
Abstract
Minimal conditioning or even no conditioning would be the preferred preparation for most gene therapy applications for nonmalignant diseases. However, reduced intensity conditioning (RIC) regimens in patients with nonhematologic malignancies have not led to long-term engraftment unless a selective advantage was present for the transplanted donor cells. Similar findings have also been observed in a number of large animal studies. Inadequate myelosuppression levels were thought to be responsible for the outcomes. To address this issue several innovative protocols in small animals have been presented with selective hematopoietic myelosuppression and less systemic toxicity. Such protocols promised to curb the transplant-related morbidity and mortality in myeloablative conditioning and provide effective long-term engraftment, especially in patients with gene-corrected autografts. In the present study we have tested some of these promising RIC regimens in nonhuman primates, a clinically relevant large animal model. Our data suggest that transient myelosuppression induced by anti-c-Kit antibody in conjunction with low-dose irradiation may lead to long-term engraftment, albeit at low levels. The animals with busulfan conditioning with or without anti-c-Kit that received gene-modified autologous transplants with green fluorescent protein expression had similar myelosuppression, but failed long-term engraftment and despite immunosuppressive treatment had all the hallmarks seen previously in similar models without immunosuppression. Our preliminary data expand current knowledge of RIC and emphasize the need to explore whether specific and directed myelosuppression alone is adequate in the absence of microenvironmental modulation, or whether innovative combinations are necessary for safe and effective engraftment.
Introduction
M
For the treatment of nonmalignant hematopoietic disorders myeloablative regimens have been used as well; however, in this setting the only purpose of the conditioning is the facilitation of engraftment. To reduce morbidity and mortality associated with myeloablative conditioning regimens, especially in patients with comorbidities, many investigators have developed and attempted to use reduced intensity conditioning (RIC) for several nonmalignant disorders (sickle cell disease, thalassemia, metabolic disorders) (Iannone et al., 2003; Hsieh et al., 2009; Anurathapan et al., 2013; Hussein et al., 2013). However, in these cases there were overall more instances of graft failures, more graft-versus-host disease (GVHD), and a reduced proportion of disease-free survival in comparison with myeloablative regimens (Bernardo et al., 2012; Tolar et al., 2012; Galambrun et al., 2013; King and Shenoy, 2014). Furthermore, in the autologous setting, where immunologically mediated graft failure and GVHD are theoretically not of concern, it is not clear what level of conditioning is required for successful engraftment of gene-corrected cells; studies so far suggest that RIC is successful only when gene-corrected cells have an advantage over the remaining nonmodified cells. In fact, as a proof of principle, several alternative nonmyeloablative approaches have been explored in preclinical models. Injection of anti-host HLA MHC class I antibody resulted in transient, partial, and reversible reduction in colony-forming unit-spleen (CFU-S) and colony-forming unit-erythroid (CFU-E) cells in bone marrow (BM) (Sadelain et al., 1990). Further insightful studies have predicted and demonstrated improved donor engraftment with low-dose irradiation, or nonmyeloablative chemotherapy (5-fluorouracil [5-FU] or cyclophosphamide [Cytoxan]) when combined with Kit ligand (KL) (Neta et al., 1993; van Os et al., 1997), granulocyte colony-stimulating factor (G-CSF), or interferon alfa-2a induction (Sato et al., 2013). Inhibition of c-Kit and its binding to stem cell factor (SCF), using monoclonal antibodies (ACK2) and multitargeted tyrosine kinase inhibitors (Sunitinib) alone, has also been demonstrated to improve engraftment in immunodeficient mice but only in combination with low-dose irradiation in immunocompetent mice (Fewkes et al., 2010; Xue et al., 2010).
The durable engraftment previously seen in mice with the combined use of anti-c-Kit antibody ACK2 and low-dose irradiation (Xue et al., 2010), and the similarly promising results using SR-1, an antibody against human c-Kit, in a xenotransplantation setting in mice (Czechowicz et al., 2011), provided promising clinical relevance for this treatment. As experiments in mice do not always predict outcomes in humans or larger animals, we decided to extend these observations by exploring some of the previously successful small animal nonmyeloablative modalities in our nonhuman primate immunocompetent model, long considered appropriate for predicting human outcomes in transplantation. Thus, in this model we tested some novel and promising regimens using low-dose irradiation or busulfan in combination with either anti-Kit antibody or a mobilizing agent that works across species. The latter two strategies tested the possibility that more “niches” would be available for the incoming donor cells, as advocated in murine experiments (Chen et al., 2006; Czechowicz et al., 2011).
Our data provide insightful information about engraftment outcomes and suggest avenues to be pursued in future experiments.
Materials and Methods
Animal care and procedures
Healthy juvenile pigtailed macaques (Macaca nemestrina), weighing between 2.5 and 6 kg, were housed at the Washington National Primate Research Center (Seattle, WA) under conditions approved by the Association for Assessment and Accreditation of Laboratory Animal Care International (Frederick, MD). All studies and procedures were approved by the Institutional Review Board and the Animal Care and Use Committees of the Fred Hutchinson Cancer Research Center (Seattle, WA) and University of Washington.
