
Abstract
The field of adenovirology is undergoing rapid change in response to increasing appreciation of the potential advantages of adenoviruses as the basis for new vaccines and as vectors for gene and cancer therapy. Substantial knowledge and understanding of adenoviruses at a molecular level has made their manipulation for use as vaccines and therapeutics relatively straightforward in comparison with other viral vectors. In this review we summarize the structure and life cycle of the adenovirus and focus on the use of adenovirus-based vectors in vaccines against infectious diseases and cancers. Strategies to overcome the problem of preexisting antiadenovirus immunity, which can hamper the immunogenicity of adenovirus-based vaccines, are discussed. When armed with tumor-associated antigens, replication-deficient and oncolytic adenoviruses can efficiently activate an antitumor immune response. We present concepts on how to use adenoviruses as therapeutic cancer vaccines and consider some of the strategies used to further improve antitumor immune responses. Studies that explore the prospect of adenoviruses as vaccines against infectious diseases and cancer are underway, and here we give an overview of the latest developments.
Introduction
A
Molecular Biology of Adenoviruses
Adenovirus structure
Adenoviruses are nonenveloped double-stranded DNA viruses with an icosahedral capsid of approximately 90 nm. The molecular weight of a functional particle is approximately 150 MDa. While a number of studies have analyzed individual parts of the adenovirus capsid using cryo-electron microscopy (cryo-EM) and X-ray diffraction (Stewart et al., 1993; Athappilly et al., 1994; Xia et al., 1994; van Raaij et al., 1999; Rux et al., 2003; Zubieta et al., 2005; Saban et al., 2006), because of the complexity of the capsid, it is only recently that the structure of the whole human adenovirus virion at 3.6 Å resolution by cryo-EM has been reported (Liu et al., 2010).
Adenoviruses consist of two major elements: the core and the capsid. The core contains the adenovirus genome in association with proteins V, VII, and X, as well as a terminal protein linked to the 5′ viral DNA terminal. The capsid consists of seven proteins: II, III, IIIa, IV, VI, VIII, and IX. The majority of the capsid is formed by hexon and penton complexes, with the size of hexon and the length of up to nine hypervariable regions present in the hexon molecule varying between different serotypes. The hypervariable regions present type-specific antigens of the hexon and are the major neutralization site for hexon (Crawford-Miksza and Schnurr, 1996; Pichla-Gollon et al., 2007). Penton base and fiber form a penton capsomere and are involved in the initial stages of adenovirus infection. A characteristic feature of all serotypes, except for 40 and 41, is the RGD motif (Arg-Gly-Asp) present in the penton base. Fiber is a homotrimer with shafts that differ in length from 5 to 22 β-repeats, depending on the adenovirus serotype, and a knob domain responsible for virus binding to the host cell (Rux and Burnett, 2004). Adenovirus fibers from different serotypes also differ in amino acid sequences that further influence their structure. The length (Shayakhmetov and Lieber, 2000; Ambriovic-Ristov et al., 2003) and the flexibility (Wu et al., 2003; Bayo-Puxan et al., 2006; Kritz et al., 2007) of the fiber shaft domain have roles in adenovirus infectivity. Additional adenovirus structural proteins are listed in Table 1.
Adenovirus genome
The adenovirus genome consists of 36 kb linear, double-stranded DNA with inverted terminal repeats (ITRs) of 100–140 bp at both ends, which act as cis elements during replication of the viral genome. The genome is organized in five early (E1A, E1B, E2, E3, and E4), four intermediary (IVa2, IX, VAI, and VAII), and one late transcriptional unit. The first viral transcriptional unit to be expressed after the virus enters the cell nucleus is the E1A unit. E1A proteins trans-activate other viral genes and are the main regulators of viral transcription. E1B proteins bind p53, Bak, and Bax proteins (Sundararajan et al., 2001) and inhibit p53-dependent apoptosis, allowing survival of infected cells. The E2A unit codes DNA polymerase, preterminal protein, and single-stranded DNA-binding protein important for replication of the viral genome. E3 proteins subvert the host immune response and allow persistence of infected cells, while the E4 transcription unit encodes proteins influencing cell cycle control and transformation. Late-region genes are transcribed in the form of long precursor transcripts from the major late promoter, and encode structural capsid proteins.
Cell binding and entry of adenoviruses
Adenoviruses bind to cellular receptors via the knob portion of the fiber protein. The primary receptor responsible for attachment of all adenovirus serotypes, except those from group B, is the coxsackie and adenovirus receptor (CAR) (Bergelson et al., 1997). Even though CAR is expressed in most tissues, there is little expression of CAR on cancer cells (Sachs et al., 2002; Fuxe et al., 2003), mature skeletal muscle cells (Nalbantoglu et al., 1999), and the apical surfaces of polarized epithelial cells (Walters et al., 1999). These exceptions, along with the fact that CAR expression is localized to tight junctions (Coyne and Bergelson, 2005), present obstacles for the use of CAR-binding adenovirus vectors.
On the basis of receptor usage, group B adenoviruses have recently been subgrouped into B1 and B2. Members of subgroup B1 (AdV11, AdV16, AdV21, AdV35, and AdV50) use CD46 as a receptor (Gaggar et al., 2003) that is expressed both apically and basolaterally on cells (Sinn et al., 2002), thus making it accessible to adenovirus infection. Members of the B2 group (AdV3, AdV7, AdV11 and AdV14) have been shown in vitro to use cell adhesion molecule desmoglein-2 (DSG2) as receptor (Wang et al., 2011). Molecules used as a receptors by group D adenoviruses are less well defined and include CAR (Roelvink et al., 1998), CD46 (Li et al., 2012), and sialic acid (Nilsson et al., 2011). Receptors that mediate cellular attachment of human adenoviruses have been reviewed recently (Arnberg, 2012). In addition to primary/direct interactions between adenovirus and cellular receptors, adenoviruses interact with blood and components of other body fluids in vivo. These interactions may have a significant role in regulating adenovirus infection pathways (Johansson et al., 2007; Waddington et al., 2008; Doronin et al., 2012; Xu et al., 2013).
After the initial interaction with CAR receptor, internalization of viral particles proceeds (with exception of AdV40 and AdV41) via endocytosis triggered by binding of an exposed RGD motif on the penton base to αv integrins on the cell surface (Huang et al., 1996; Davison et al., 1997, 2001). Adenovirus uptake usually occurs via dynamin and clathrin-dependent receptor-mediated endocytosis (Gastaldelli et al., 2008), while adenoviruses belonging to group B use macropinocytosis as an infectious pathway (Amstutz et al., 2008; Kalin et al., 2010). It has been reported that adenovirus can also use lipid rafts and caveolae as routes of entry in plasmocytic cell lines (Colin et al., 2005) and human corneal cells (Yousuf et al., 2013).
