris running buffer (Biorad), transferred to polyvinylidene fluoride membrane, and probed with either primary monoclonal antibody anti-NP (Southern Biotech, Birmingham, AL) or primary monoclonal anti-SIV gag p27 (NIH AIDS Research and Reference Reagent Program) at 4°C overnight. Horseradish peroxidase-conjugated goat antimouse secondary antibody (KPL, Inc., Gaithersburg, MD) was added, and protein expression was detected by autoradiography using ECL substrate kit (Thermo Fisher Scientific). β-Actin was used as a protein-loading control and probed with an antimouse β-actin primary antibody (Sigma-Aldrich, St. Louis, MO).
Animals and immunization
Four- to six-week-old female C57Bl/6 mice were purchased from the National Cancer Institute, and ICR mice were purchased from ACE Animals (Boyertown, PA). Groups of four mice were immunized intramuscularly with 109 or 1010 vp of various AdC7 vectors diluted in phosphate-buffered saline (PBS) and administered into the tibialis anterior muscle of each hind limb. Mice were housed at the Animal Facility of the Wistar Institute, and all procedures used were approved by institutional protocols.
Preparation of samples
Blood was collected by submandibular bleeding and placed into 4% sodium citrate. Peripheral blood mononuclear cells were harvested as described (Santra et al., 2009). Serum was prepared by allowing blood to clot at room temperature for 30 min. Cells were then pelleted, and serum was isolated by centrifugation in an Eppendorf microcentrifuge for 30 min at 5000 rpm.
Enzyme-linked immunosorbent assay
Sera of individual mice were pooled and tested for Gag- or NP-specific antibodies by enzyme-linked immunosorbent assay. Briefly, 96-well plates were coated overnight at 4°C with the Gag protein at 10 μg/ml or A/PR8 virus at 32 HAU/ml. Wells were washed with PBS/0.05% Tween-20, followed by overnight blocking at 4°C with PBS/3% BSA/0.05% Tween-20. Wells were then washed and incubated with serial-diluted samples (in duplicates) for 2 hr at room temperature. Wells were subsequently washed, and bound IgG was detected with a goat antimouse IgG alkaline phosphatase conjugate (Sigma-Aldrich). Bound enzyme was detected with DEA substrate (KPL, Inc.). Plates were read on a microplate reader at 405 nm.
Tetramer and intracellular cytokine staining
Intracellular cytokine staining to detect Gag- and NP-specific CD8+ T cell responses was performed as follows. Lymphocytes were stimulated either with a peptide (2 μg/ml) encoding the immunodominant H2b-restricted class I epitope of NP (ASNENMETM) or with a peptide pool (3 μg/ml) of 15-mers overlapping by 11 amino acids covering the entire SIVmac239 Gag sequence. A rabies virus glycoprotein peptide or SIINFEKL were used as a negative stimulation controls. Lymphocytes were stimulated in the presence of Golgi Plug (BD Biosciences, Franklin Lakes, NJ) for 5 hr in a 10% CO2 37°C incubator before surface staining with anti-CD8 and anti-CD44 antibodies and with a stain identifying dead cells. Cells were stained for 30 min at 4°C, then washed twice with PBS, and permeabilized with Cytofix/Cytoperm (BD Biosciences) for 30 min at 4°C. Cells were then washed with 1×Perm/Wash (BD Biosciences) and stained for 30 min at 4°C with APC-labeled anti-IFN-γ, Alexa700-labeled anti-IL-2, and PE-Cy7-labeled anti-TNF-α. All antibodies (Biolegend, San Diego, CA) were diluted at 1:100 in 1×Perm/Wash. After washing, cells were fixed with Fixative Buffer (BD Biosciences). Cells were run on an LSRII (BD Biosciences) and data were analyzed with FlowJo (TreeStar, Ashland, OR).
Statistical analysis
Experiments were conducted repeatedly using 3–4 mice per group. Results show the median±interquartile range. Significances between groups were analyzed by one-way ANOVA.
Results
Construction of vectors
pShuttle vectors were constructed to insert an expression cassette into the E3 locus of an E1- and E3-deleted AdC7 vector. Specifically, E3 pShuttle vectors contained either the RSV or the CB promoter and the rabbit globin poly(A) tail. Vectors were designed to insert the expression cassette into the deleted E3 domain of the AdC7 viral molecular clone in the clockwise or counterclockwise orientation. Additional E3 pShuttle vectors lacked the intron, the enhancer, or both. pShuttle vectors for insertion of the expression cassette into E1 of the viral molecular clone contained the CMV promoter and the bovine growth hormone poly(A) tail with or without enhancer or intron. The two E1 and E3 pShuttle vectors were used to construct recombinant AdC7 molecular clones. Virus was rescued upon transfection of molecular clones into HEK 293 cells. Virus was deemed unfit if three attempts to recue failed to yield viral clones that could be expanded further. The design of the E1 and E3 pShuttle vectors and a representative Ad vector are shown in Fig. 1. A list of the different types of vectors and their characteristics, including their ability to rescue and their genetic integrity upon expansion, is shown in Table 1.