Lentiviral vectors
The human immunodeficiency virus-based lentiviral vectors were self-inactivating pRRL vector backbones containing a central polypurine tract and a woodchuck posttranscriptional regulatory element. The internal promoters were either a short internal elongation factor-1α promoter expressing a P140K variant of methylguanine methyltransferase (P140K-MGMT) for animals 1, 2, and 3 or a spleen focus-forming viral promoter expressing P140K-MGMT and a phosphoglycerate kinase promoter driving the expression of green fluorescent protein (GFP) for animals 4 and 5. The vectors were pseudotyped with the vesicular stomatitis virus G (VSVG) envelope and produced as described previously (Trobridge et al., 2008).
Primate CD34+ cell isolation, lentivirus transduction, and transplantation
CD34+ cell enrichment was performed as previously described (Trobridge et al., 2008). Briefly, BM samples were subjected to red cell lysis in an ammonium chloride-buffered solution, and the resulting white blood cells were stained with a CD34 antibody (clone 12.8, produced in-house) and subsequently with anti-IgM microbeads (Miltenyi Biotec, Auburn, CA). CD34+ cells were enriched by magnetic-activated cell sorting on LS columns (Miltenyi Biotec) with purities ranging from 70 to 90%. After enrichment, cells were cultured for 15–18 hr in either Iscove's modified Dulbecco's medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (Atlas Biologicals, Fort Collins, CO) or StemSpan serum-free expansion medium (SFEM) (StemCell Technologies, Vancouver, BC, Canada). Medium was supplemented with penicillin–streptomycin (100 μg/ml; Life Technologies) and a combination of recombinant human interleukin (rhIL)-3, rhIL-6, thrombopoietin (rhTPO) (all from PeproTech, Rocky Hill, NJ), and Flt3 ligand (rhFlt3 ligand; Miltenyi Biotec), granulocyte colony-stimulating factor (rhG-CSF), and stem cell factor (rhSCF) growth factors (Amgen, Thousand Oaks, CA), each at a concentration of 100 ng/ml. Cells were then transduced with lentiviral vectors in the presence of protamine sulfate (4 μg/ml) and cyclosporine (1 μg/ml) in flasks precoated with the CH296 fragment of RetroNectin at a multiplicity of infection of 10 for 6–8 hr followed by a second virus addition for 15–18 hr overnight. Transduced cells were harvested and the infusion product was either cryopreserved for 5–10 days or infused as a fresh product into nonmyeloablatively conditioned animals.
Conditioning regimen
Macaques 1, 2, and 3 were treated with nonmyeloablative doses of total body irradiation (TBI) at 300 cGy 7 days before cell infusion. In addition to TBI, macaque 1 received two administrations of the anti-c-Kit antibody SR-1 at doses of 50 mg on days 12 and 9 before cell infusion. Macaque 2 received a single bolus dose of AMD3100 at 4 mg/kg 6 hr before cell infusion. Macaques 4 and 5 both received nonmyeloablative conditioning with busulfan (Sigma-Aldrich, St. Louis, MO) at doses of 8 mg/kg administered 1 and 2 days before cell infusion. Macaque 5 additionally received two doses of the anti-c-Kit antibody SR-1 at doses of 50 mg 10 and 7 days before cell infusion. All monkeys receiving the anti-c-Kit antibody were pretreated with steroids to prevent reactivity against a foreign protein.
Antibody purification
For in vivo studies, Harlan Bioproducts for Science (Indianapolis, IN) purified the mouse monoclonal anti-c-Kit antibody (SR-1; Broudy et al., 1992) and tested endotoxin levels, which were <0.2 endotoxin units (EU)/mg. Some in vitro studies used anti-c-Kit antibody affinity purified by the University of Nebraska Monoclonal Antibody Core Laboratory (Omaha, NE).
Colony-forming unit assay
Transduced, mock-transduced, and BM white blood cell samples from transplanted macaques were cultured in semisolid methylcellulose medium for 12–14 days in the presence of rhIL-3, recombinant human erythropoietin (rhEPO), rhSCF, and recombinant human granulocyte-macrophage colony-stimulating factor (rhGM-CSF) (100 ng/ml each; ReachBio, Seattle, WA). Colonies were enumerated and scored on the basis of morphology. Gene marking was assessed in colonies by PCR analysis for lentiviral integration. Individual colonies from CFU-C (colony-forming unit in culture) assays for each macaque were picked and transferred into 90 μl of water supplemented with 1.7 U of proteinase K from Engyodontium album (formerly Tritirachium album; Sigma-Aldrich). DNA was isolated from individual colonies as described previously (Beard et al., 2006) and analyzed by PCR to determine the percentage of colonies positive for lentiviral integration.
Gene-marking assessment
Heparinized peripheral blood (PB) and BM collected at various time points after transplantation were subjected to red cell lysis with ammonium chloride-buffered solution. The resulting leukocytes were either prepared for DNA extraction and then analyzed for lentiviral integration by qPCR or subjected to flow cytometric analysis for GFP expression on a BD FACSCanto I flow cytometer (BD Biosciences, San Jose, CA). GFP expression was determined in granulocytes and lymphocytes on the basis of gating in forward and right angle light scatter. Lineage-specific markers for granulocytes (CD13, clone L138), lymphocytes (CD3, clone SP34-2 and CD20, clone L27), monocytes (CD14, clone M5E2), and stem cells (CD34, clone 563) were also used. All antibodies were purchased from BD Biosciences.