After being liberated from the endosome, adenovirus interacts with cytoplasmic dynein and microtubules and moves toward the nucleus. The adenovirus capsid docks to the nuclear pore complex protein (NPC), nuclear histone H1 attaches to the hexon (Trotman et al., 2001), and microtubule motor kinesin-1 disrupts both NPC-docked capsids and the NPC, thus opening access for the viral genome to enter the nucleus (Strunze et al., 2011).
Recombinant Adenoviruses as Vaccine Vectors
Adenoviruses have proven to be highly efficient vehicles for introducing foreign DNA into target cells. Additionally, adenoviruses intrinsically induce host immune responses upon cell infection. Both of these features, accompanied by extensive knowledge of adenovirus molecular biology and methods for manipulating the viral genome that are now available, make adenoviruses attractive candidates for vaccine vector development.
Recombinant adenovirus vector development
There are several ways to create space for inserting foreign DNA (i.e., the antigen of interest) into adenoviruses. Most of the currently investigated recombinant adenovirus-based vaccine vectors are vectors of first and second generation where foreign DNA can be inserted into at least three regions of the adenoviral genome: the E1, E3, and the short region between E4 and right ITR. E1 deletion impairs viral replication and renders these adenoviruses replication deficient. Replication-deficient E1/E3-deleted vectors can accommodate approximately 6.5 kb of foreign DNA and can be propagated in E1-complementing producer cell lines, such as HEK-293 (Graham et al., 1977), 911 (Fallaux et al., 1996), or PER.C6 (Fallaux et al., 1998). Additional deletions in the E2 and E4 coding sequences provide a larger capacity for transgene insertion. Helper-dependent adenovirus vectors containing only ITR repeats, a packaging signal, and no viral structural genes are less often used as vaccines. Nevertheless, they have been shown to stimulate superior transgene-specific immune responses, both T cell and antibody responses, when compared with first-generation adenovirus vectors (Harui et al., 2004; Kron et al., 2011; Weaver et al., 2013). This suggests that helper-dependent adenoviruses could be considered as suitable vaccine vectors. Besides replication-deficient adenovirus vectors, conditionally replication-competent or oncolytic adenovirus vectors (OAds) have also been developed, which can selectively replicate in cancer cells but not in normal cells.
Recombinant adenovirus vectors transduce both quiescent and actively dividing cells, can be produced at very high titers, have relatively high capacity for transgene insertion, and allow high expression of the recombinant protein. After entering the nucleus, adenoviruses do not integrate into the host genome, avoiding the risk of insertional mutagenesis and increasing the safety of these vectors. First-generation adenovirus vectors provide only transient transgene expression, and while this is of limited benefit for gene therapy, it is nonetheless adequate for cancer treatment and vaccination.
Immunogenicity of adenovirus-based vectors
Adenoviruses are highly immunogenic and induce both innate and adaptive immune responses in mammalian hosts. Even though the repertoire of the innate immune response differs between different cell types, adenovirus infection induces production of numerous chemokines and cytokines that modulate the initiation of inflammation. Activated molecules include cytokines such as tumor necrosis factor α (TNF-α), IL-6, IL-1, IL-12, and type I interferons (IFNs), and chemokines such as macrophage inflammatory protein 1, RANTES, IL-8, and monocyte chemoattractant protein 1 (MCP-1). Immune responses to adenovirus vectors can be induced by the presence of virus itself, via recognition through pathogen recognition receptors (PRR), or as a consequence of interactions with cell surface receptors. Signaling triggered by fiber/CAR interactions activates downstream signaling of ERK 1/2, JNK, MAPK, and NF-κB, leading to upregulation of IL-8, GRO-α, GRO-γ, or RANTES (Tamanini et al., 2006). After internalization into the endosome, adenoviral DNA can be recognized by Toll-like receptors (TLR), namely, TLR9 (Iacobelli-Martinez and Nemerow, 2007), whose activation results in MyD88-dependent and -independent IFN responses (Basner-Tschakarjan et al., 2006; Zhu et al., 2007). Because of endosome membrane penetration, adenovirus activates NLRP3 inflammasome and IL-1β release via TLR9 sensing and the release of cathepsin B into the cytoplasm (Barlan et al., 2011). The escape of adenovirus into the cytoplasm also results in type I IFN activation that can be IRF3 dependent (Nociari et al., 2007) or IRF7 dependent (Fejer et al., 2008). Very recently, cyclic-GMP-AMP synthase (cGAS), part of the cGAS/STING/TBK1 DNA sensing cascade, has been reported as a dominant cytosolic DNA sensor responsible for detection of internalized adenovirus leading to induction of type I IFN (Lam et al., 2014). Alternatively, IFN-γ and TNF-α induced by recombinant adenovirus administration have been shown to inhibit transgene expression from adenovirus vectors in vitro by a transcription-related mechanism. Early induction of those cytokines after vector administration in vivo suggests that innate immunity contributes to the regulation of adenovirus-mediated transgene expression (Sung et al., 2001). Additionally, recombinant adenoviruses from different serotypes induce different IFN pathways. For example, in contrast to recombinant AdV5, immunization with recombinant AdV28 and AdV35 induced production of IFN-α that skewed the central/effector memory distribution and functional profile of the CD8+T cell response, and limited transgene expression by these vectors (Johnson et al., 2012). These findings should be considered in the design of adenovirus vaccine vectors. Even though the induction of innate immune responses may hamper adenovirus vaccine-based antigen expression, it nonetheless may be advantageous in that it provides the characteristics of a natural adjuvant. For more details regarding innate immune responses to adenovirus, see Hendrickx et al. in this issue (2014).
Adaptive immune responses against adenoviruses are directed toward early and late adenoviral antigens and comprise B cell, CD4+ (Onion et al., 2007), and CD8+ T cell responses (Yang et al., 2003). Adenovirus-specific T cells can be quickly activated upon adenovirus vector administration, with hexon protein being the major target for CD4+ and CD8+ T cells (Olive et al., 2002; Leen et al., 2004). There is evidence that coordinated CD4+ and CD8+ T cell responses specific for human adenovirus hexon epitopes contribute to the control of adenovirus infection in healthy individuals (Zandvliet et al., 2010). It has been shown that CD4+ T cells cross react between different adenoviruses serotypes, and that T cell frequency decreases with age (Olive et al., 2002). Similarly, adenovirus-specific CD8+ T cells derived from humans and mice cross react between human and mouse adenovirus serotypes (Fitzgerald et al., 2003). As in the case of eliciting innate immune response, adenovirus vector-based vaccines may benefit from vector-induced adaptive immune responses, which can enhance vaccine efficacy by providing an adjuvant effect.
