
Abstract
Despite nearly three decades of research, a safe and effective vaccine against human immunodeficiency virus type 1 (HIV-1) has yet to be achieved. More recently, the discovery of highly potent anti-gp160 broadly neutralizing antibodies (bNAbs) has garnered renewed interest in using antibody-based prophylactic and therapeutic approaches. Here, we encoded bNAbs in first-generation adenoviral (ADV) vectors, which have the distinctive features of a large coding capacity and ease of propagation. A single intramuscular injection of ADV-vectorized bNAbs in humanized mice generated high serum levels of bNAbs that provided protection against multiple repeated challenges with a high dose of HIV-1, prevented depletion of peripheral CD4+ T cells, and reduced plasma viral loads to below detection limits. Our results suggest that ADV vectors may be a viable option for the prophylactic and perhaps therapeutic use of bNAbs in humans.
Introduction
S
Highly potent human monoclonal antibodies that recognize HIV envelope component gp120 or gp41 of a broad range of virus clades have been identified. These broadly neutralizing antibodies (bNAbs) have a number of unusual characteristics, including a high degree of somatic mutation, extended CDR H3 regions, and poly- and autoreactivity, making them difficult to elicit by immunization or natural infection. 5 –7 As an alternative approach, delivery of bNAbs by viral vectors has been investigated, as this approach may require less frequent dosing compared with passive immunization schemes for inducing high levels of transgene expression and sustained production of bNAbs in vivo. Previously, antibody-like immunoadhesins were encoded on adeno-associated viral (AAV) vectors for sustained gene production and long-term protection against simian immunodeficiency virus (SIV) challenge in rhesus macaques 8 ; more recently, AAV has been used to encode both the heavy and light chains of full-length bNAbs, and intramuscular administration of AAV-vectorized bNAbs afforded protection against intravenous as well as mucosal challenges with HIV-1 in humanized mice. 9,10 AAV, however, has some limitations including a small carrying capacity of 4 kb and difficulties in virus propagation and purification. 11 –13
In the present study, we sought to vector bNAbs in a first-generation adenoviral serotype 5 (FG ADV5) vector. Compared with AAV, FG ADV vectors have an increased carrying capacity of 5–8 kb and are relatively easy to genetically manipulate, propagate, and purify to high-titer stocks. 14,15 Because of their favorable safety profile, ADV-based vectors are among the most frequently used vehicles for clinical gene delivery to date, making up almost one-quarter of all gene therapy trials targeting cardiovascular disease, cystic fibrosis, and cancer, among others. 16 –18
Here, the bNAbs VRC03 and PG16 were separately encoded in an FG ADV5 vector and evaluated for their ability to prevent HIV acquisition in humanized mice. VRC03 was isolated from a panel of broadly neutralizing antisera, using envelope antigen probes, and it targets the CD4-binding site of gp120. 19 PG16 was identified in secreted culture supernatants of memory B cells from a clade A HIV-1-infected subject and binds to quaternary epitopes on the V1V2 loops of gp120. 20 We show here that a single intramuscular injection of these FG ADV5 viral particles (VPs) produced high serum titers of bNAb in Hu-PBL humanized mice 21 and protected against HIV-1 infection, despite multiple repeated intraperitoneal challenge with HIV-1.
Materials and Methods
Cells, plasmids, and antibodies
293T, HEK 293, and TZMBL cells were originally obtained from the American Type Culture Collection (ATCC, Manassas, VA). Unless otherwise specified, all cells were maintained in complete Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Grand Island, NY) supplemented with 1% penicillin–streptomycin (Life Technologies), 1% amphotericin B (Sigma-Aldrich, St. Louis, MO), and 10% heat-inactivated fetal bovine serum (FBS; Life Technologies) in a 37°C humidified 5% CO2 incubator. VRC03- and PG16-encoding plasmids were kind gifts of J. Mascola (Vaccine Research Center, NIAID, NIH, Bethesda, MD) and H. Gottlinger (Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA), respectively. Monoclonal bNAbs and infectious molecular clones of HIV were obtained through the AIDS Repository and Reagent Program.
Construction of FG ADV5 vectors
FG ADV5 vectors were produced with the pAdeno-X expression system (Clontech, Mountain View, CA) according to the manufacturer's instructions. Using standard cloning techniques, transgene-encoding expression cassettes were constructed and ligated into the pShuttle2 vector, using the restriction endonuclease sites NotI and AflII. pShuttle-encoded expression cassettes were used to generate the final ADV5 constructs, using the PI-SceI and I-CeuI homing mega-endonuclease sites. All plasmids were purified with plasmid purification kits (Qiagen, Valencia, CA) according to the manufacturer's instructions. PCR was performed with Takara PrimeSTAR DNA (Clontech), using the manufacturer's listed thermocycling conditions, and the resulting product was gel-purified with a Qiagen gel extraction kit. All restriction endonuclease digestions were performed with NEB restriction endo- and meganucleases, according to the specified temperatures and conditions. Ligations were performed at 16°C overnight and transformations were performed with the STBL3 Escherichia coli strain.
Before making ADV VPs, bNAb expression by the individual pShuttle HC and LC plasmids as well as recombinant ADV expression plasmids was first confirmed by transient transfection of 293T cells, using Lipofectamine 2000 (Life Technologies).