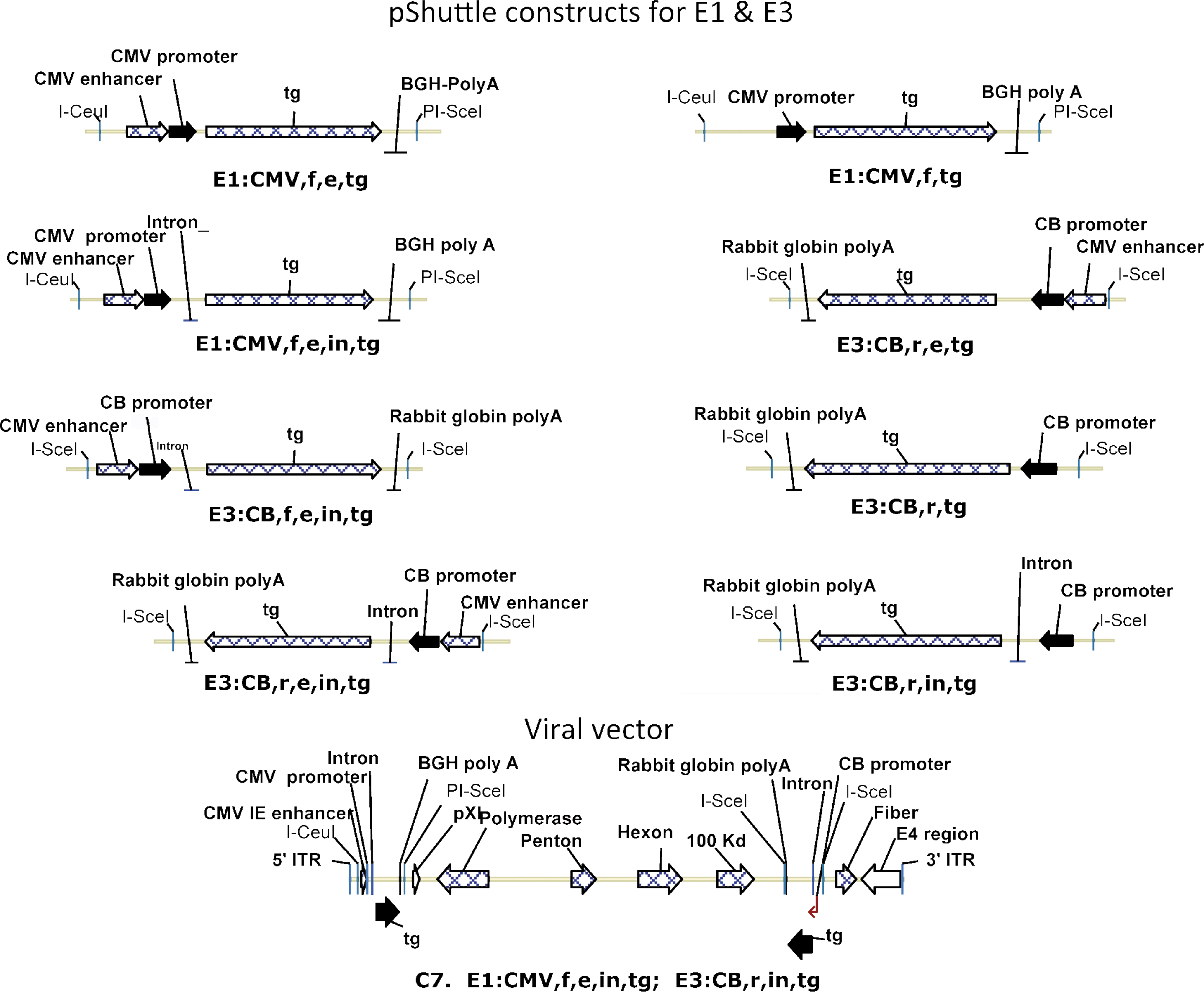
Diagrams of shuttle vectors and the vector genome. BGH, bovine growth hormone; CB, chicken β-actin; CMV, cytomegalovirus; e, enhancer; f, forward orientation; in, intron; r, reverse orientation; tg, transgene. Color images available online at
CB, chicken β-actin; CMV, cytomegalovirus; F, forward; N, no; R, reverse; RSV, respiratory syncytial virus; Y, yes; in, intron; tg, transgene; e, enhancer.
None of the recombinant molecular clones that contained the RSV promoter in E3 with the expression cassette in the forward (5′–3′) orientation, regardless of the presence of an expression cassette in E1, could be rescued. Vectors with the RSV promoter in E3 with the expression cassette in the reverse (3′–5′) orientation or in E1 in the forward orientation resulted in infectious virus. Two out of four molecular clones with the CB promoter in an E3 expression cassette in the forward orientation failed to rescue, while most of the clones containing the CB promoter in an E3 expression cassette in the reverse orientation could be expanded.