Preexisting antiadenovirus immunity
As virtually every individual will be infected by one or more adenovirus at some point in life, often at an early age, most populations display preexisting immunity to the most common adenovirus serotypes. As a consequence, the prevalence of neutralizing antibodies against AdV5 and other common serotypes is high in humans (Mast et al., 2010; Barouch et al., 2011; Zhang et al., 2013b). Adenovirus-neutralizing antibodies (mainly IgG) are mostly targeted against the surface loops of the hexon capsid protein. However, antibodies against the penton base and fiber knob can also neutralize adenoviruses (Wohlfart, 1988; Hong et al., 2003; Bradley et al., 2012). For AdV5, the specificity and immunogenicity of neutralizing antibodies differ according to whether infection is induced by natural infection or immunization. Neutralizing antibodies elicited by natural infection are directed largely at the AdV5 fiber and hexon, while exposure to recombinant AdV5 through vaccination elicits antibodies primarily to hexon and other capsid proteins rather than fiber (Cheng et al., 2010). Adenovirus-neutralizing antibodies were found to be adenovirus serotype-specific, with minimal or no cross-reactivity between different species (McCoy et al., 2007; Sharma et al., 2010). This should be considered when evaluating outcomes of clinical trials using vectors based on adenoviruses from different species (Calcedo et al., 2009).
Strategies to Overcome Antiadenovirus Vector Immunity
Activation of host innate immune responses and preexisting immunity to common adenovirus serotypes may limit the efficacy of adenovirus-based vaccine vectors. Adenovirus vector-specific cytotoxic T cells and neutralizing antibodies may impede the induction of immune responses to the vaccine-encoded antigens, as they may reduce the dose and duration of exposure of target cells to the vaccine antigens. These features have forced the development of new strategies to evade undesired antivector host immune responses, including the search for other types of adenoviruses that occur at low prevalence in human populations.
Different human and nonhuman adenovirus serotypes as a platform for vaccine development
The high prevalence of anti-AdV5 immunity in the developing world (50–90%) (Holterman et al., 2004) has made the identification of novel and rare adenovirus serotypes a priority in the field of vaccine development. Some of the rare human adenovirus types that are under evaluation include AdV11, AdV26, AdV28, and AdV35 (Vogels et al., 2003; Holterman et al., 2004; Lemckert et al., 2006; Abbink et al., 2007; Kahl et al., 2010). The seroprevalence of AdV11, AdV35, AdV50 (group B), AdV26, AdV28, AdV48, and AdV49 (group D) was found to be less than 10% in healthy adult blood donors from Sub-Saharan Africa and Belgium (Abbink et al., 2007; Vogels et al., 2003). These rare serotypes were also immunogenic in the presence of anti-AdV5 immunity (Lemckert et al., 2005; Geisbert et al., 2011). It was shown that vaccination with AdV35-, AdV26-, and AdV48-based vectors, which are thought to utilize CD46 as their primary cellular receptor, induced significantly greater innate cytokine responses than AdV5, which uses the CAR receptor (Teigler et al., 2012). AdV26 is highly immunogenic as a single vector, grows to high titers in AdV5 E1-complementing cell lines, and has shown promise as a vaccine vector because its immunogenicity appeared favorable in rhesus monkeys. A first-in-human evaluation of the safety and immunogenicity of a recombinant AdV26-based vaccine demonstrated that this vector elicited broad and diverse antigen-specific humoral and cellular immune responses in humans (Baden et al., 2013; Barouch et al., 2013). Additionally, AdV26 could be used repeatedly and the humoral immune responses could be boosted in the face of antivector immunity. While memory T cells elicited by AdV5 vectors were high in magnitude, they exhibited functional exhaustion and decreased anamnestic potential following secondary antigen challenge compared with AdV26, AdV35, and AdV48 vectors (Penaloza-MacMaster et al., 2013).
Another approach to circumvent the challenges of preexisting immunity to adenovirus vectors is to use vectors derived from nonhuman adenoviruses. Vectors based on simian adenoviruses (Tatsis et al., 2006; Colloca et al., 2012; Sheehy et al., 2012a; Antrobus et al., 2013; Cervasi et al., 2013), an ovine adenovirus (Bridgeman et al., 2009), and a replication-defective bovine adenovirus (Singh et al., 2008) are some examples that have shown promise as vaccine vectors. In particular, vaccine vectors based on ape adenoviruses have recently gained interest because they are largely similar to human adenoviruses and have no, or only low seroprevalence in human populations from different geographical areas (Xiang et al., 2006; Dudareva et al., 2009; Colloca et al., 2012).
Capsid-modified adenovirus-based vaccine vectors
Modifying fiber, protein IX, or hexon proteins is an attractive strategy to improve adenovirus vectors in terms of increasing infection rates (Kurachi et al., 2007), but also for allowing evasion of preexisting antiadenovirus immunity. Neutralizing antibodies against AdV5 mostly target the hexon protein (Bruder et al., 2013); thus, hexon modification allows virus to escape neutralizing antibodies, subsequently leading to efficient induction of adenovirus-coding antigen-specific immune responses (Abe et al., 2009; Bruder et al., 2012). Indeed, recombinant AdV5-based vectors have been developed that have undergone a complete hexon exchange (Youil et al., 2002). In the case of AdV5, the preferred location to insert foreign peptides is the hypervariable region 5 of hexon loop L1, which avoids interfering with the function and efficacy of AdV5. Replacing seven short hypervariable regions on the surface of the AdV5 hexon with the corresponding hypervariable regions from AdV48 protected this chimeric recombinant AdV5 vector from preexisting anti-AdV5 immunity (Roberts et al., 2006). Another example is an adenovirus vector with a chimeric Ad5–Ad12 hexon that was not neutralized by plasma from C57BL/6 mice immunized with AdV5. The same vector was also capable of transducing the livers of C57BL/6 mice previously immunized with AdV5 (Roy et al., 1998).
Alternatively, inserting epitopes of interest into one of the adenovirus capsid proteins can ensure a specific antiepitope immune response. Krause and colleagues (2006) compared the influence of the site of insertion (hexon, penton base, fiber knob, or protein IX) on epitope immunogenicity and concluded that the best humoral response was obtained when an epitope was inserted into the fiber protein. Antiepitope humoral responses are influenced not only by the number of epitopes per capsid, but also by preexisting antiadenovirus immunity (Lanzi et al., 2011). The differential immune response might also be influenced by the position, composition, and structural properties of the inserted antigens. Recently, chimpanzee-derived AdC68 bearing a B cell epitope within the hexon variable region 1 induced epitope-specific antibody responses of higher magnitude and avidity than those carrying epitope within variable region 4, or vectors expressing the epitope as part of a transgene product. Furthermore, specific antibody responses could be boosted by a second dose of the variable region 1 hexon-modified vector, but not by repeated immunization with the variable region 4 hexon-modified vector (Zhou et al., 2013). However, it is worth noting that epitope display on the surface of the adenovirus particle is limited in regard to the length of peptide that can be incorporated.