Quantitative PCR analysis
Gene marking in leukocytes isolated from peripheral blood and bone marrow of transplanted macaques was analyzed by TaqMan 5′-nuclease quantitative real-time PCR assay as described previously (Beard et al., 2010). Sample DNA was analyzed in duplicate with a lentivirus-specific primer–probe combination (forward, 5′-TGAAAGCGAAAGGGAAACCA; reverse, 5′-CCGTGCGCGCTTCAG; probe, 5′-AGCTCTCTCGACGCAGGACTCGGC [Integrated DNA Technologies, Coralville, IA]) and in a separate reaction with a β-globin-specific primer–probe combination (forward, 5′-CCTATCAGAAAGTGGTGGCTGG; reverse, 5′-TTGGACAGCAAGAAAGTGAGCTT; probe, 5′-TGGCTAATGCCCTGGCCCACAAGTA [Integrated DNA Technologies]) to adjust for equal loading volume of genomic DNA per reaction.
Results
Similar staining patterns of anti-c-Kit antibody with human and nonhuman primate CD34+ cells
We have previously shown immunophenotypic and functional data with anti-c-Kit, using human cells (Broudy et al., 1992). In the present study, we first tested the expression of c-Kit (CD117) in nonhuman primate (NHP) BM samples, using the anti-c-Kit antibody (SR-1). Although the proportion of cells labeled with anti-c-Kit (SR-1) among CD34-enriched cells from macaque was low, it was significantly increased after culturing with cytokines (IL-3, SCF, EPO) (Fig. 1A, left). A similar upregulation of c-Kit expression is seen in human CD34+ cells after culture under the same conditions (Fig. 1A, right). Prior studies have also concluded that this antibody inhibits c-Kit signaling and the clonogenicity of human cells in vitro (Broudy et al., 1992) and in vivo in a xenogeneic model (Czechowicz et al., 2011). To test the functional significance of anti-c-Kit antibody (SR-1) in NHP cells we cultured BM cells at various concentrations of anti-c-Kit antibody in comparison with similarly treated human cells. As seen in Fig. 1B and Supplementary Table S1 (supplementary data are available online at

c-Kit expression by monkey or human bone marrow (BM) cells.
Effect of low-dose TBI±additive treatments on engraftment of gene-modified cells
Previous data have documented that low-dose irradiation (200–300 cGy) given alone was not sufficient for engraftment in immunocompetent mice (Xue et al., 2010). In addition, low engraftment was seen long term in rhesus monkeys with 500 cGy (Huhn et al., 1999) or with 320–400 cGy (Rosenzweig et al., 1999), using in vitro gene-modified autologous cells. Therefore in monkeys transplanted with autologous lentivirus-transduced BM CD34+ cells we administered low-dose irradiation (300 cGy) in combination with either AMD3100 a few hours before infusion, or the anti-c-Kit antibody SR-1. Both of the latter treatments were aimed at freeing more stem cell “niches” either through mobilization (Chen et al., 2006) or through elimination of stem cell occupancy, respectively (Czechowicz et al., 2011). The mobilization effectiveness of AMD3100 in primates was previously shown by our group (Bonig et al., 2009). The timeline of conditioning protocols and the PB recovery kinetics for these three animals are depicted in Fig. 2A and B.
