
Abstract
Angiostatin and other plasminogen derivatives exhibit antitumor activities directly or indirectly, have demonstrated promising anticancer effects in preclinical studies, but have mostly failed in clinical trials partly due to their short serum half-lives. Our previous studies demonstrated that recombinant human plasminogen kringle 1–5 (K1–5) has superior antitumor activity compared with angiostatin. In addition, optimization of recombinant K1–5 with three amino acid substitutions enhances its antitumor effect. The current study was thus undertaken to evaluate prolonged expression of optimized K1–5 as cancer gene therapy. The recombinant adeno-associated virus (AAV) vector was used to express a secreted form of the optimized K1–5 (AAV-sK15tm) to improve its pharmacokinetic profile, which was considered to be the hurdle in angiostatin treatment of cancer. We successfully generated high-titer recombinant AAV vectors and observed sustained transgene expression for 567 days after a single injection of virus. The treated animals did not display any visible signs of abnormalities and showed normal serum biochemistry. The therapeutic potential of this treatment modality was demonstrated by both a strong inhibition of lung metastasis in the mouse B16F10 melanoma model and significant growth retardation of Lewis lung carcinoma xenografts in C57BL/6N mice as well as human A2058 melanoma xenografts in NOD/SCID (nonobese diabetic/severe combined immunodeficient) mice. Taken together, our results suggested that AAV-sK15tm produced long-term suppressive effects on cancer growth in vivo and should warrant serious consideration for clinical development.
Introduction
T
Angiostatin is the first four kringles of a 38-kDa internal proteolytic fragment of plasminogen. O'Reilly and colleagues first discovered angiostatin and demonstrated that primary tumor suppresses its remote metastases through the generation of angiostatin. 5 Although recombinant angiostatin exhibited excellent antiangiogenic and anticancer effects in preclinical studies, it has performed poorly in clinical trials, likely due to its short serum half-life. 6 –8 Given the extremely short half-life of recombinant angiostatic proteins such as angiostatin and endostatin in the blood stream, their efficacy is limited in in vivo settings. 9 Given their promising potential in the inhibition of tumor angiogenesis and metastasis, several approaches were adapted to develop an optimized cancer therapy using angiostatin or its mimetics. 10 –16 Previously, we demonstrated that urokinase-activated plasmin can process human plasminogen to release an angiogenesis inhibitor, plasminogen kringle 1–5 (K1–5). 17 K1–5 exhibits similar antiangiogenic activity as angiostatin and appears to be 50-fold more effective than angiostatin in suppressing the proliferation of endothelial cells, likely due to the synergistic effect of the kringle 5 domain. 17 In addition, we demonstrated that optimized K1–5 that has three amino acid substitutions (K15tm), including N289A, T346A, and L532R, shows greater antiangiogenesis and antitumor activity compared with wild-type K1–5 because of the increase in the binding ability of K15tm to endothelial cells. 11 Although the administration of purified or recombinant angiostatic proteins is capable of inducing a significant tumor-suppressive effect in cell models and animal models, the limitation of their short serum half-life and high cost of manufacturing continues to pose major obstacles to their clinical application. With the advances in vector technologies, gene therapy may provide a solution to these difficulties. 18
Various strategies for cancer gene therapy have been developed. 19 For instance, a suicide gene expression vector that encodes a pro-drug-converting enzyme can be delivered to cancer cells, so as to induce tumor cell death directly and specifically. The efficacy of this strategy depends on highly efficient transduction of the cancer cells, which may be difficult to accomplish with many cancer types. Alternatively, antiangiogenesis therapy can be developed by introducing angiostatic genes via viral or nonviral vectors. The targeted cells for transgene expression in antiangiogenesis gene therapy can be cancer or noncancer types. What is important for the success of such approaches is likely going to be the long-term presence of antiangiogenic factors at sufficiently high concentrations. It will therefore require vectors that are capable of continuous and long-term expression with low toxicity and immunogenicity. The adeno-associated viral (AAV) vector system has the ability to efficiently transduce a wide range of cells, including postmitotic, terminally differentiated cells, and is known to be relatively safe and nonpathogenic with low immunogenicity when compared with other viral vectors. 20
The present study evaluated recombinant pseudotyped AAV serotype 2/8 vector expressing a secreted form of K15tm, AAV-sK15tm, in various cancer gene therapy settings. The dose-dependent and sustained expression of K15tm in mouse serum after AAV-sK15tm transduction was observed. AAV-sK15tm conferred a protective and therapeutic effect against experimental metastasis of mouse melanoma cells. This effect was associated with lower proliferative and higher apoptotic indices in metastatic foci of AAV-sK15tm-injected mice. Moreover, significant suppression of human melanoma xenografts in NOD/SCID (nonobese diabetic/severe combined immunodeficient) mice was also demonstrated.