Evading neutralizing antiadenovirus antibodies in vivo can also be achieved by physically shielding virus particles. Fully detargeted PEGylated adenovirus vectors were able to escape from preexisting antiadenovirus antibodies and induced strong cellular and humoral immune responses to vector-encoded transgene products, which qualifies them as a novel and safe vector format for vaccination (Wortmann et al., 2008).
Heterologous prime-boost regimens utilizing adenovirus vectors
Most vaccines require more than one immunization (prime-boost) to be effective; however, this strategy also allows development of antivector immunity. Heterologous prime-boost strategies that use different vaccine components containing the same antigen are another means by which induced antivector immunity may be overcome. This strategy involves priming with one vector, thus focusing the immune system on the required immunogen, and then boosting with another vector. The booster dose provides the same antigen, leading to an increase in the magnitude and breadth of specific T and B cell responses, while avoiding antivector immunity conferred by the prime immunization (Lu, 2009). It has been observed that the magnitude of the immune response following prime-boost regimens can be different depending on the immunization route used (Lambe et al., 2013). The immunogenicity of AdV5, AdV35, and AdV11 was evaluated in different prime-boost regimens in mice with preexisting anti-AdV5 immunity. Interestingly, AdV35–AdV11 and AdV11–AdV35 combinations were more immunogenic than any regimen containing AdV5 (Lemckert et al., 2005). Since cross-reactive AdV35/AdV11-specific humoral and cellular immune responses were detected, this strategy should be evaluated in combination with additional vectors that are immunologically distinct from either AdV11 or AdV35: namely, vector combinations of rare human adenovirus serotypes such as AdV35 and AdV49 (Thorner et al., 2006) or AdV26 and AdV35 (Zahn et al., 2012). There are also reports of heterologous prime-boost schedules using chimpanzee adenoviruses AdVC6 and AdVC68. These are very distinct, non–cross-reacting serotypes that were highly efficient in increasing the transgene product-specific CD8+ T cell response (Pinto et al., 2003). Variations on adenovirus heterologous prime-boost regimens under evaluation combine adenovirus with DNA, poxvirus-derived vectors like modified vaccinia Ankara (MVA), or protein subunit vaccines. AdVC63-MVA heterologous prime-boost immunization induced strong, long-lasting multifunctional CD8+ and CD4+ T cell responses that exhibited a central memory-like phenotype regardless of prior immunization with adenovirus vectors of alternative human or simian serotypes (Draper et al., 2010).
Adenovirus Vector Vaccines Against Infectious Diseases
Adenovirus-based vectors have been and are being investigated as vaccines targeting a broad range of viral, bacterial, and protozoan pathogens. They are particularly used in disease areas where classical vaccination strategies have proven ineffective, difficult, or technically impossible. Additionally, recombinant adenovirus-based vectors are promising vaccination vehicles especially in the area of infectious diseases where protection is associated with cellular responses. This is because adenovirus-based vectors induce strong and sustained cellular responses, particularly CD8+ T cell responses (Suleman et al., 2011). The proposed mechanism of action of adenovirus-based immunization after intramuscular administration is illustrated in Fig. 1.
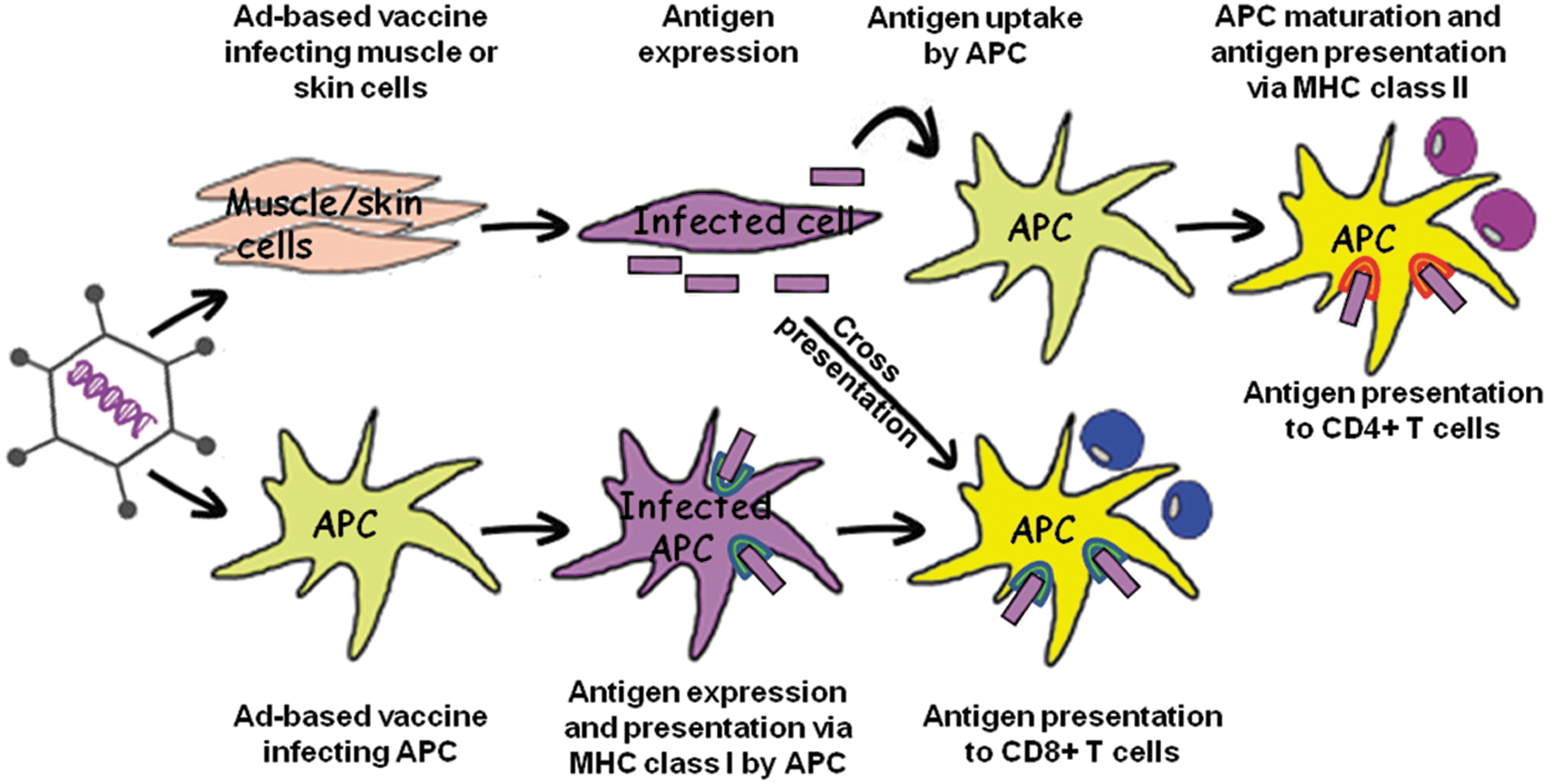
Mechanism of action of adenovirus-based vaccine after intramuscular administration. The antigen encoded by the adenovirus-based vaccine can be presented to T cells by antigen presenting cells (APCs) via two distinct routes. The indirect route involves antigen expression by muscle cells or keratinocytes that have been transduced after vaccine injection in skin or muscle. Antigen expressed by infected muscle cell or keratinocyte can be presented by APCs to CD4+ T cells or via cross-presentation to CD8+ T cells. The direct route occurs by direct transduction of APC and subsequent expression of antigen by APC. These antigens are presented to CD8+ T cells.