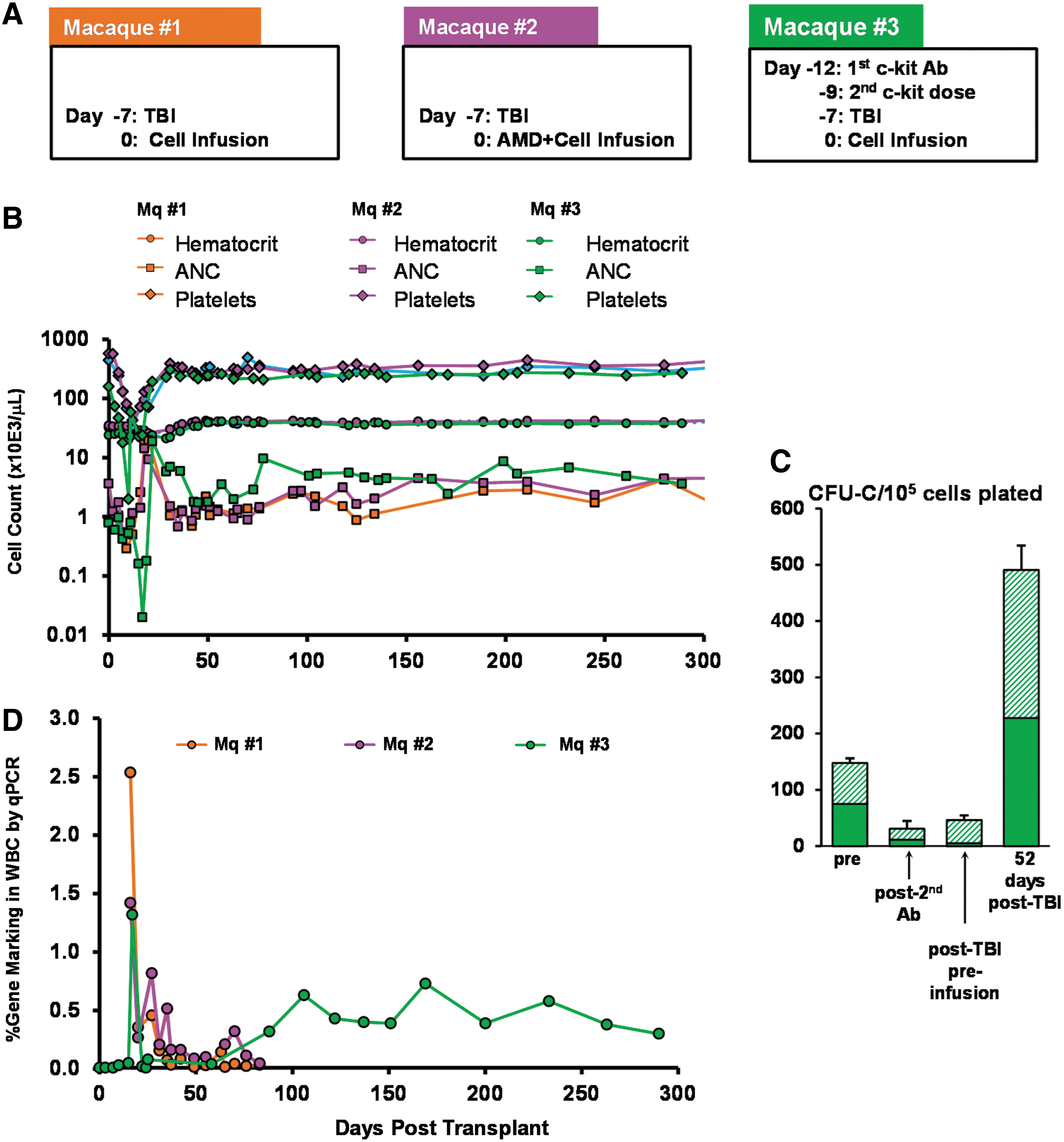
Engraftment and gene-marking levels in animals conditioned with 300 cGy total body irradiation (TBI)±AMD3100 and treatment with anti-c-Kit antibody, SR-1.
Animals treated with 300 cGy±AMD3100 (macaques 1 and 2) recovered uneventfully without critical hematocrit or platelet reduction and with moderately severe neutropenia. Days with absolute neutrophil counts (ANCs) <500 were 16 and 12, respectively. In contrast, the animal treated with 300 cGy and anti-c-Kit antibody (macaque 3) showed more severe cytopenias (19 days with ANC <500) requiring two blood transfusions at 7 and 11 days posttransplantation, G-CSF treatments, and with more delay in reaching pretreatment levels. Thereafter the animal recovered completely in the next 2–3 weeks. Bone marrow analyses by clonogenic assays were carried out in macaque 3 before any treatment, after the second anti-c-Kit infusion, and after TBI before cell infusion (Fig. 2C). A significant reduction of ∼85% was seen only in erythroid colonies after the second anti-c-Kit antibody infusion and an additional ∼50% reduction of nonerythroid colonies was seen post-TBI and preinfusion, compared with baseline values. These data were fairly consistent with the in vitro data presented in Fig. 1 and Supplementary Table S1. A quick rebound at 52 days postinfusion ensued (Fig. 2C).
To test whether recovery was occurring with genetically modified cells, proviral marking was assessed by qPCR in PB leukocytes at regular time points posttransplantation. Apart from a single spike of positivity in the two monkeys with 300 cGy±AMD3100, a progressive and relatively stable gene marking in PB was present only in the animal with 300 cGy plus anti-c-Kit antibody exceeding 300 days (Fig. 2D). Of note, while the cell dose for infusion in macaques 1 and 2 (300 cGy±AMD3100) was ∼14 million cells/kg, the cell dose for macaque 3 (300 cGy plus anti-c-Kit antibody) was only ∼6.1 million cells/kg. Levels of gene modification in the infusion products were similar (26–37%) as determined by percentage of lentivirus-positive colonies in CFC assays. Therefore anti-c-Kit treatment combined with low-dose irradiation yielded a modest but persistent engraftment of gene-modified cells not present in the other two animals.
Effect of busulfan±anti-c-Kit antibody treatment on engraftment of gene-modified cells
Prior efforts in our laboratory to use nonmyeloablative doses of busulfan (4 mg/kg ×2) for autologous transplants yielded low engraftment (Beard et al., 2010). Therefore, we doubled the busulfan level within a nonmyeloablative range to encourage engraftment in macaque 4. Furthermore, we combined this busulfan treatment with anti-c-Kit antibody in macaque 5. Gene marking in the infusion product was 44 and 45%, respectively, as determined by flow cytometric analysis for GFP expression.