Materials and Methods
Preparation of AAV-sK15tm
AAV-sK15tm virion was produced at the Research Vector Core, Harvard Gene Therapy Initiative (Harvard Medical School, Boston, MA). Briefly, the K15tm expression virion was generated by tripartite transfection (AAV vector plasmid encoding the five kringle domains of plasminogen with three amino acid substitutions, N289A, T346A, and L532R; AAV helper plasmid pLT-RC08 encoding AAV Rep proteins from serotype 2 and Cap proteins from serotype 8; and adenovirus helper miniplasmid pHGTI-Adeno1) into 293A cells as previously described. 21 The transfection was performed according to a protocol developed as previously described. 22
ELISA
The serum samples for detection and measurement of K15tm expression were obtained by submandibular blood collection or intracardiac blood collection performed after sacrifice of the animals. The blood samples were allowed to clot at room temperature, followed by centrifugation at 1,564×g for 15 min to remove the clot. The serum aliquots were stored at −80°C until analysis. The sandwich ELISA used to detect K15tm expression was developed with a capturing antibody against human plasminogen developed in our laboratory, a detection antibody purchased from Abcam (ab48364; Abcam, Cambridge, UK), and recombinant K15tm proteins from a yeast protein expression system. 11 The antibody against human plasminogen was generated by immunizing BALB/c mice with human plasminogen purified from human plasma by passage through a lysine Sepharose-4B column. Briefly, the capturing antibody (160 ng/well) was coated onto the ELISA plate (Nunc, Rochester, NY) in phosphate-buffered saline (PBS) overnight at 4°C followed by blocking with PBS containing 1% bovine serum albumin. Serum samples or various concentration of recombinant K15tm used as standards were mixed with detection antibody in PBS containing 0.5% bovine serum albumin and 0.025% Tween 20 and were added into the ELISA plate. After washing with PBS containing 0.025% Tween 20, the tetramethylbenzidine (TMB) substrate was added onto the ELISA plate and the reaction was terminated with 1 N sulfuric acid 10 min after adding the substrate. The absorbance at 450 nm was measured with a multiple-well ELISA reader.
Experimental metastasis
All animal experiments were performed in accordance with the guidelines and regulations of the Institutional Animal Care and Use Committee, National Cheng Kung University (Tainan, Taiwan). Experimental metastasis was performed in C57BL/6N mice inoculated intravenously with murine B16F10 melanoma cells. The B16F10 melanoma cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), penicillin–streptomycin, and 25 mM glucose. When B16F10 melanoma cells reached 80% confluence, the cells were detached from the culture dish by incubation with nonenzymatic cell dissociation buffer (Life Technologies, Carlsbad, CA) and neutralized with medium containing 10% FBS, followed by washing in PBS. The cells, in 100 μl of PBS, were then injected into the tail vein of each mouse. The AAV-sK15tm virion was prepared and diluted in Dulbecco's phosphate-buffered saline (DPBS; Life Technologies) and was given to each mouse via intravenous injection 1 day before or after tumor inoculation according to the experimental protocol shown in the figures. In some studies AAV-Luc, which expresses luciferase, was used as control. 21 At the end of each experiment, the animals were killed by CO2 inhalation. Lung tissue was collected from the animals and maintained in PBS containing 4% paraformaldehyde. Metastatic nodules on the lung surface were enumerated.
Tumor xenografts
Subcutaneous injection of Lewis lung carcinoma (LLC) cells into the dorsum of male C57BL/6N mice was performed to evaluate the effect of AAV-sK15tm treatment on tumor growth in vivo. LLC cells were purchased from the ATCC and maintained in the DMEM containing 10% FBS, penicillin–streptomycin, and 25 mM glucose. When LLC cells reached 80% confluence, the cells were detached from the culture dish by incubation with nonenzymatic cell dissociation buffer and neutralized with medium containing 10% FBS, followed by washing with PBS. The LLC cells (5×105 cells) in 100 μl of PBS were then injected into each mouse subcutaneously. AAV-sK15tm virion was given to each mouse by intravenous injection 6 days after tumor inoculation. Tumor volume was measured at the indicated time points by using a scale ruler, and tumor weight was measured after the mice were killed. At the end of the experiment, the animals were killed by CO2 inhalation. Tumor tissue was collected from the tumor-bearing animals and maintained in PBS containing 4% paraformaldehyde.
Histology
Lung tissue or tumor tissue from the animals was processed for O.C.T. compound embedding and cryosectioning. The tissues were washed with PBS and then embedded in O.C.T. compound. Tissue sections (4–5 μm thick) were collected and processed for immunofluorescence staining for the proliferation marker Ki67 (ab15580; Abcam) and for terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay (Roche, Indianapolis, IN). Intratumoral vessel density was measured by CD31 (ab7388; Abcam)-positive staining. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI). Micrographs were taken with an inverted microscope (Leica DMI6000 B; Leica Microsystems, Wetzlar, Germany) equipped with a digital EMCCD (electron-multiplying charge-coupled device) camera (iXon DU-897; Andor/Oxford Instruments, Belfast, Northern Ireland) and an HCX PL FLUOTAR L 40×/0.6 objective lens (Leica Microsystems) at 25°C with LAS AF software (Leica Microsystems). Positive results for Ki67 staining or the TUNEL assay were calculated with Photoshop and normalized to the DAPI stain.
Statistics
Data are expressed as means±standard error mean. Statistical analyses were performed by Student t tests, one-way analysis of variance (ANOVA), or two-way ANOVA followed by Bonferroni multiple comparison tests when there were more than two groups to compare. p<0.05 was considered statistically significant.
Results
Generation of research-grade high-titer recombinant AAV carrying optimized K1–5
To achieve long-term transgene expression, we took advantage of a recombinant AAV system to express K15tm. Previously, we successfully generated a recombinant AAV serotype 2/8 vector carrying the human thrombomodulin (TM) gene and examined its liver-specific expression of transgene. 21 In this study, we used the same expression system to generate a sustained liver-based secretory system of K15tm in vivo. The expression of transgene was under the control of the cytomegalovirus promoter, and the K15tm gene was fused to a secretory signal peptide, which allows for secretion of mature K15tm protein outside of the transduced cells (Fig. 1a). Viral particles were generated by tripartite transfection of AAV Rep (serotype 2) and Cap (serotype 8) expression plasmid, K15tm-expressing AAV ITR vector, and adenovirus helper miniplasmid into human 293A cells (Fig. 1b). Recombinant AAV vector particles were purified by iodixanol density gradient and Q-Sepharose column chromatography. Purified virus was dialyzed extensively against DPBS, concentrated by passage through an Amicon spin column, and titered by dot-blot hybridization. The titer of AAV-sK15tm was 1.8×1013 genome copies/ml.