One of the most important viral pathogens against which adenovirus vectors may prove successful is the prevention or treatment of human immunodeficiency virus (HIV) infections. AdV5 vectors encoding the gag, pol, and env HIV proteins have been developed and clinically evaluated in mice and nonhuman primates. A replication-deficient AdV5 vector vaccine encoding the gag, pol, and nef genes of HIV-1 was developed by Merck and evaluated in a phase 2b trial (STEP study) (Buchbinder et al., 2008). Although this vaccine succeeded in inducing CD8+ T cell responses in homosexual men, it failed to protect against HIV infection and appeared to increase the risk of infection in uncircumcised men who were seropositive for AdV5 (Gray et al., 2010). This finding has precluded the use of AdV5 vector-based vaccines in AdV5 seropositive volunteers with HIV. Mechanisms that could explain the enhanced HIV-1 acquisition in these trials include the possible formation of immune complexes of AdV5 with vector-specific antibodies, which in turn activate dendritic cells (DCs) and CD4+ T cells (Perreau et al., 2008), or the homing of vector-specific CD4+ T cells to mucosal surfaces that could serve as targets for HIV-1 infection (Benlahrech et al., 2009). A more recent study in which an AdV5-based HIV vaccine was tested as a booster vaccination after DNA prime vaccination in AdV5 seronegative circumcised men (HVTN505) also failed to reduce HIV-1 acquisition or viral-load (Hammer et al., 2013).
Alternative serotype vectors or vectors based on nonhuman adenoviruses are now under investigation as potential alternative HIV-1 vaccine platforms. The majority of these vaccine vector candidates have distinct biological properties compared with AdV5, exemplified by the use of different cellular receptors and the induction of distinct innate and adaptive immune profiles. These differences with AdV5 warrant their investigation as vaccines to prevent or treat HIV, other infections, and noninfectious diseases (Barouch, 2010). AdV26 encoding HIV env has been investigated in phase 1 clinical trials and was found to activate significant cellular and humoral response in a dose-dependent manner. Recombinant AdV26 HIV-1 Env vaccine (Ad26.ENVA.01) elicited a dose-dependent expansion of the magnitude, breadth, and epitopic diversity of Env-specific binding antibody responses, with induction of multiple CD8+ and CD4+ T cell memory subpopulations and cytokine secretion phenotypes (Baden et al., 2013; Barouch et al., 2013). AdV35 has similar potential to AdV26, and has also been evaluated in phase 1 clinical trials with evidence of activation of HIV-specific cellular and humoral responses, with no reports of vaccine-associated severe adverse events (Keefer et al., 2012). An accelerated heterologous prime-boost regimen involving administration of AdV35 at birth and AdV26 encoding SIVmac239 Gag at 4 weeks of life elicited potent and durable Gag-specific cellular and humoral immune responses in neonatal rhesus monkeys, including mucosal responses that remained detectable at 1 year of age (Liu et al., 2012). Induction of simian immunodeficiency virus-specific cellular immune responses in foreskin by AdV26 and AdV35 vaccine vectors suggests that alternative serotype adenovirus vectors induce potentially important immune responses in foreskin (Balandya et al., 2014). Replication-competent AdV4 vectors expressing HIV-1 (Env and mosaic Gag) immunogens (Alexander et al., 2013) are currently under evaluation in a phase 1 safety and immunogenicity trial (
Adenovirus vectors are also being explored as innovative vaccine concepts to protect against influenza infection. An adenovirus-based influenza A virus vaccine directed against the hemagglutinin (HA) protein of an H5N1 strain induced both HA-specific antibodies and a cellular immune response (Van Kampen et al., 2005; Gao et al., 2006). Replication-defective AdV5-based multivalent vaccines have been evaluated against H5, H7, and H9 avian influenza viruses in a mouse model, with induction of high levels of humoral and cellular immune responses. Mice were protected against virus replication after challenge with H5, H7, and H9 avian influenza virus subtypes (Vemula et al., 2013). Adenovirus group D-based influenza vaccines were equally efficient as group C vaccines when delivered mucosally by the intranasal route. At doses as low as 108 virus particles per mouse, group D vaccines induced complete protection against a stringent lethal challenge dose of influenza, supporting the further investigation of group D adenoviruses as mucosal vaccines (Weaver and Barry, 2013). A first-in-man clinical trial demonstrated that an orally delivered AdV5 vector vaccine expressing HA from avian influenza induced immune responses to antigen with a favorable safety profile (Peters et al., 2013). At the same time, the first clinical trial of a replication-competent AdV4 vector vaccine (AdV4-H5-Vtn) expressing HA from avian influenza A was reported. Orally administered AdV4-H5-Vtn induced a cellular immune response, and when boosted with an inactivated H5N1 vaccine, led to robust hemagglutination inhibition and H5N1-neutralizing antibody responses (Gurwith et al., 2013). A dose escalation study in humans using a replication-deficient chimpanzee adenovirus vector vaccine expressing conserved influenza nucleoprotein and matrix protein 1 antigens, ChAdOx1 NP+M1, induced strong T cell immune responses, making it a promising influenza vaccine vector for universal vaccination against multiple influenza strains (Antrobus et al., 2013).