Genetic integrity of the rescued virus was assessed by isolation of the viral DNA followed by restriction enzyme digestion using two sets of enzymes that identify the expression cassettes. Loss of the E1 transgene was observed in three viruses containing the CB promoter; in one, the expression cassette was placed in the forward orientation, and in two, in the reverse orientation. All the vectors contained Cre-recombinase fused to the ligand-binding domain of a mutated estrogen receptor within E3 and a floxed gene within E1; loss of the transgene was most likely caused by the recombinase function of the E3 transgene product rather than other characteristics of the virus. All other recombinant viruses showed the expected banding patterns.
After expansion in 109 HEK 293 cells, vectors were titrated with regard to vp and multiplicity of infection (MOI) (Fig. 2). Yields for vectors carrying the CB promoter in either orientation with or without an enhancer or intron in the E1 or E3 expression cassette ranged between 0.1 and 2×1014 vp per 109 HEK 293 cells and were largely comparable to those obtained with traditional vectors containing a single-expression cassette with a CMV promoter, an enhancer and an intron in E1 (Fig. 2a). Vectors containing the RSV promoter within the E3 expression cassette had comparable yields. Vector infectivity, reflected by the ratio of vp to MOI, was in general similar to that seen with traditional AdC7 vectors (<500); although, this ratio was highly variable for some of the constructs and was influenced by the transgene (Fig. 2b).

Growth characteristics and infectivity of vectors.
Genetic stability of some of the vectors was tested for by serially passaging the virus on HEK-293 cells. After the 1st and 10th passage, genomic DNA was isolated from the rescued virus followed by restriction enzyme digests and gel electrophoresing. As shown for E1303, both passages showed the identical banding pattern, suggesting that the vector was genetically stable (Fig. 3).
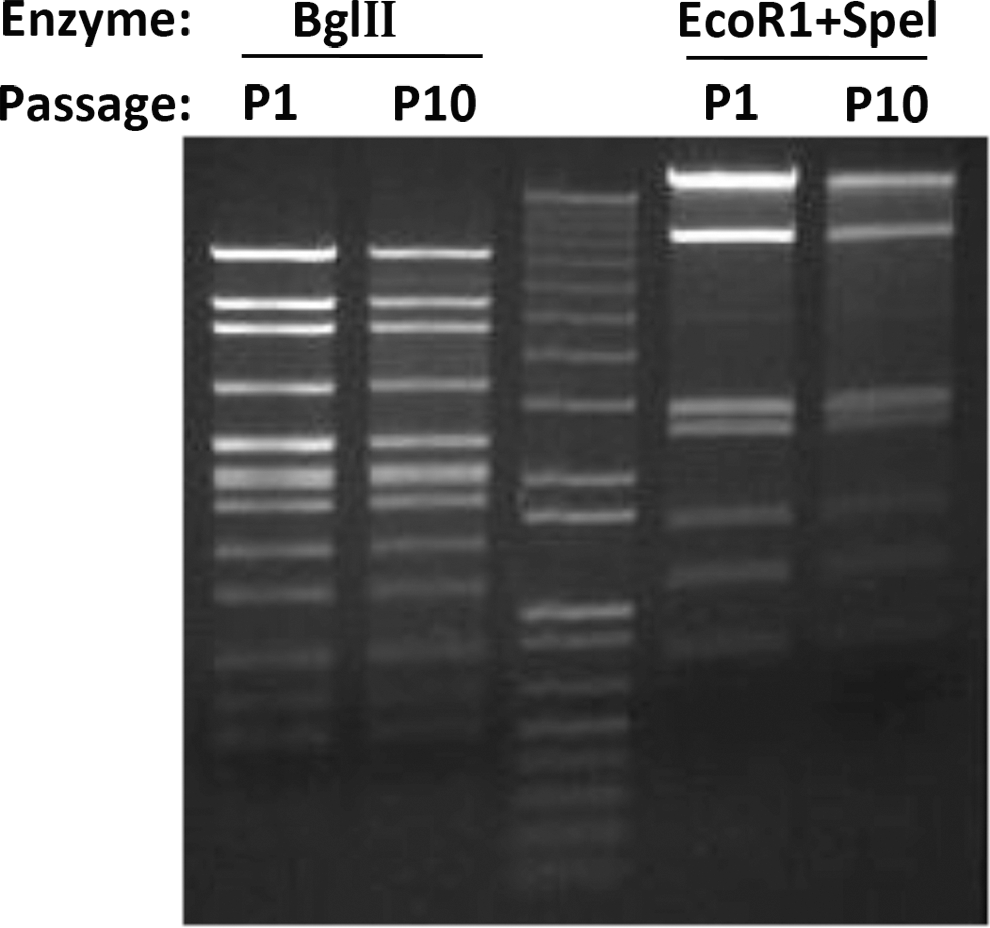
Genetic stability of the vector genome. Shown is the banding pattern of the genome of vector E1301 after the 1st and 10th passage on HEK 293 cells upon digestion with BglII and EcoRI together with SpeI.