Construction of pAAV-sK15tm and generation of AAV-sK15tm virion.
Long-term expression and safety of AAV-sK15tm
As characterized in our previous report,
21
intravenous administration of the AAV2/8 vector resulted predominantly in liver transduction. To assess the capacity of the AAV2/8 vector to confer long-term gene expression, we next examined the expression profile of the AAV2/8 sK15tm vector by monitoring the serum K15tm concentration in mice over time, after injection of various amounts of AAV-sK15tm virion. Four different doses of AAV-sK15tm virion (1010, 2×1010, 5×1010, or 1011 genome copies/mouse) were given to mice via intravenous injection. Serum samples from mice injected with virus or vehicle control were collected at the indicated time points. To differentiate the transgene product, human K15tm protein, from the endogenous mouse plasminogen, a sandwich ELISA was successfully developed by using an antibody we developed in-house against human plasminogen as the capturing antibody and a commercially available antibody against mouse plasminogen as the detection antibody. This ELISA method exhibited extremely low background detection of mouse plasminogen and high sensitivity for K15tm (Fig. 2a), indicating that our in-house antibody is specific to human plasminogen and its derivatives. By using this ELISA method, the expression kinetics of AAV-sK15tm were depicted (Fig. 2a and b). Dose-responsive expression of K15tm was detected in serum as soon as 2 days after intravenous virus injection. Remarkably, with a single injection, expression was sustained for 567 days (Fig. 2a) at the conclusion of this experiment. The expression of K15tm was found to rise quickly, increase over time, and reach its steady state serum concentration 3 weeks after virus injection (Fig. 2b). The virus-injected mice did not exhibit any visible signs of abnormality and displayed normal levels of serum hepatic enzymes (Fig. 2c), C-reactive protein, and blood urea nitrogen (Supplementary Fig. S1a; supplementary data are available online at
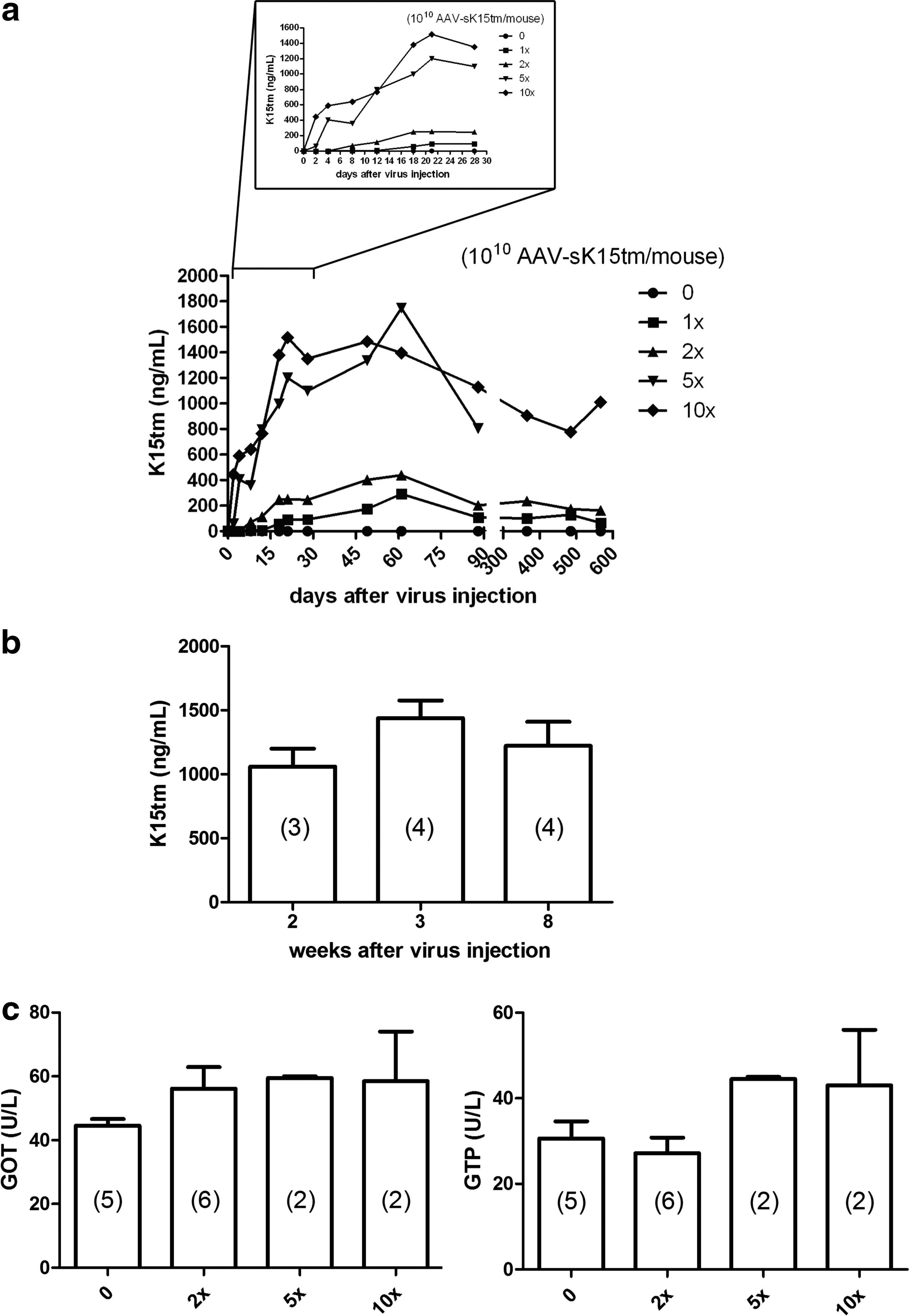
Serum concentration of K15tm and hepatic enzymes after injection of AAV-sK15tm virion. Serum samples were obtained at the indicated time points. A sandwich ELISA was developed to detect human K15tm.