Many attempts have been made to develop viral vectors against malaria, aiming to activate the host immune system by expressing pathogenic malaria-specific antigens expressed at various stages of the parasite life cycle. Both humoral and cellular immune responses are needed for effective protection against Plasmodium falciparum, and results obtained with adenovirus-based vaccine vectors are encouraging. AdV5 vectors expressing Plasmodium falciparum apical membrane antigen 1 or merozoid surface protein 1 antigens stimulated effector memory T cell responses when administered alone, but immunogenicity was improved when prime-boost vaccination was performed with DNA encoding both antigens (Chuang et al., 2013). AdV26 and AdV35 vectors encoding Plasmodium yoelii circumsporozoite protein given as a booster after circumsporozoite protein immunization elicited an even more robust and sustainable IFN-γ+ CD8+ T cell response than one- or two-component regimens (Radosevic et al., 2010). Antibodies and CD8+T cells producing IFNγ and TNFα with specificity for P. falciparum circumsporozoite were induced by an AdV35-based vaccine, with modest neutralizing antibody responses induced against the vector itself (Ouedraogo et al., 2013). Combinations of chimpanzee-derived simian AdV63 in a heterologous prime-boost regimen with MVA expressing the same antigen in nonhuman primates (Capone et al., 2010) and humans (O'Hara et al., 2012; Ewer et al., 2013; Ogwang et al., 2013) were shown to induce protective CD8+ T cell immunity to human malaria. However, the utility of T cell-inducing blood-stage malaria vaccine is questionable, since it does not impact parasite growth rates in the blood, suggesting that the focus for vaccination should remain on high-titer antibody induction (Sheehy et al., 2012b).
Adenovirus-based vaccines have also been designed to combat emerging viruses such as hantavirus and ebola virus. Nonreplicating AdV5 vectors expressing viral antigens were able to elicit active cellular immune response and protect hamsters and mice from lethal hantavirus infection (Safronetz et al., 2009). AdV26 and AdV35 vaccine vectors expressing ebola virus glycoproteins generated robust antigen-specific cell-mediated and humoral immune responses that were not affected by AdV5 immune status. Partial protection against ebola virus was demonstrated after a single administration of an AdV26 vaccine, with complete protection after boosting with AdV35 one month later (Geisbert et al., 2011). Airway vaccination of nonhuman primates with AdV5-based Ebola virus vaccine can efficiently bypass preexisting immunity to AdV5 and induce protective immune responses, although at lower efficacy than that using an intramuscular vaccine delivery route (Richardson et al., 2013).
Adenovirus-based vaccines have been investigated for the use against bacterial pathogens such as Mycobacterium tuberculosis, Bacillus anthracis, Clostridium difficile, and other pathogens for which current vaccines are either lacking or suboptimal. In a phase 1 study, the safety and immunogenicity of AdHu5Ag85A, a human AdV5-based vaccine expressing an immunogenic M. tuberculosis antigen Ag85A, was evaluated in healthy adults. Although AdHu5Ag85A was immunogenic in all adults, polyfunctional CD4+ and CD8+ T cell booster responses were more potent in those previously vaccinated with Bacillus Calmette–Guérin. Furthermore, there was little evidence that preexisting anti-AdV5 immunity significantly dampened the potency of the AdHu5Ag85A vaccine (Smaill et al., 2013). It was recently reported that intranasal instillation of a nonreplicating adenovirus vector encoding B. anthracis protective antigen could confer rapid and sustained protection against inhaled anthrax in mice in a single-dose regimen in the presence of preexisting adenovirus immunity (Zhang et al., 2013a). An adenovirus-based vector encoding pathogenic C. difficile CdTA and CdTB antigens generated rapid and robust humoral as well as cellular immune responses in mice, and protected against lethal challenge from C. difficile–associated diarrhea (Seregin et al., 2012).
The data from published preclinical and clinical trials using adenovirus vector-based vaccines against other pathogens are briefly summarized in Table 2.
Adenovirus Vectors as Cancer Vaccines
The rationale underlying cancer vaccines is that they should enable the immune system to recognize cancer cells and influence their growth or lead to their eradication. One approach is immunization against tumor-specific antigens, which can be achieved via adenovirus-mediated delivery of tumor-associated antigens (TAAs) or immunomodulatory molecules.
Repication-deficient adenoviruses expressing tumor-associated antigens or immunomodulatory molecules
While CD4+ cytotoxic and helper T cells appear to be effective in de-bulking tumor mass in animal models (Schultz et al., 2000; Cho et al., 2003), CD8+ cellular activation is considered to be equally important for successful immunotherapy. Skin or lymph node resident DC subsets are capable of cross-priming CD8+ cells through cross presentation of the exogenous antigens (Rock and Shen, 2005). Adenoviruses have been shown to generate antitumor immunity in animal models by transfer of TAA to DCs (Lotem et al., 2006; Backer et al., 2010; Hangalapura et al., 2011). TAA can be introduced to DCs either directly by infecting them with adenovirus, or by indirect TAA uptake from TAA-coding adenovirus infected cells. Melanoma-specific antigen coding adenovirus-transduced DCs induced broader and stronger immunity against various peptides of the vaccinated antigen in comparison to a direct peptide-loaded DC vaccine (Tuettenberg et al., 2003). Despite promising induction of immune responses, clinical results indicate that DC-adenovirus vaccines still need improvement (Kuball et al., 2002; Todorova et al., 2005; Lubaroff et al., 2009).
Recent studies, some of which were clinical trials in humans, reported positive outcomes when replication-deficient adenovirus vectors were used as cancer immunotherapeutics. It has been shown that immunization of mice with AdV5-PSA/PSCA vaccine, an adenovirus carrying prostate-specific antigen (PSA) and prostate stem cell antigen (PSCA), induced strong antitumor immunity when challenged with mouse prostate tumor cell lines expressing human PSA (Karan et al., 2011). AdV5/F35 (chimeric AdV5 with AdV35 fibers), encoding Epstein-Barr virus (EBV) nuclear antigen-1 fused to multiple CD8+ T cell epitopes from the EBV latent membrane proteins, LMP1 and LMP2, was investigated in a phase 1 clinical study as a therapeutic tool for recurrent or metastatic nasopharyngeal carcinoma. The study reported stabilization of disease and possible delayed tumor progression with prolonged survival due to AdE1-LMPpoly-generated T cells (Smith et al., 2012). Recently, a replication-deficient AdV40-based intravenous vaccine expressing mouse mesothelin was shown to be an effective prophylactic cancer vaccine against metastatic lesions of pancreatic cancer in mice due to antigen and tumor-specific cytotoxic lymphocyte-mediated immunity (Yamasaki et al., 2013). As for adenovirus vector-based vaccines targeting pathogens, preexisting antiadenovirus immunity may present an obstacle for TAA-expressing adenovirus cancer vaccine vectors. However, in a phase 1/2 study where cohorts of patients with advanced colorectal cancer were immunized with escalating doses of AdV5 encoding the tumor antigen carcinoembryonic antigen (CEA) (Ad5 [E1-, E2b-]-CEA[6D]), CEA-specific cell-mediated immune responses were observed despite the presence of preexisting AdV5 immunity in the majority of patients (Morse et al., 2013).