Expression of transgene products
Expression of the transgenes from either cassette was measured by real-time RT-PCR and by Western blot of lysates of HEK 293 cells infected with 103 vp per cell. We focused on vectors that encoded gag of SIV and/or NP of influenza A/PR8 virus in either E1 or E3. Quantitative analysis of mRNA for the transgenes showed as expected that vectors that lacked the NP transgene were negative for NP RNA transcripts (Fig. 4a) and vectors that lacked gag failed to amplify a sequence with the gag-specific primers (Fig. 4b). Levels of NP and gag transcript expression varied. While this was in part a reflection of the differences in vp-to-MOI ratios of the different vector batches, the composition of the expression cassettes also contributed to expression levels. The amount of NP RNA was highest from vectors encoding NP in E1 under the control of the CMV promoter with both an enhancer and an intron and either no expression cassette within E3 (E1288) or a forward-oriented E3 expression cassette containing the CB promoter, an intron, and an enhancer (E1310). Placing the E3 expression cassette in the reverse orientation appeared to dampen NP RNA levels from the E1 expression cassette. Vectors that carried the same enhancer in the reverse-oriented E3 cassette and in the E1 cassette (E1174) resulted in lowered levels of NP expression from the E1 cassette, while the presence or absence of the intron within both cassettes did not have this effect (E1302 and E1303, respectively). Deletion of the enhancer and intron elements from the NP-encoding E1 cassette also lowered levels of NP transcripts (E1301). Moving NP from the E1 cassette to the E3 cassette also resulted in lower NP expression (E1492, E1374, E1219). These vectors also tended to have higher vp-to-MOI ratios (376–2940) than vectors encoding NP in E1 (48–549).
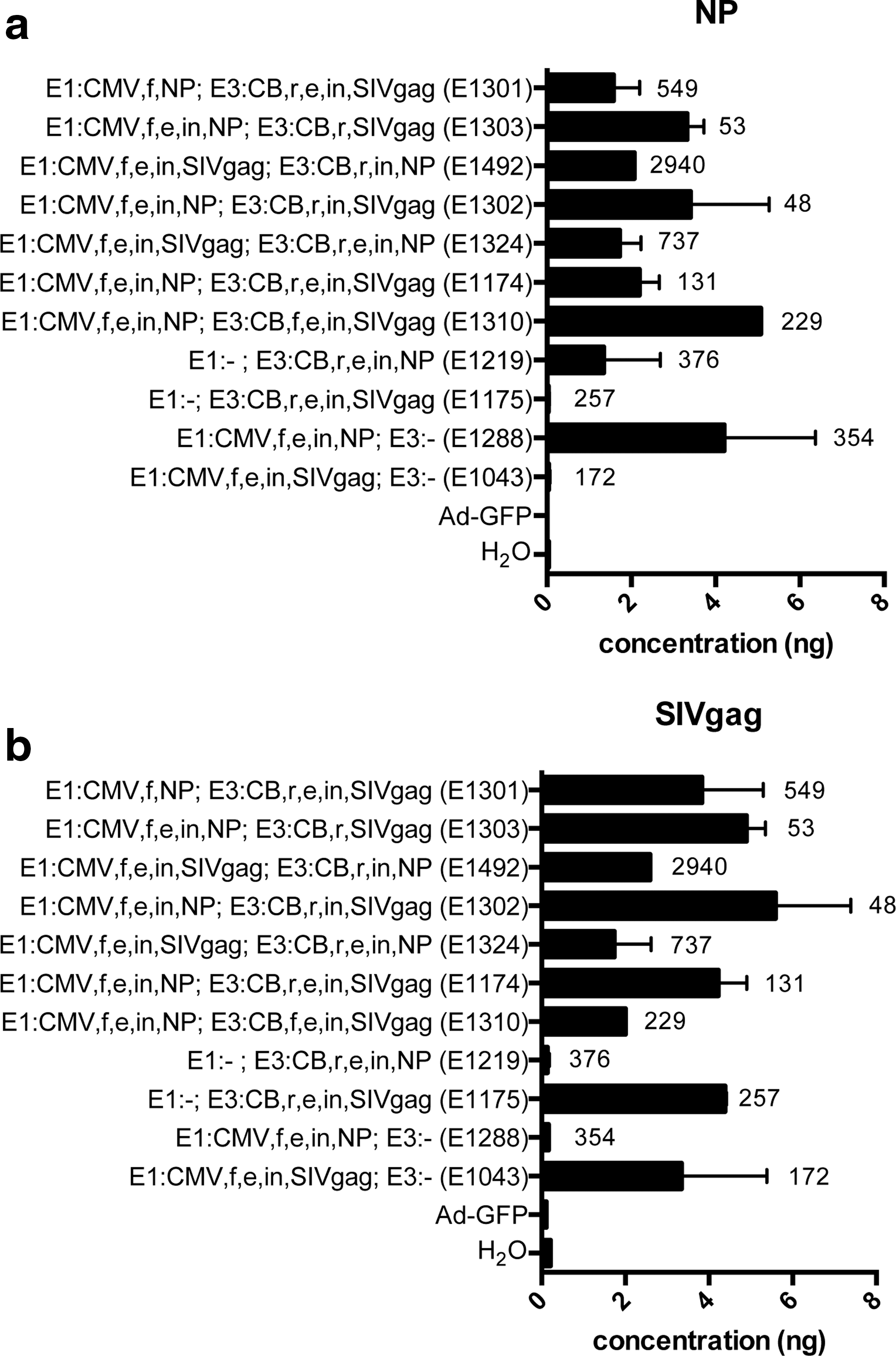
Levels of transcript expression. Graphs show levels of RNA transcript expression achieved within 24 hr by 106 HEK 293 cells transduced with 1010 vp of vectors. The experiments were conducted twice for most vectors, and bars show median levels+range.