Pretreatment of AAV-sK15tm protects mice against lung metastasis
Because AAV-sK15tm exhibits a fast, high, and sustained expression pattern, as well as being apparently harmless to mice, we proceeded to study its efficacy as cancer therapy, using an experimental metastasis model. The mice first received a single injection of DPBS (vehicle control), AAV-Luc, or AAV-sK15tm intravenously 1 day before intravenous injection of B16F10 melanoma cells (Fig. 3a). Two weeks after tumor injection, the lung surface of mice was occupied by metastatic foci (Fig. 3b). The number of metastatic foci was significantly reduced in the mice injected with AAV-sK15tm as compared with the control groups (Fig. 3c). This result indicated that AAV-sK15tm possessed a protective antitumor effect.
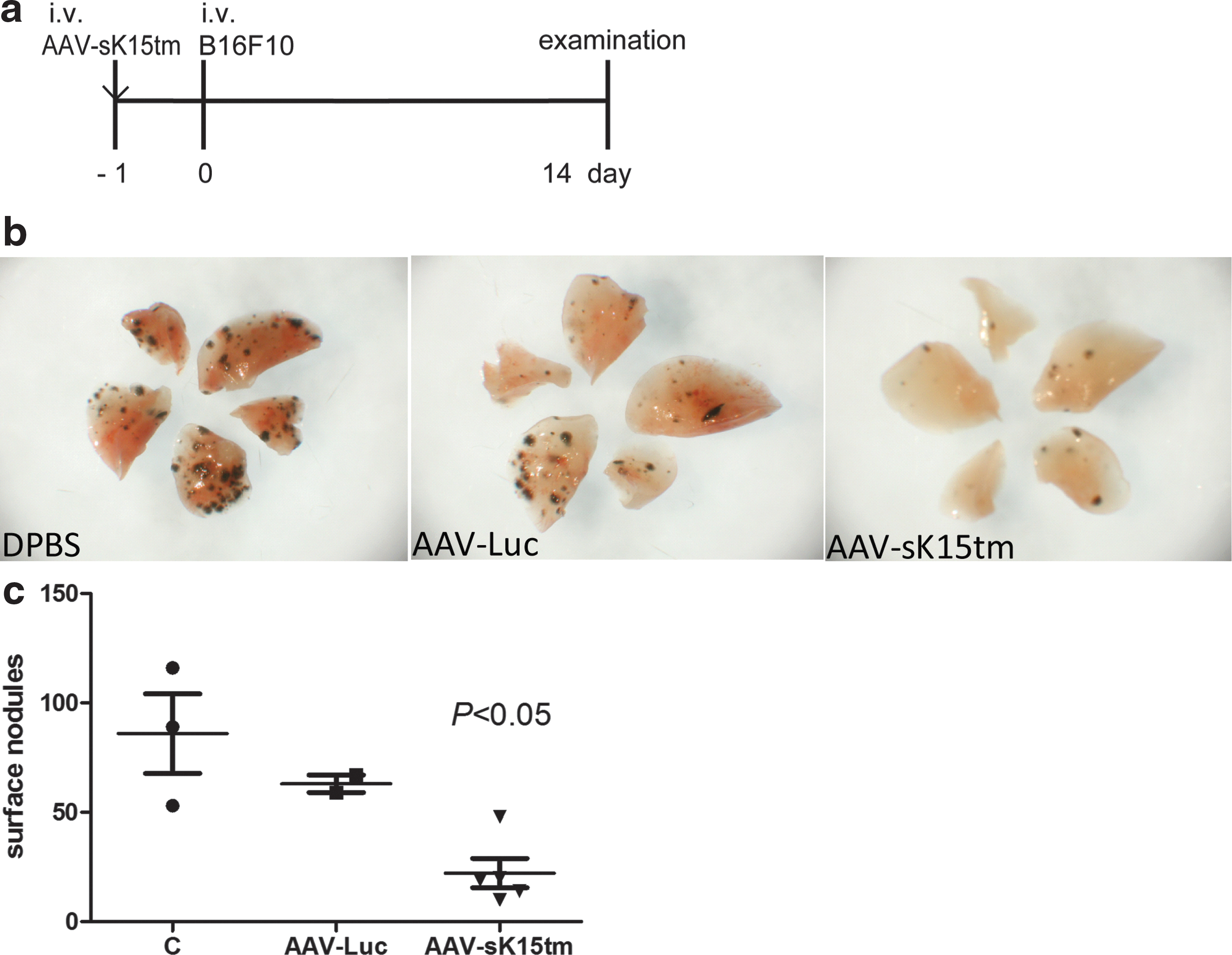
Pretreatment of AAV-sK15tm protects mice against lung metastasis.