The timeline for busulfan and antibody administration for these two animals along with their PB recovery kinetics are shown in Fig. 3A and B. BM studies in macaque 5 before and after busulfan plus anti-c-Kit antibody treatment, and before cell infusion, showed >90% suppression of clonogenic progenitors of all types (BM before: 66 BFU-E±2.0 and 400 nonerythroid colonies per 100,000 cells plated; BM after: 7±0.9 BFU-E and 16±3.0 nonerythroid colonies per 100,000 cells plated).
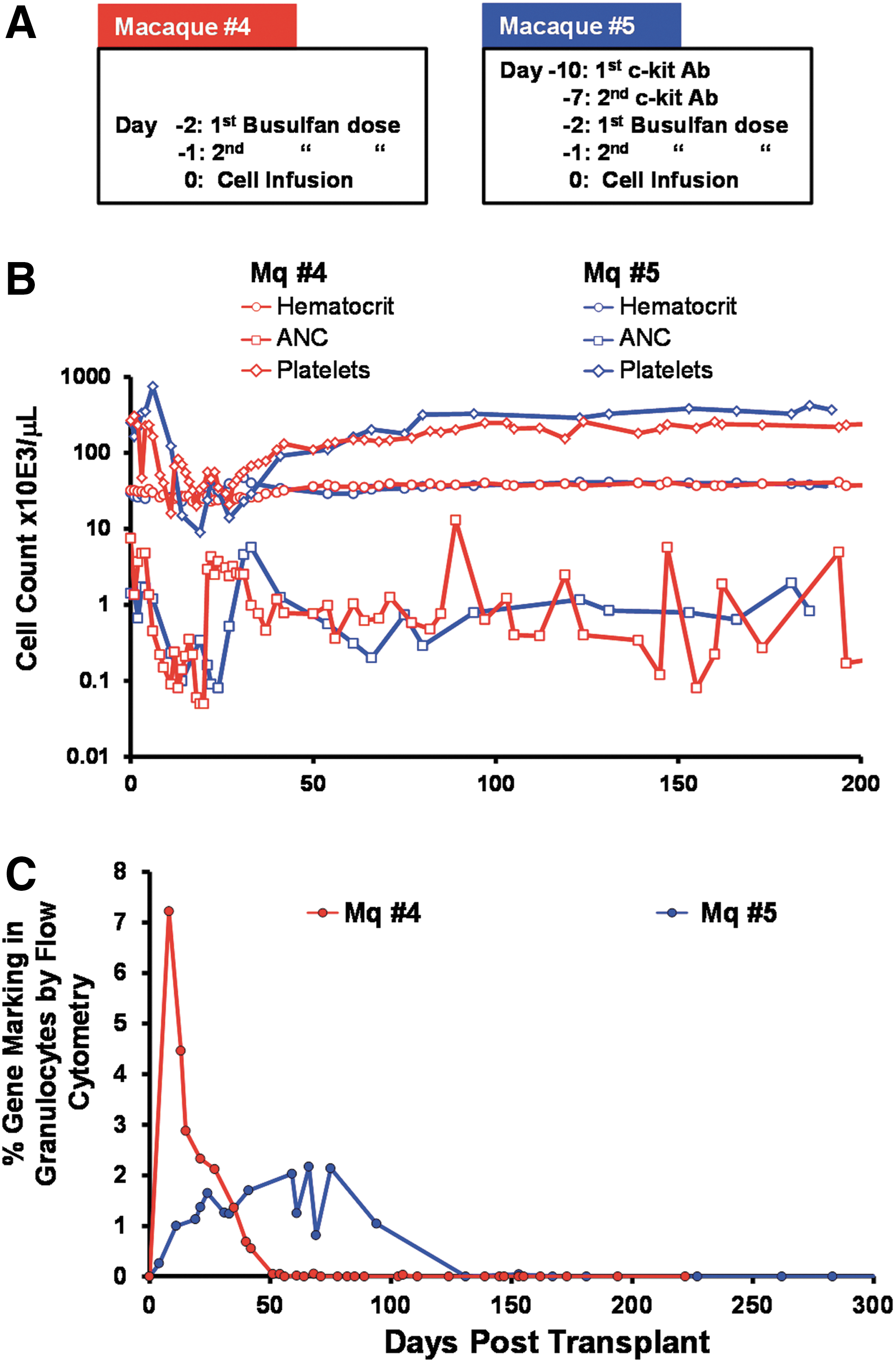
Engraftment and gene-marking levels in animals conditioned with busulfan±anti-c-Kit antibody (SR-1).
Because we used GFP as a marker for gene modification in these animals, we included immunosuppressive therapy after infusion of cells with tacrolimus at doses of 1.5 mg/kg twice a day to maintain levels between 5 and 15 ng/ml in PB for 6.5 months posttransplantation. The animal with the combined treatment (macaque 5) required significant support including three transfusions of whole irradiated blood at 17, 18, and 26 days posttransplantation and G-CSF treatments until 27 days posttransplantation. ANC levels were <500 for 21 days in the busulfan-conditioned and 27 days in the busulfan plus anti-c-Kit antibody-conditioned animals, respectively. In macaque 5, a brisk recovery ensued after the third week of cell infusion. However, the platelet and polymorphonuclear leukocyte (PMN) recovery were protracted compared with macaque 4.