Results differed for gag transcripts; vectors expressed similar or even higher amounts of gag when this gene was encoded in E3 (E1043, E1775, E1310, E1174, E1302, E1303, E1301) compared with those that had the gag transgene in E1 (E1043, E1324, E1492). Vectors that expressed gag from an E3 cassette placed in the forward orientation showed lower levels of gag transcript expression (E1310) compared with those that carried the same cassette in the reverse orientation (E1174), although this difference could in part reflect differences in vp-to-MOI ratios between the two vectors. All vectors expressing gag from the CB-controlled reverse-orientation E3 cassette expressed approximately equal levels of gag transcripts regardless of the presence of intron and enhancer within E3 (E1301, E1174), lack of the enhancer (E1302), or lack of both the enhancer and the intron within E1 (E1303). Also, expression of gag from E3 was not reduced in vectors that shared enhancer and intron elements between the E1 and E3 expression cassettes (E1174). Overall, these results suggest that expression from the E1 cassette was influenced by the composition of the E3 cassette, while transgene expression from an E3 reverse-oriented cassette was relatively unaffected by the presence and characteristics of an E1 expression cassette.
Some of the vectors were tested further for protein expression levels by Western blot; specifically, E1219, E1288, E1174, E1302, E1302, and E1301 were tested for expression of NP (Fig. 5a) and E1043, E1288, E1175, E1219, E1310, E1174, E1324, E1302, E1492, E1303, and E1301 were tested for expression of Gag (Fig. 5b). Western blotting showed only subtle differences in NP expression; however, expression of Gag was more variable. As expected, the two vectors that did not carry the gag gene (E1288, E1219) did not express the Gag protein on the Western blot when probed for p27. Expression levels were reduced in cells infected with E1310, a vector that expressed Gag from a forward-oriented