AAV-sK15tm treatment decreases lung metastasis
Encouraged by the demonstration of the protective effect of AAV-sK15tm in tumor metastasis, we further evaluated the therapeutic potential of AAV-sK15tm in the same model. In this treatment protocol, animals were given two different doses of AAV-sK15tm intravenously 1 day after intravenous injection of B16F10 melanoma cells (Fig. 4a). A higher serum concentration of K15tm was observed, as expected, in mice injected with the higher dose of AAV-sK15tm (Fig. 4c). Metastatic tumor nodules appeared on the lung surface in each group (Fig. 4b). Compared with the control group, the number of metastatic foci was dramatically reduced in mice receiving the high dose of AAV-sK15tm (5×1010 genome copies), whereas the effect was intermediate and more variable in mice receiving the low dose of AAV-sK15tm (2×1010 genome copies) (Fig. 4d). This result indicated that AAV-sK15tm has therapeutic potential, acting in a dose-dependent manner, in treating tumor metastasis.
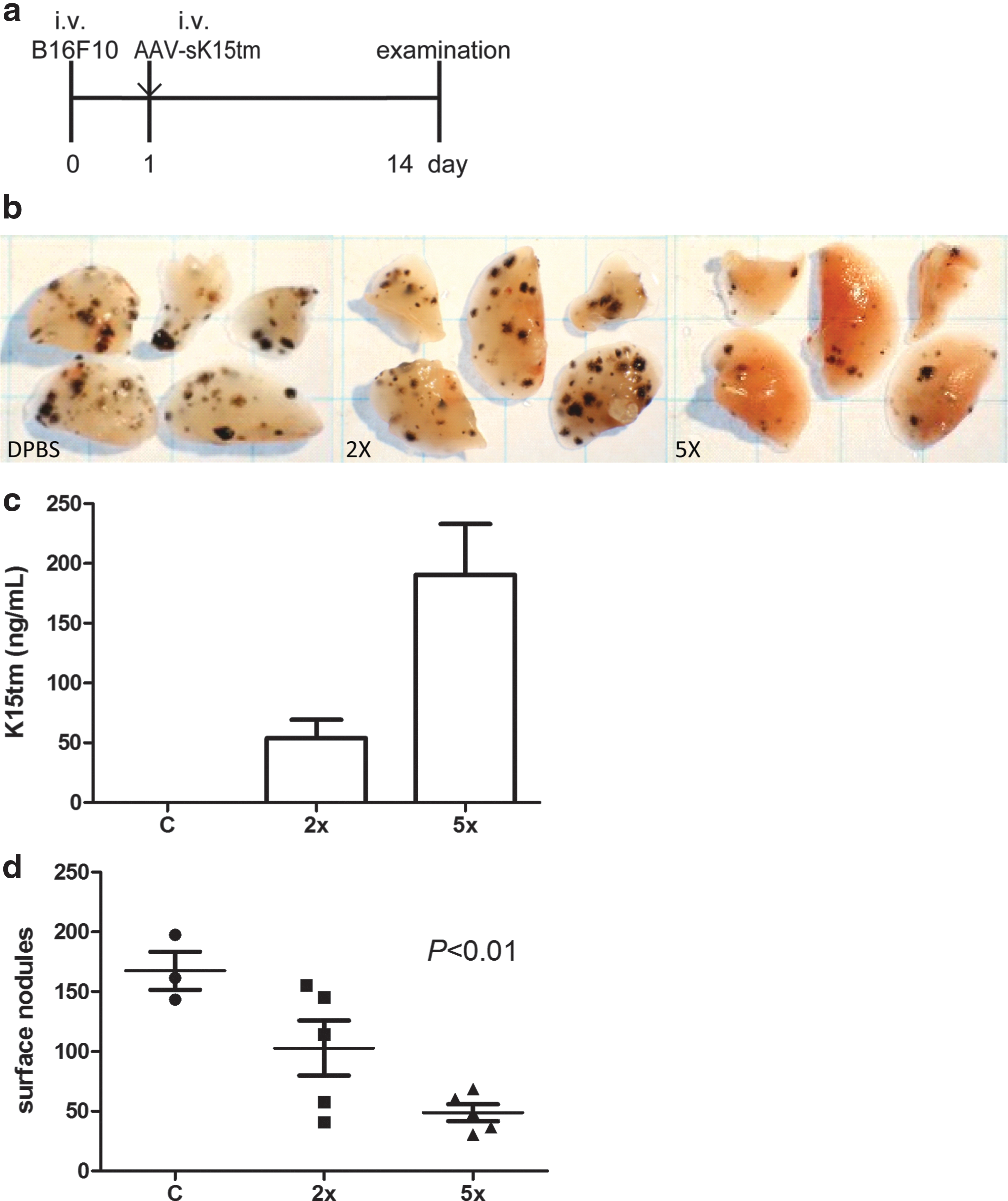
AAV-sK15tm treatment decreases lung metastasis.