Active “immunoediting” allows tumors to grow in immune-competent hosts (Hanahan and Weinberg, 2011), and strategies that tumors use to evade immunity have been described (Croci et al., 2007). To target this immune suppressive microenvironment, adenovirus vectors expressing GM-CSF, CCL19 (de Gruijl and van de Ven, 2012), HLA costimulatory ligands such as CD80 (Gilligan et al., 1998), IL-12 (Sangro et al., 2004), and IL-24 (Tong et al., 2005) have been used, but without evidence of clinical efficacy. One of the approaches evaluated in a preclinical model combined a replication-deficient adenovirus expressing the soluble Programed Death-1 (PD-1) receptor and the herpes simplex virus thymidine kinase (HSVtk) to induce both direct cytotoxicity and antitumor immunity. This dual-module adenovirus significantly enhanced CD8+ T cell-mediated tumor rejection and allowed complete suppression of secondary tumor challenge at a distal site (Shin et al., 2013).
Oncolytic adenovirus-based vaccines
Even though replication-competent OAds are rapidly cleared from the bloodstream and the tumor microenvironment, they still have potential to induce innate and adaptive immunity against tumors. Oncolytic adenoviruses offer multimodal mechanisms of tumor killing: an initial oncolytic event elicited by the virus resulting in cancer cell lyses, followed by induction of an antitumor immune response as a result of this oncolytic process, or OAd-mediated expression of therapeutic antigen. In this regard the oncolytic virus can be considered as a vaccine. Most of the arguments for this perspective rely on the fact that adenovirus infections and adenovirus-mediated killing of tumor cells result in highly immunogenic responses. Oncolysis leads to the accumulation of TAAs, pathogen-associated molecular pattern (PAMPs), damage-associated molecular patterns (DAMPs; also known as “danger signals”), and proinflammatory cytokines, which are thought to aid in breaking tumor tolerance and enhancing antitumor immunity. In addition, OAds can be armed with TAAs or immunomodulatory transgenes. A summary of the mechanism of action of oncolytic adenovirus-based cancer vaccine is depicted in Fig. 2.
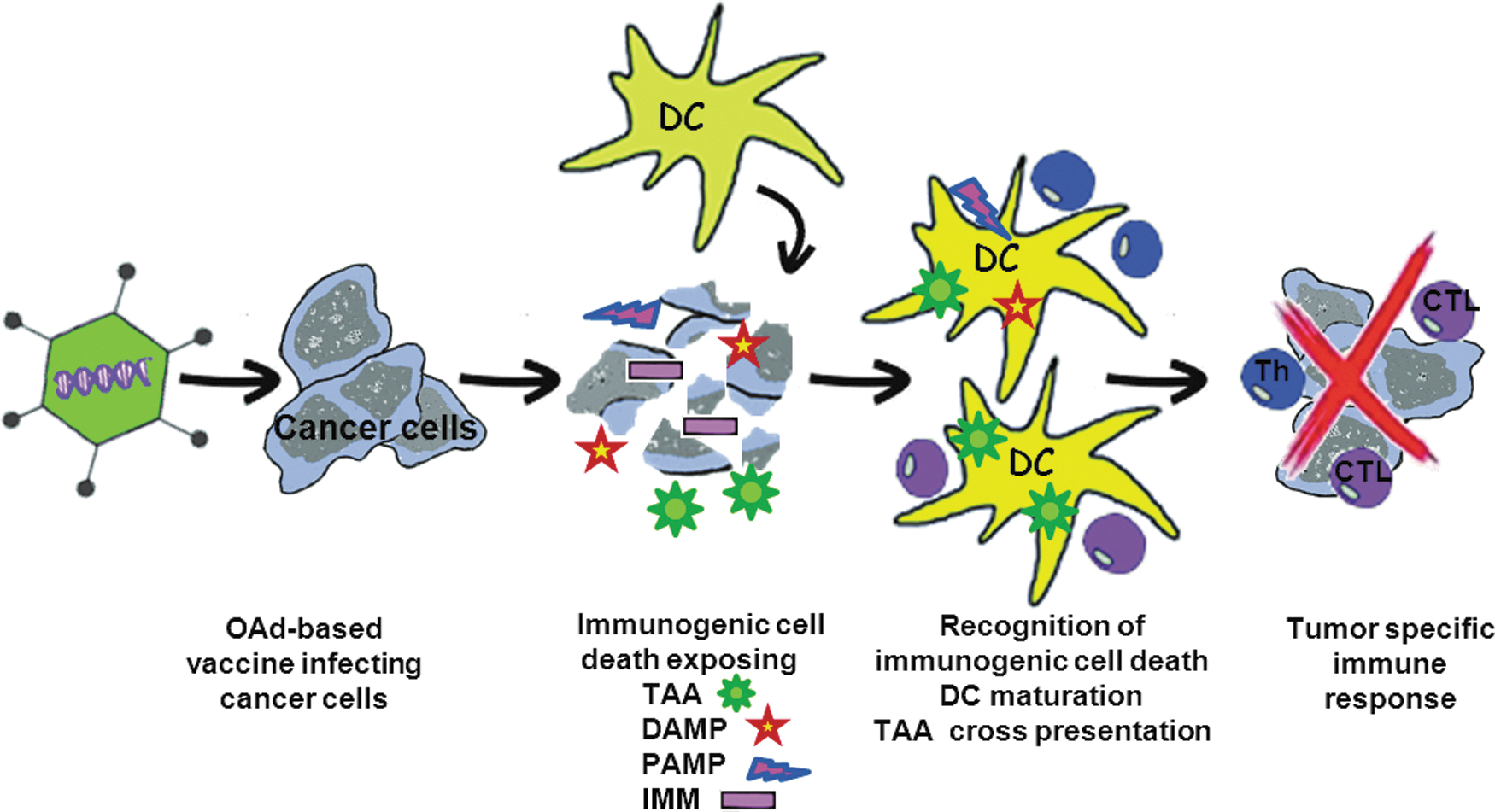
Mechanism of action of oncolytic adenovirus-based cancer vaccine. After being delivered either intratumoraly or systemically, oncolytic adenovirus vector (OAd)-based vaccine armed with immunomodulatory transgene reaches tumor tissue and begins to replicate selectively in cancer cells, leading to expression of immunomodulatory transgene as well as death of infected cells. Due to immunogenic cell death, different danger signals such as damage-associated molecular pattern (DAMP) and OAd-derived pathogen-associated molecular pattern (PAMP) as well as tumor-associated antigens (TAA) are liberated from dying OAd-infected cancer cell. The released DAMPs and PAMPs activate and mature dendritic cells (DC), and TAAs are processed and cross-presented to naive T cells. Together with action of immunomodulatory transgene, antitumor immune response is elicited, involving both CD4+ and CD8+ T cells, which may lead to eradication of tumor.