Gene-marking levels were monitored closely after transplantation once a week in the PB (Fig. 3) and at 1, 3, and 6–8 months posttransplantation in the BM. GFP+ gene-modified cells were detected in multiple lineages in the PB (Supplementary Table S2) and BM (Fig. 4 and data not shown) at 1 month posttransplantation in both animals. Whereas the marking level within BM CD34+ cells was 3.5% in macaque 4 (busulfan alone), the levels in macaque 5 were 17% (busulfan plus anti-c-Kit antibody; Fig. 4B). By 5 months posttransplantation, however, these levels decreased to 0.2 and 0.8%, respectively. At 5.5 months posttransplantation a course of G-CSF was administered for 5 days to test whether any dormant and undeclared donor cells in BM could be awakened, expanded, and mobilized. Furthermore, such a treatment provokes redistribution of all engrafted donor clones and safeguards against sampling errors in BM samples (Verovskaya et al., 2014). Levels of GFP+ cells in the PB and BM assessed after treatment showed that although mobilization of CD34+ cells into the PB was successful, gene-marking levels remained the same posttreatment (Fig. 5). Nevertheless, CFU-C assays performed from BM at 6–8 months posttransplantation revealed only ∼3% lentivirus-positive colonies in both animals (data not shown).

Engraftment of gene-modified CD34+ cells in the bone marrow (BM) of monkeys 4 and 5 four weeks after transplantation. Flow plots depict the percentage CD34+ cells in total bone marrow cells and the proportion of these cells that are positive for GFP in macaques conditioned with busulfan alone
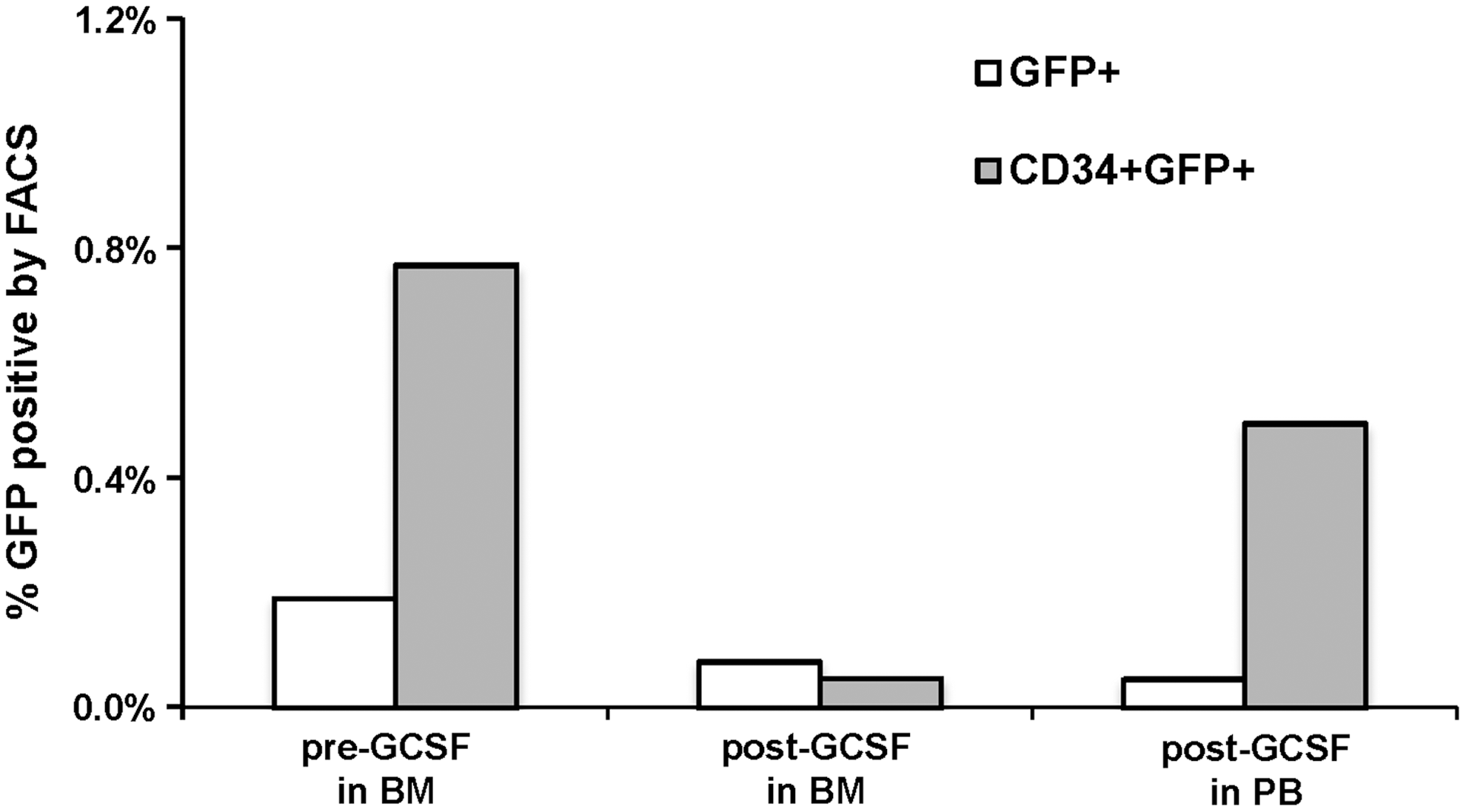
Effect of G-CSF-mediated mobilization of gene-modified CD34+ cells from bone marrow to peripheral blood. The graph represents the percentage of GFP+ cells within the peripheral blood and bone marrow cells (open columns) and CD34+ cells (shaded columns) before and after a 5-day G-CSF treatment regimen at ∼5 months posttransplantation in macaque 5. Significant mobilization is seen, but no change is seen in marking among CD34+ cells.