Metastatic foci in mice injected with AAV-sK15tm show lower growth index and more apoptosis
Because the tumor foci on the lung surface of mice injected with AAV-sK15tm were fewer in number and smaller than those of the control mice in our study, we further examined the proliferative and apoptotic indices of tumor foci by means of immunofluorescence staining of the proliferation marker Ki67, and by TUNEL assay. Positive staining of Ki67 was detected heterogeneously both in the metastatic foci of control mice and AAV-sK15tm-injected mice. The proportion of Ki67-positive cells correlated well with the sizes of metastatic foci in both groups. In other words, the Ki67-positive signal was lower in the smaller foci of AAV-sK15tm-injected mice when compared with vehicle control mice. This observation suggested that tumor foci on the lung surface of AAV-sK15tm-injected mice exhibited less proliferative activity as demonstrated by less Ki67 staining when compared with the vehicle control group (Fig. 5a). On the other hand, apoptotic cell death was barely detected in the control group. In contrast, pronounced apoptotic cell death was induced in the AAV-sK15tm-injected group (Fig. 5b). Together, these results indicated that the growth-suppressive effects of AAV-sK15tm on B16F10 melanoma resulted from a combination of lower proliferation and higher programmed cell death of the cancer cells.
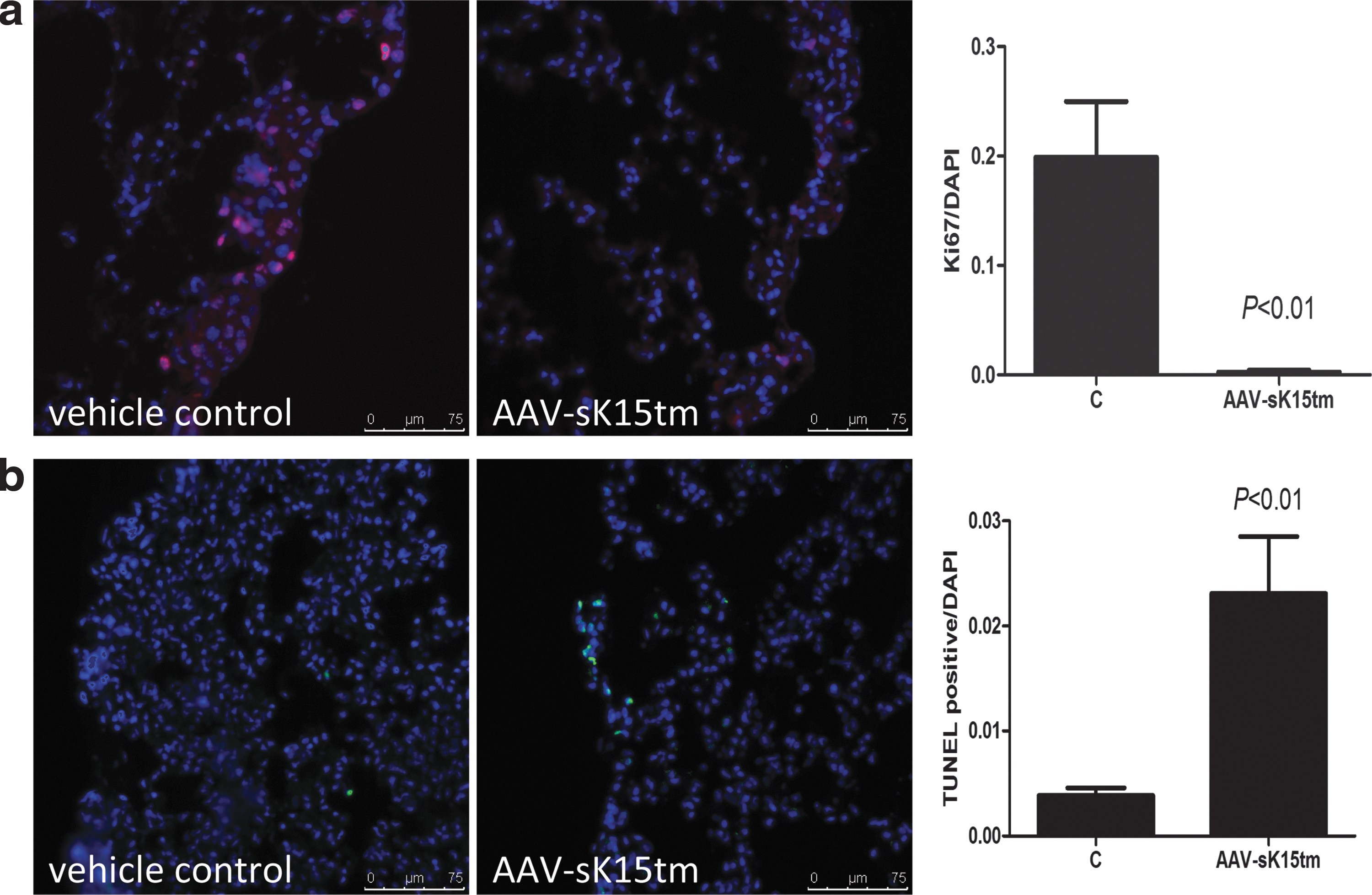
Growth suppression of metastatic foci in the AAV-sK15tm-treated group. The data shown on Ki67 and terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end labeling (TUNEL) staining are representative of three experiments.
Treatment of AAV-sK15tm suppresses tumor growth and tumor-induced angiogenesis in mice
To build on the observation that AAV-sK15tm exhibited an anticancer effect in the mouse melanoma model, we further examined whether AAV-sK15tm was able to hinder the growth of cancer in vivo by using a tumor xenograft model. In this model, cancer cells were first injected subcutaneously into the dorsum of experimental mice, followed by AAV-sK15tm injection intravenously when tumor became palpable (Fig. 6a). Tumor volume was monitored at the indicated time points (Fig. 6b), and tumor weight was measured at the end of the experiment (Fig. 6c). Intratumoral vessels were identified by CD31-positive staining (Fig. 6d). Results demonstrated that the growth of LLC cells in vivo and intratumoral vessel density were significantly inhibited by the administration of AAV-sK15tm. Similar results were obtained in human xenografts model in NOD/SCID mice (Supplementary Fig. S3). Taken together, these results indicate that AAV-sK15tm may be a strong candidate for the development of cancer therapy.