A potential limitation to vaccination with OAds is that of immunodominance (Restifo, 2001; Kedl et al., 2003), in which adenovirus epitopes might mask TAA epitopes, a behavior which has been identified in adenovirus-based vaccination approaches (Schirmbeck et al., 2008). A recent clinical trial evaluating Ad5/3-Δ24-GMCSF (granulocyte macrophage colony-stimulating factor-coding AdV5 capsid chimeric adenovirus) reported first-in-human immunologic data suggesting that the induction of antiviral immunity might correlate with the induction of antitumor T cell immunity (Kanerva et al., 2013).
Several groups have validated OAds expressing GM-CSF as potent inducers of antitumor immunity in preclinical studies (Bristol et al., 2003; Ramesh et al., 2006). In a phase 1 clinical trial with the GM-CSF-armed OAd CG0070, 48.6% of bladder cancer patients demonstrated a complete response (Burke et al., 2012). IL-12 is a potent proinflammatory cytokine bridging innate and adaptive immune responses by activating NK, CD4+, and CD8+ cells (Del Vecchio et al., 2007). OAds carrying the IL-12 gene either at the E1A or E3 regions of the genome have shown enhanced antitumor efficacy in vivo (Lee et al., 2006; Bortolanza et al., 2009). The promising results obtained with IL-12- and GM-CSF-armed OAds encouraged others to generate OAd expressing both molecules simultaneously. Coexpression of both IL-12 and GM-CSF from oncolytic adenovirus Ad-ΔB7/IL12/GMCSF significantly enhanced antitumor efficacy of this virus in an immunocompetent murine melanoma model. Combination regimes of this OAd with DC vaccination promoted DC recruitment, maturation, and migration to the draining lymph nodes, thereby reinforcing the potential of this approach to improve antitumor immunity. (Zhang et al., 2011).
Chemokines are key regulators in the recruitment of immune cells to the tumor site (Franciszkiewicz et al., 2012). Of particular interest is the protein CCL5/RANTES, a key chemokine implicated in the regulation of antitumor immunity (Lapteva and Huang, 2010). OAds expressing CCL5/RANTES significantly enhanced tumor regression in murine tumor models, which correlated with recruitment of DCs, NK cells, and antigen-specific CD8+ T cells (Lapteva et al., 2009).
An OAd has been engineered to express a soluble version of CD40L, a T cell-derived molecule that interacts with the CD40 receptor (CD40R) in antigen presenting cells (APCs), thereby triggering antigen presentation and T cell activation (Elgueta et al., 2009). This virus, called Ad5/3-hTERT-E1A-hCD40L, is a chimeric adenovirus coding for CD40L in the E3 region and its replication is controlled by the tumor-restricted human telomerase reverse transcriptase (hTERT) promoter (Diaconu et al., 2012). Besides showing efficacy and immunomodulatory properties in preclinical settings, Ad5/3-hTERT-E1A-hCD40L has been tested in nine patients with advanced solid tumors (Pesonen et al., 2012). Disease control was observed in majority of the patients, and high levels of virus, CD40L, RANTES, and tumor-specific CD8+ T cells were detected in tumor biopsies. Similarly to APCs, MHC-dependent T cell activation requires co-stimulatory signals present on either APCs or tumor cells that need to be recognized by the CD28 co-stimulatory receptor on the T cell surface (Chen and Flies, 2013). Co-stimulatory CD28 ligands expressed on the APC surface, such as B7-1 and B7-2 (also known as CD80 and CD86, respectively), are, however, not present in tumor cells. In an effort to solve this problem, OAds have been engineered to coexpress B7-1 ligands with immunomodulatory molecules (i.e., IL-12 or GM-CSF). Notably, enhanced survival and antitumor efficacy were observed in B16-F10 immunocompetent tumor models as a result of immune-mediated cell cytotoxicity and CD4+ and CD8+ T cell infiltration at the tumor site (Choi et al., 2006; Lee et al., 2006).
Outlook and Conclusions
Adenovirus vectors are capable of inducing potent cellular, humoral, and mucosal immunity, which makes them attractive candidates for vaccine development. In comparison to traditional vaccines, adenovirus-based vaccines can be armed with different antigens and used to elicit immune response against wide spectrum of pathogens, acting at the same time as an adjuvant for the encoding antigens. Challenges to vaccine development have included the need to identify serotypes that bind to target tissue receptors, and the need to overcome preexisting immunity to common serotypes. Use of rare serotypes circumvents the issue of preexisting immunity to common serotypes and prime-boost regimens of alternative adenovirus vectors have been found to be safe and immunogenic. Adenovirus-based cancer vaccines, either as nonreplicating vectors or as oncolytic adenoviruses, have shown potential in preclinical and clinical settings. Oncolytic adenovirus-based vaccines presenting TAAs or armed with immunostimulatory genes have been shown to induce strong antitumor immunity, and to result in eradication of some primary and metastatic cancers in animal models and in humans, earning further investigation.
There has been substantial progress in the field of adenovirus vector vaccine development, and some adenovirus-based vaccines have been tested extensively in clinical trials in humans. While we still cannot declare complete success, adenovirus vectors have great potential and remain a key platform for vaccine development. Exciting years are ahead in the field, as results from current clinical trials will further enlarge our knowledge and understanding of adenovirus vectors themselves and how to use them to achieve the desired immune response. In addition, key hurdles in manufacturing, formulation, and delivery have largely been solved and allow large-scale deployment of adenovirus-based vaccines and therapies.
Footnotes
Acknowledgments
We would like to acknowledge the FP7-PEOPLE-2011-ITN Marie-Curie Action “Initial Training Networks” for research support: ADenoViruses as novel clinical treatments (ADVance), Project reference: 290002.
Author Disclosure Statement
D.M. and J.C. are employees of Crucell Holland BV, and H.C. is an employee of PsiOxus Therapeutics Ltd. This does not alter the authors' adherence to all Human Gene Therapy policies on sharing data and materials.