E3 cassette containing the CB promoter together with the intron and enhancer, both of which were also present in the E1 cassette. Of note, this vector also showed reduced levels of gag transcripts (Fig. 3). Expression levels were highest for E1302, a vector that contained the standard E1 cassette encoding NP, whose expression was driven by the CMV promoter and the intron and enhancer, and a reverse-oriented E3 cassette with gag, whose expression was driven by CB promoter and the intron. Again, this construct also achieved the highest levels of RNA transcripts as shown in Fig. 4.
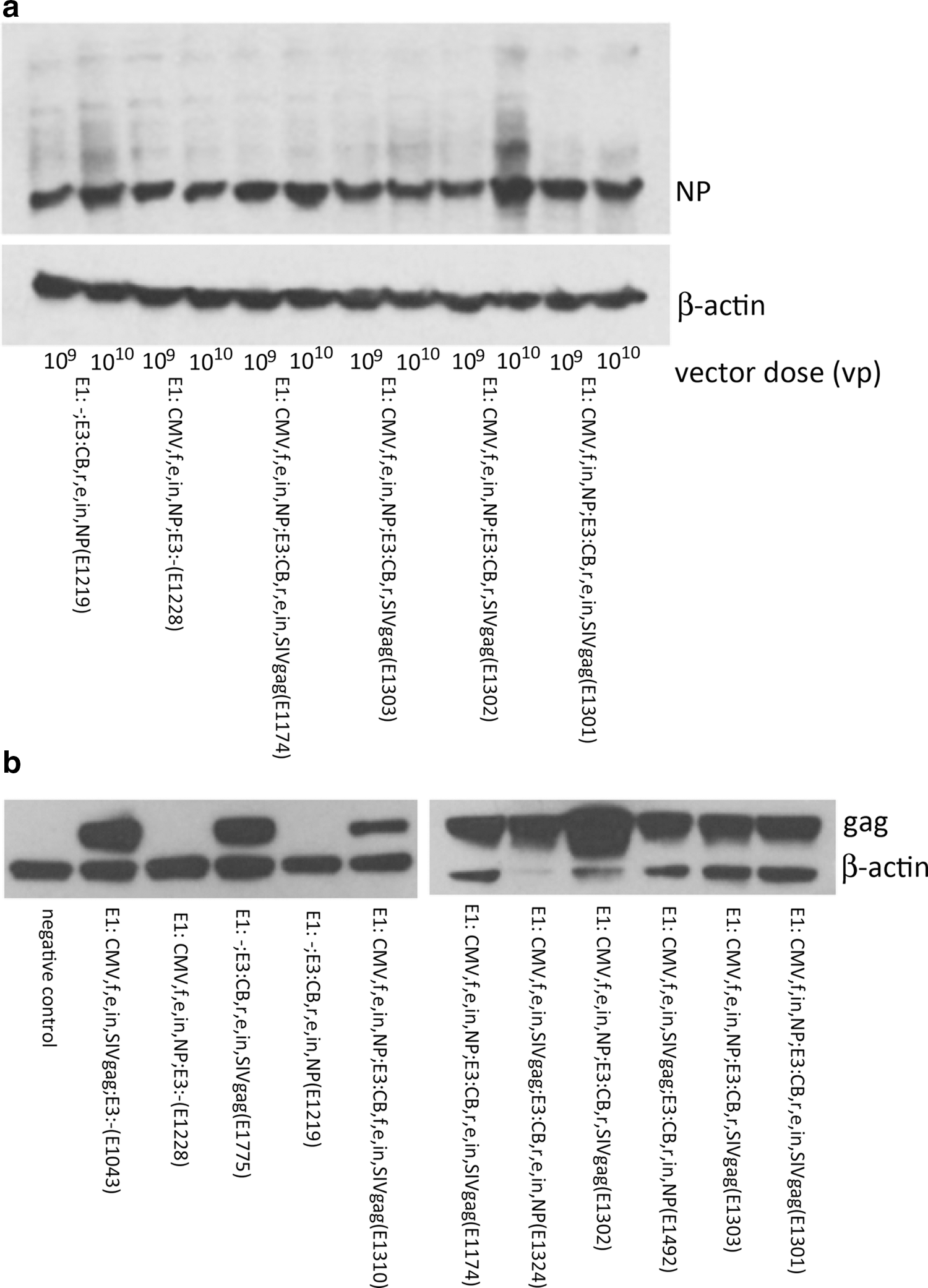
Protein expression. Shown are Western blots for NP
Induction of antibody responses
To test immunogenicity of some of the more promising dual-expression vectors, outbred ICR mice were initially immunized with 1010 vp of vectors expressing NP and SIVgag. All of the vectors induced antibodies against NP and Gag when compared with background levels in naïve control mice (Fig. 6a and b). Responses to NP (Fig. 6a) were highest in mice immunized with E1303 and lowest in mice immunized with E1302. Both vectors carried NP in E1 under the control of the CMV promoter, an enhancer, and an intron. E1302, unlike E1303, also contained the same intron in E3. E1301, a similar vector that contained neither enhancer nor intron in the E1 expression cassette and the intron but no enhancer within the E3 cassette, induced intermediate titers of NP-specific antibodies that were comparable to those achieved by the single-expression vectors E1219 or E1288. Again, all of the vectors induced Gag-specific antibodies (Fig. 6b). Responses to E1303 again were highest, followed by responses to E1302. Responses to the other vectors were largely comparable. It is noteworthy that single-expression vectors carrying the NP- or gag-encoding sequences in E1 induced similar antibody titers to the corresponding vectors carrying either transgene within E3.
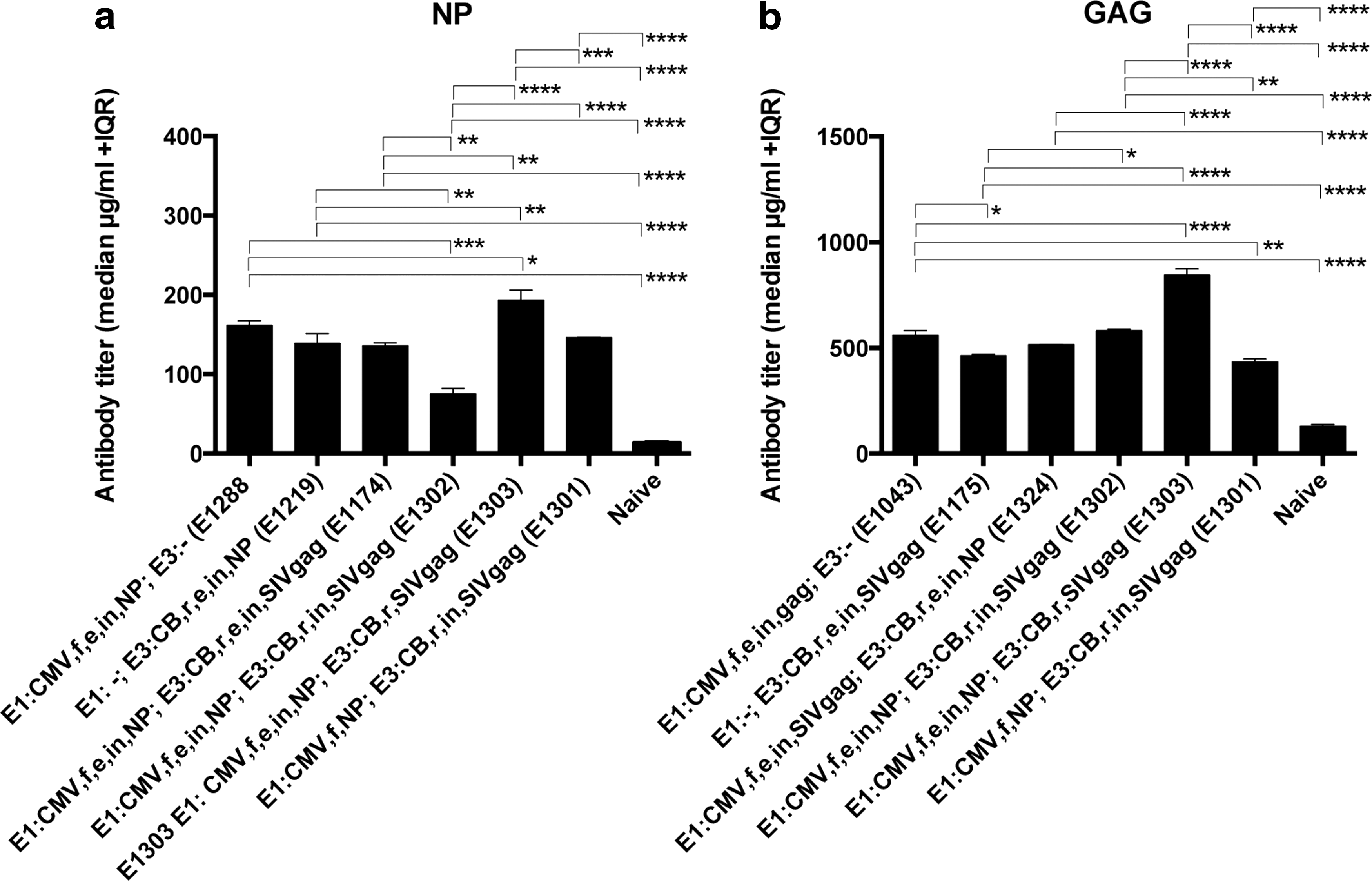