Proviral integration in PB leukocytes, measured by qPCR analysis, was detected only up to 50 days posttransplantation in macaque 4 (busulfan alone), similar to GFP expression, but was undetected throughout for macaque 5 (busulfan plus anti-c-Kit antibody; data not shown).
Discussion
Although reduced intensity conditioning (RIC) regimens have been hailed as appropriate and acceptable for nonfatal hematologic diseases and gene therapy, many attempts to use these conditioning regimens have not been encouraging. Sustained engraftment in larger animals and in clinical gene therapy studies using RIC has been disappointing unless the gene-corrected cells had a selective advantage (Aiuti et al., 2002; Bauer et al., 2008; Hsieh et al., 2009; Andrade et al., 2011; Uchida et al., 2014). Given the success of full myeloablative conditioning the results have been attributed to insufficient myeloablation of host cells. To enhance the level of myeloablation without the accompanying undesirable systemic effects, chemo/irradiation-reduced regimens have been combined with myelospecific treatments (Fewkes et al., 2010) or interferon induction (Sato et al., 2013) to reduce host stem cells, and promising results using these strategies have been documented in mice. In the present study we attempted to incorporate similar strategies in an autologous hematopoietic stem cell transplantation (HSCT) setting in the nonhuman primate model using pigtailed macaques.
In comparing myelosuppression in animals conditioned with low-dose TBI and busulfan, it is seen that low-dose TBI results in higher nadirs of ANC in comparison with busulfan unless combined with the anti-c-Kit antibody (SR-1). Likewise, recovery in busulfan-treated animals was much slower than in low-dose TBI-treated animals. In comparison with myeloablative conditioning with 1020 cGy (Supplementary Fig. S1), the busulfan animals had higher nadirs (20 vs. 70 cells/μl, respectively) but took a longer time to reach pretreatment levels. Although the ANC never went below 100 for low-dose TBI-treated animals without c-Kit antibody treatment, the days on which the ANC was less than 100 in animals treated with TBI plus anti-c-Kit, busulfan alone, and busulfan plus anti-c-Kit was 17, 12, and 24, respectively.
Therefore, under both low-dose TBI and busulfan background, the anti-c-Kit treatment was responsible for enhancing the myelosupppressive effect. These differences in myelosuppression were combined with significant differences in engraftment of gene-modified autologous cells compared with non–antibody-treated controls or to prior data using low-dose TBI levels (500 cGy) as a monotherapy in the primate model (Huhn et al., 1999). Thus our data do establish the principle that transient and reversible c-Kit inhibition can enhance myelosuppression and engraftment of ex vivo genetically modified cells in the primate model.
Engraftment of gene-modified cells in the animal with 300 cGy plus anti-c-Kit antibody was stable for more than 300 days posttransplantation. Although these data are encouraging, the levels of engraftment were lower than an average of ∼20% gene-marking levels expected with full myeloablation with freshly prepared infusion products (Beard et al., 2010). Nevertheless, despite the low-level engraftment, in cases where the endogenous cells are at a competitive disadvantage compared with gene-corrected cells, progressive engraftment of these cells can prevail. This is illustrated by the successful treatment of dogs with canine leukocyte adhesion deficiency (CLAD) receiving only 200 cGy and overnight transduced cells with sustained, low-level, but long-term engraftment (Bauer et al., 2008), and it may reflect scenarios expected with thalassemia recipients given autologous gene-corrected cells.
The pattern of engraftment in the busulfan-treated animals was different. GFP was used as a marker of gene-modified cells to track engraftment in a more sensitive way in multiple lineages over time. Because GFP can induce immune responses against gene-modified cells (Lutzko et al., 1999; Rosenzweig et al., 2001) and thus cause their elimination, these animals were given immunosuppressive treatment with tacrolimus. GFP+ cells were detected at a low level of in various lineages early after transplantation, with the highest levels seen in CD34+ cells. However, both GFP and gene-marking data in PB PMNs were short-lived in both busulfan-treated animals.