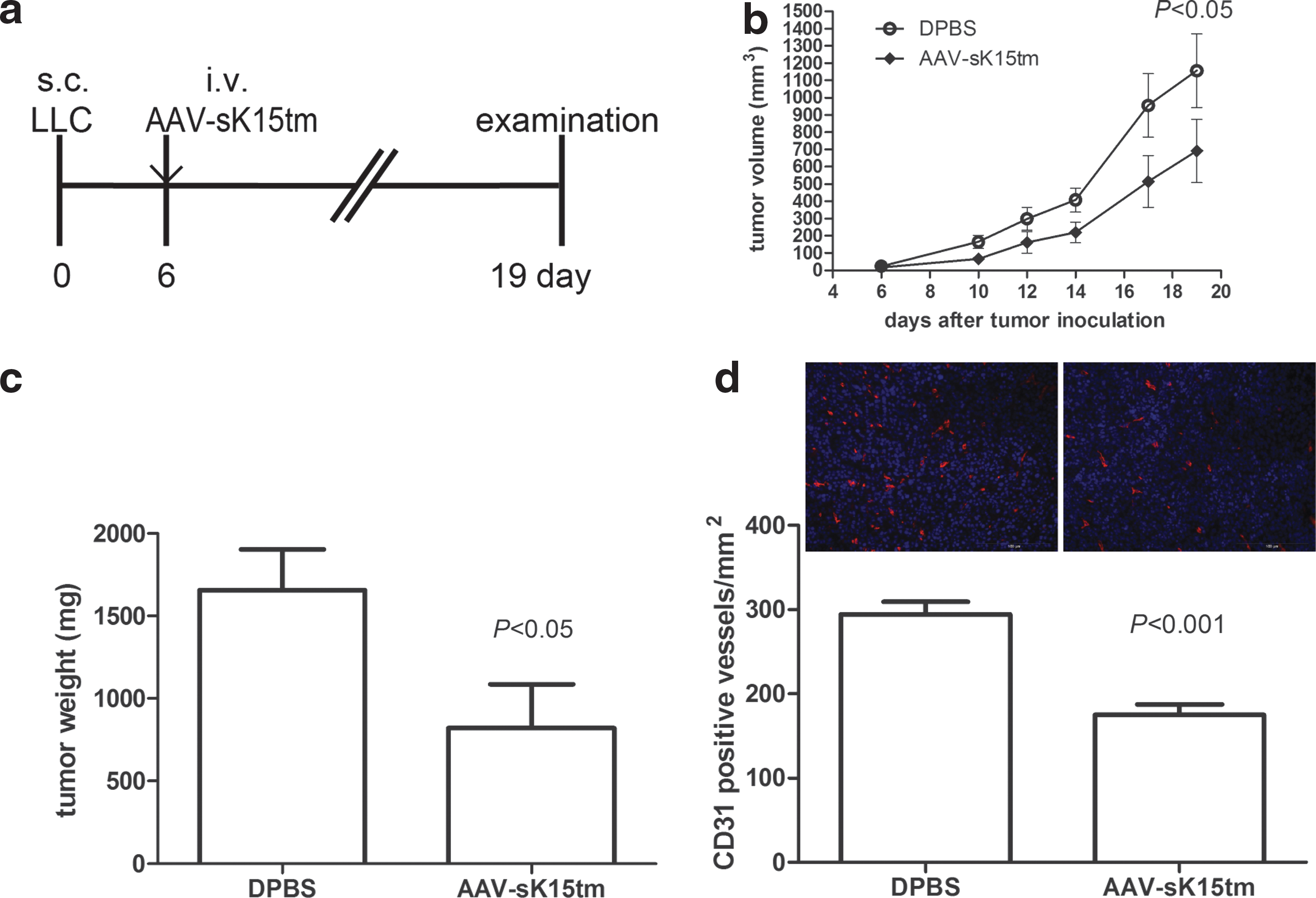
Treatment of AAV-sK15tm suppresses the growth of LLC xenografts and tumor-induced angiogenesis in mice.
Discussion
The major objective of this study was to generate a long-term expression system carrying an angiostatic factor that has superior potential overall in cancer therapy, especially in tumor metastasis, which remains the main challenge to current treatment and the major cause of death among patients with cancer. Antiangiogenic therapy has been developed in many forms and has been thought to be beneficial for suppression of tumor growth and its remote dissemination. 23 However, several angiostatic factors that were promising in preclinical studies were found to have limited or no benefits in clinical trials, partly due to limitations in dosing and preparation. The need for continued administration of costly protein drugs to maintain a serum level high enough to offset the opposing, proangiogenic activities in the milieu of cancer growth may be the major limitation of such approaches. Gene therapy, with the ability to convert host organs into depots of therapeutic protein production, may provide a solution to this problem.