Antibody responses. Pooled sera from mice immunized with the indicated vectors were tested for antibodies to NP
Induction of CD8+ T cell responses
T cell responses were tested in C57Bl/6 mice immunized with 1010 vp of the different vectors. Mice were bled 2 and 6 weeks after immunization together with a group of naïve mice, and specific CD8+ T cell responses to Gag and NP were measured by intracellular cytokine staining for IFN-γ, TNF-α, and IL-2 upon a brief in vitro stimulation with the corresponding peptides. The sum of all possible cytokine combinations, as determined by Boolean gating, was calculated and compared with background responses in naïve control mice. Significant responses to NP were obtained with the control vector E1288, in which NP is encoded in E1 and which lacks an E3 expression cassette. Of the dual-expression vectors, E1310, which contains the E3 expression cassette in the forward orientation and identical introns and enhancers in the E1 and E3 expression cassettes, and E1302, which contains a reverse-oriented E3 expression cassette and the same intron in the E1 and E3 expression cassette, achieved statistically significant frequencies of NP-specific CD8+ T cells at 2 weeks after immunization. By week 6, NP-specific CD8+ T cell responses to E1303, which contains the same E1 expression cassette as E1302 but lacks the intron in E3, reached significance. E1301, which lacks the enhancer and intron in E1, and E1174, which contains the same enhancer and intron in E1 and E3, failed to induce detectable T cell responses to NP (Fig. 7a). Gag-specific CD8+ T cell responses were overall lower (Fig. 7b). By 2 weeks after immunization, responses were significant in mice immunized with E1174, E1302, or E1303. Responses to E1301 reached significance 6 weeks after immunization. The E1310 vector failed to induce a detectable response. The E1302 and E1303 vectors were thus the only dual-expression vectors able to induce significant CD8+ T cell responses to both transgene products.