The findings in these two animals are of interest and in need of interpretation. It is unclear whether we are dealing with only short-term repopulation, or ultimate silencing/elimination of any long-term repopulating cells infused. We can only speculate about the putative reasons leading to loss of persisting engraftment in these animals. It is possible that despite the immunosuppressive treatment given, some anti-GFP immunoreactivity was present (not tested). What is intriguing and rather supportive of this hypothesis is the fact that our flow cytometric data are similar to data provided by previously described animals given GFP-marked cells but no immunosuppression (Lutzko et al., 1999; Rosenzweig et al., 2001), that is, higher levels were detected in CD34+ cells than in any other maturing cells and maintained longer in CFU-Cs tested from BM. It is possible that CD34+ cells were maintained or eliminated last, because of lower levels of GFP expression than in maturing cells, similar to low levels of vector DNA persisting in CFU-Cs even after loss of transduced cells in PB as shown in previous studies (Lutzko et al., 1999; Rosenzweig et al., 2001). Although immunosuppressive treatment with tacrolimus was maintained within therapeutic levels (i.e., between 5 and 15 ng/ml), its effectiveness in preventing GFP immunoreactivity in a nonmyeloablative transplant setting has yet to be validated.
Several other theoretical possibilities can be cited, although we consider them less likely. It is possible that the anti-c-Kit antibody did not influence the levels of many primitive HSCs in the macaque BM. If repopulating HSCs have low levels of c-Kit expression in quiescent versus proliferating cells (Ogawa et al., 1991; Gunji et al., 1993; Shin et al., 2014) and the antibody dose given was not sufficient to have a significant impact on these cells, engraftment cannot be sustained long term. In this context data have shown that c-Kitlo HSCs are more primitive than c-Kithi HSCs and lead to long-standing, durable engraftment levels in serial transplantation assays compared with c-Kithi HSCs (Shin et al., 2014). Thus, according to this scenario, the blocking antibody treatment in this animal likely suppressed c-Kithi HSCs more than the c-Kitlo long-term HSCs (Gunji et al., 1993). Experiments in mice in which only repeated doses of anti-c-Kit antibody (ACK2) could influence stem cells, not just progenitor cells (Ogawa et al., 1991; Gunji et al., 1993), could support this argument. Although further studies are needed to confirm the functional differences between c-Kitlo and c-Kithi cells, especially in the setting of different conditioning regimens, this reasoning should have equally affected macaque 3 in our study with the stable engraftment (Fig. 2).
A consideration unrelated to c-Kit expression in stem cells that may have impacted engraftment is the delay in cell infusion after antibody treatment necessary for antibody clearance. This delay may have allowed the recovery of many more host cells, reducing the myeloablating impact of antibody treatment. Nevertheless, busulfan was given 2 days before cell infusion and may have counteracted this recovery. It is also likely that the remaining endogenous cells had a competitive advantage over ex vivo-cultured cells (either GFP+ or GFP−). Numerous previous studies have shown the functional impairment of cultured cells compared with fresh cells (especially more than 1–2 days) in terms of homing, early proliferative expansion, and long-term engraftment (Szilvassy et al., 2001; Yong et al., 2002; Liu et al., 2003; Ahmed et al., 2004; Mazurier et al., 2004). However, despite the above-cited theoretical arguments, we think it is unlikely that homing per se is critical here, as it can be predicted that homing in a nonmyeloablative setting is not worse than in fully myeloablated recipients. Numerous prior studies have shown that homing in unconditioned recipients is better than in fully conditioned recipients (Hendrikx et al., 1996; Jiang et al., 2009), and it is the engraftment potential due to the altered microenvironment that is different between the two settings. It is also unlikely that the infusion cell doses were inadequate, as similar numbers were sufficient in fully ablated animals (although the true number of repopulating cells in each infusion product is unknown).
Taken together, our data show that the effectiveness of any RIC regimen should be tested in large animal models before any clinical application. Furthermore, it will be important to determine in future experiments whether host myelosuppression alone (through antibody treatments or other specific approaches) at any level is sufficient to ensure high and durable engraftment. In fact, the requirement of “niche” availability has been challenged when selective ablation of stem cells failed to achieve engraftment (Gerbaulet et al., 2013). Although homing and BM lodgment of infused HSCs can be achieved irrespective of host HSC ablation, the microenvironmental conditions necessary for their initial expansion in BM may not be present (Andrade et al., 2011). These concepts should be tested in future experiments in which specific myelosuppression (antibodies or otherwise) protocols are used alone or in creative combinations with microenvironment-modulating parameters to enhance engraftment. In addition, parallel approaches to selectively enhance gene-modified cell engraftment (prostaglandin E2 [Porter et al., 2011, 2013], rapamycin [Rohrabaugh et al., 2011], or dipeptidylpeptidase-4 inhibition ex vivo [Broxmeyer et al., 2012]) or the use of a cell-selectable agent for their in vivo expansion (Beard et al., 2010) should also be explored, if they are reproducibly and consistently effective.
Footnotes
Acknowledgments
The authors thank the staff at the University of Washington National Primate Center (Seattle, WA) and thank Veronica Nelson, Erica Wilson and Kelvin Sze for providing technical assistance. The authors also thank Grace Choi for help with preparing the manuscript and Tatiana Ulyanova for assistance with FACS. H.-P. Kiem is a Markey Molecular Medicine Investigator and the recipient of the José Carreras/E.D. Thomas Endowed Chair for Cancer Research. This work was supported in part by the National Institutes of Health (Bethesda, MD) grants HL084345, HL053750, and DK056465.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.