Among the various viral vectors developed for gene therapy, AAV vectors have been generally considered safe 24 and ideal for cancer gene therapy because of their lack of pathogenicity and toxicity, their ability to infect both dividing and nondividing cells of various tissue origins, and their low host immune response and natural persistence. 25 The safety of recombinant AAV2 as a transgene delivery vector has been documented in many human clinical trials and many preclinical studies. 26 –28 In addition, the cell/tissue tropism, cytotoxicity, and immunogenicity of AAV vectors have been extensively characterized. 29 One minor question about the AAV2 vector concerns the cellular immune responses against the capsid protein of this human-derived AAV virus. This immune reaction would diminish transgene expression, thereby decreases therapeutic efficacy. 30 Cross-packaging the AAV2 capsid with various AAV serotypes has been developed and results in increased tissue tropism and transgene expression. 28 Among the AAV serotypes, AAV8 exhibits lower immunogenicity because of its nonhuman origin and strong tropism to the liver. 31 It also transduces multiple organs when high-dose vector is systemically administered, thus allowing transgene expression to be specific or widespread with low immune targeting. 32 Moreover, an inducible system of therapeutic gene expression carried by AAV vector has been proposed and demonstrated to be a safe and efficient way for treating liver cancer. 33 In this study, we used recombinant AAV2/8 as the delivery vector for K15tm, which exhibits the greatest effect in inhibiting endothelial cell proliferation, inducing endothelial cell apoptosis, and suppressing angiogenesis and tumor growth in mice when compared with wild-type K15. 11 Consistent with what is known about the vector system, mice injected with AAV-sK15tm showed sustained transgene expression for more than 500 days after virus injection, with no detectable signs of side effects or abnormalities. Moreover, biochemical analyses showed comparable levels of serum hepatic enzymes (Fig. 2c), blood urea nitrogen, and C-reactive protein (Supplementary Fig. S1) in mice injected with AAV-sK15tm compared with the control animals, indicating that neither AAV2/8 transduction nor K15tm expression induced significant immune/inflammatory responses, or damage to the transduced hepatocytes (Fig. 2c) and kidney health. The expression of K15tm was detected in mouse serum as early as 2 days after injection in a dose-dependent manner, and its expression reached its steady state serum concentration 3 weeks after virus injection (Fig. 2a). In addition, sustained high-level expression of serum K15tm was observed 2 weeks after virus inoculation (Fig. 2b), indicating little or no turnover of hepatocytes during virus transduction and transgene expression. It suggested that no cytotoxic effect was induced in hepatocytes by either AAV2/8 or K15tm. This result is consistent with the previous study in nonhuman primates using AAV2/8, showing long-term and sustained expression of transgene with no cytotoxicity. 26
The feasibility of using AAV vectors for gene therapy in correcting genetic defects has been successfully demonstrated in many animal models and in clinical trials. 27,34,35 Cancer gene therapy has been gaining more and more attention, as it holds promising potential to enable long-term, sustained expression of therapeutic proteins while reducing the treatment costs over the long run. However, the widely variable nature of disease progression in cancer will likely pose significant additional challenges to gene therapy approaches than many hereditary conditions. To meet these challenges, an ideal vector system needs to be capable of directing therapeutic protein expression in a rapid, robust, and controllable fashion. Several studies have demonstrated proof-of-the-concept results, in which antiangiogenic gene therapy pretreatment, using viral or nonviral vectors carrying angiostatic factors, showed significant suppression on tumor growth and metastasis. 14,15,36 However, the limitation on the timing of transgene expression impeded the critical evaluation of real therapeutic effects of these approaches. In this study, we evaluated the antitumor potential of AAV-sK15tm in a pretreatment protocol as well as in a treatment protocol. In the pretreatment protocol, the AAV-sK15tm dose of 2×1010 genome copies/mouse was effective in preventing tumor metastasis (Fig. 3). However, in the treatment protocol setting, this dose showed no or borderline suppressive effect (Fig. 4). In addition, serum K15tm was less than that of non-tumor-bearing mice with the same dose of virus infusion (Figs. 2a and 4c). This result suggested that tumor burden in the host may negatively impact the gene expression potential from the gene therapy vectors. Taken together, our result indicated that a higher dose of vector may be needed in the treatment protocol.
Our gene delivery strategy essentially turned the host liver into the primary site of therapeutic protein production. In terms of cell division and turnover, the liver is a relatively static organ in adult animals as well as in human beings. At the same time, the liver is also metabolically active and the major organ for plasma protein synthesis. By exploiting the natural advantage of long-lived hepatocytes for transgene expression, we successfully demonstrated that mice receiving a single injection of recombinant AAV2/8 continued to express the angiostatic factor K15tm at high levels without any display of adverse reactions for up to 1.5 years. The therapeutic efficacy of AAV-sK15tm was demonstrated by suppression of tumor growth and metastasis, partly through induction of tumor apoptosis and reduction of tumor-induced angiogenesis. Although treatment with AAV-sK15tm did not affect wound recovery (Supplementary Fig. S2), its possible effects on normal female reproductive cycles and other angiogenesis-dependent events remain to be clarified before clinical applications. The significant inhibition of tumor growth and metastasis by a single administration of the optimized AAV-sK15tm vector in this study provides a critical proof-of-concept validation of its application as cancer gene therapy.
Footnotes
Acknowledgments
This work was supported by the National Science Council, Taiwan (NSC 99-2320-B-006-008-MY3, NSC 99-2628-B-006-003-MY3, and NSC 100-2325-B-006-003-MY3 to H.-L.W. and NSC 102-2917-I-564-018 to C.-H.K.) and by a grant (“Aim for the Top University Plan,” National Cheng Kung University) from the Ministry of Education, Taiwan.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.