CD8+ T cell responses. PBMCs from individual mice were tested 2 and 6 weeks after immunization or NP-specific
Discussion
E1-, E3-, or E1- and E3-deleted Ad vectors were initially explored as gene transfer vehicles for permanent replacement of faulty or missing genes (Kozarsky and Wilson, 1993; Chengalvala et al., 1994). They elicited potent immune responses to both the transgene product and Ad antigens (Jooss et al., 1998), which prevented sustained expression of the therapeutic protein. Ad vectors were then developed as vaccine carriers for a number of infectious agents (Tatsis and Ertl, 2004; Ledgerwood et al., 2010; Barnes et al., 2012; Hoft et al., 2012; Sheehy et al., 2012; Gurwith et al., 2013) as well as for cancer immunotherapy (Aurisicchio and Ciliberto, 2011). Using Ad vectors derived from a variety of serotypes, families, or species showed consistently that they induced potent transgene product-specific B and CD8+ T cell responses. Several Ad vectors are in clinical trials as vaccine carriers for antigens of HIV-1 (Catanzaro et al., 2006; Baden et al., 2012), Mycobacterium tuberculosis (Hoft et al., 2012), hepatitis C virus (Barnes et al., 2012), Ebola virus (Ledgerwood et al., 2010), influenza virus (Gurwith et al., 2013), and Plasmodium falciparum (Sheehy et al., 2012). One Ad vaccine vector, based on a replication-competent E3-deleted Ad vector of human serotype 5 (HAdV-5) expressing the rabies virus glycoprotein for immunization of wildlife animals, has been licensed (Mainguy et al., 2013).
Space constraints within the Ad capsid limit the size of the genome that can be packaged. Insertion of very long transgenes, even if they do not effect vector fitness, results in poor protein expression, which, in turn, affects the vector's ability to induce potent immune responses to the encoded antigen (Tatsis et al., 2005). For complex pathogens, this necessitated the use of multiple vectors expressing different proteins, the use of inserts separated by internal ribosomal entry segments (Ye et al., 2007; Qiu et al., 2012), or the use of the so-called dual-expression vectors. Such vectors could also be useful to express a vaccine antigen together with an immunomodulator. Early versions of dual-expression vectors carried two versions of the same cassette within E1 that differed in the transgene but contained the identical (Zinn et al., 2002) or different (Liu et al., 2012) regulatory elements. Others used bidirectional promoters to control up- and downstream transgenes placed into E1 (Chai et al., 2011) or two expression cassettes with different promoters placed in tandem into E1 (Irie et al., 2005). Others, similar to our approach, inserted one transgene under the control of a CMV promoter into E1 and a second transgene under the control of the RSV promoter into E3 with both expression cassettes in the same orientation (Niu et al., 2006). Our attempts to construct stable SAdV-24 vectors with a forward-oriented expression cassette containing the RSV promoter in E3 failed regardless of the presence of an expression cassette in E1. While the same cassette placed in the reverse orientation resulted in fit vectors, yields were below average. Vector growth characteristics were improved when the CB promoter was used in the E3 cassette. Some of the vectors that contained the CB promoter in an E3 cassette in forward orientation could be rescued; others failed. Those that could be expanded had average yields and vp-to-MOI ratios, but RNA and protein expression levels of the encoded transgene were low. Better results were obtained with vectors that contained the CB promoter in the reverse orientation within the E3 domain. All vectors could be rescued and yields were comparable to those obtained with standard vectors that carried only an expression cassette within E1.
Expression of the two transgenes used from comparison, that is, gag of SIVmac239 and NP of influenza A/PR8 virus, was affected by their assignment within the Ad genome; NP was expressed at higher levels if placed into E1, while Gag expression was similar or higher from cassettes within E3. The presence of other regulatory sequences, that is, introns and enhancers, shared between the two cassettes affected expression from E1 cassette but not from a reverse-orientation E3 cassette. RNA and protein expression levels in vitro in some but not all cases predicted the vectors' ability to induce immune responses. For example, E1310, which carried gag in a forward-orientation E3 cassette achieved only low levels of gag RNA transcripts and the Gag protein and accordingly induced comparatively low Gag-specific antibody responses and no Gag-specific CD8+ T cells. In contrast, E1302 induced a markedly lower antibody response to NP and Gag as compared with E1303 even though RNA expression levels of both transgenes were comparable between vectors, and protein expression was higher from E1302 than from E1303. Shared intron and enhancer elements within the E1 cassette and the forward- or reverse-orientation E3 cassette were especially detrimental to the vectors' ability to induce T cell responses. In the end, only one dual-expression vector, that is, E1303, which carried the intron and enhancer within E1 but neither within E3, induced potent B and T cell responses to both transgene products.
In summary, dual-expression Ad vectors are useful but require careful design to achieve stable vectors that express both antigens at levels that suffice to induce immune responses. In our experience, placing the E3 cassette in a reverse orientation combined with an E1 cassette in a forward orientation results in stable vectors with higher levels of protein expression and superior immunogenicity of the E3-derived transgene product, compared with vectors that contain both cassettes in the forward orientation. Furthermore, shared sequences between the two cassettes such as intron and enhancer have detrimental effects on the vectors' ability to induce immune responses in vivo in spite of comparable protein expression in vitro.
Footnotes
Acknowledgments
This work was supported through NIAID/NIH grant U19AI07407805 HIV-1 Vaccine Based on Chimp Serotypes of Adenovirus.
Author Disclosure Statement
The authors have no conflict of interest in the reported results. J.C.S., R.K.K., X.Z., A.B., E.C., Y.L., Z.Q.X., and H.C.J.E. have no interests to declare: not competing, personal, funding, employment, or other competing interests to disclose.