FG ADV VP production and purification
The initial production of ADV and subsequent harvest and high-titer amplifications and purifications were performed as previously described 22 with the following changes: twenty-five 15-cm plates of HEK 293 cells were infected, harvested by low-speed centrifugation, and lysed. Only a single round of step-gradient ultracentrifugation was performed, using an SW41 rotor for 1 hr at 35,000 rpm at 16°C. After dialysis, purified ADV VPs were syringe-filtered through a 0.45-μm polyvinylidene difluoride (PVDF) membrane before the addition of glycerol to a final concentration of 20% for long-term cryostorage. To confirm that no genomic rearrangements had occurred during ADV amplification, adenoviral genomic DNA was extracted from purified VPs and subjected to restriction enzyme digestion as described. 22
In vitro transduction with FG ADV VPs
Purified ADV VPs were used to transduce 293T cells at a multiplicity of infection (MOI) of 10. Supernatants (SNs) were harvested 48 hr after transduction, syringe-filtered through a 0.45-μm filter to remove culture debris, and stored at −20°C.
Quantification of bNAb production by Western blot
Culture SNs or mouse sera diluted in phosphate-buffered saline (PBS) were mixed 1:1 with Laemmli sample buffer (Bio-Rad, Hercules, CA), supplemented with 2-mercaptoethanol, and heated at 95°C for 5 min. Samples were loaded onto 10% Tris-HCl gels (Bio-Rad) and run for 1 hr at 120 V. Gels were transferred onto PVDF membranes for 1 hr at 100 V. Membranes were blocked in 5% milk made in 0.05% PBS–Tween (PBS-T), washed three times with 0.05% PBS-T, immunoblotted with secondary goat anti-human whole IgG conjugated with horseradish peroxidase (HRP; Sigma-Aldrich), washed three times, allowed to react with HyGLO quick-spray chemiluminescent substrate (Denville Scientific, Metuchen, NJ), and developed with autoradiographic film.
Quantification of bNAb production by sandwich ELISA
Ninety-six-well polystyrene plates (Corning, Corning, NY) were coated overnight at 4°C with goat-anti-human Fc antibody (Bethyl Laboratories, Montgomery, TX) at 100 ng/well, diluted in PBS. The next day plates were washed five times with 0.05% PBS-T, blocked at room temperature for 1 hr with 5% milk and 2% bovine serum albumin (BSA) in PBS, and washed five times. Two-fold serial dilutions of standards, PBS-diluted mouse sera, or bNAb-containing supernatants were added for 1 hr at room temperature. Plates were washed five times and goat anti-human lambda (for detecting PG16) or kappa (for detecting VRC03) HRP-conjugated antibody (Bethyl Laboratories) was added for 1 hr at room temperature. After five washes, tetramethylbenzidine (TMB) substrate (KPL, Gaithersburg, MD) was added for 10 min and the reaction was stopped by the addition of 1 M H3PO4 before reading the absorbance at 450 nm.
Env-pseudotype HIV neutralization assays
Two-fold serial dilutions of bNAb-containing supernatants, mouse sera diluted in complete media, or purified recombinant bNAbs were added to 96-well plates in duplicate. One hundred microliters of high-titer Env-pseudotyped HIV stocks (generally resulting in a viral input of 50,000–100,000 relative light units [RLUs] in the absence of bNAb) or infectious HIV-1 at an MOI of 1 was added per well and the plates were incubated at 37°C for 30 min. After incubation, 10,000 TZMBL cells were added to each well and the plates were incubated for 48 hr. Luciferase assays were then performed as described 23 with the following changes: culture medium was aspirated and wells were washed twice with PBS. For each well, 100 μl of Tropix lysis solution (Life Technologies) supplemented with 1 mM dithiothreitol (DTT) was added and incubated for 30 min at room temperature. Ninety microliters of each lysate was transferred to 96-well flat-bottom optical white plates (Corning). Fifty microliters of luciferase assay buffer and 15 μl of luciferin substrate were added for luminometer reading. Neutralization data were plotted in GraphPad Prism (GraphPad Software, San Diego, CA) and 50% inhibitory concentration (IC50) values were determined with the nonlinear regression curve-fitting feature.
HIV-1 production and titering
For the production of Env-pseudotyped HIV, 100 μg each of Env-deficient HIV-1 backbone and Env were used to transfect 15-cm plates of 293T cells seeded at 50–75% confluence, using standard calcium phosphate coprecipitation methods. Sixty to 72 hr after transfection, SNs were harvested and syringe-filtered through a 0.45-μm filter before being aliquoted and stored at –80°C. Infectious HIV was generated and titered as described 24 with the following modifications: 10-cm dishes were plated with 2×106 293T cells the day before transfection. Twenty-five micrograms of cDNA encoding an infectious molecular clone of HIV-1 was used for transfection. Culture medium was replaced 4 hr posttransfection. To determine the titer of infectious HIV, a β-galactosidase assay was performed on TZMBL cells to calculate the 50% tissue culture infective dose (TCID50) expressed as focus-forming units (FFUs).
Animals
NSG mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred and maintained at Yale University (New Haven, CT) according to guidelines established by the Institutional Animal Committee. All experiments were performed according to protocols approved by the Institutional Review Board and the Institutional Animal Care and Use Committee of Yale University.
NSG-Hu-PBL mice were engrafted as described. 21 Briefly, 5–10×106 peripheral blood mononuclear cells (PBMCs), purified by Ficoll density gradient centrifugation of healthy donor blood (obtained from the New York Blood Bank) were injected intraperitoneally in a 100-μl volume into 5- to 12-week-old NSG mice (Jackson Laboratory), using a 1-cm3 syringe and 25-gauge needle. Cell engraftment was tested 10 days postinjection as described. 21,25 Briefly, mice were anesthetized with an isoflurane chamber, and 50–100 μl of whole blood was collected by retro-orbital bleeding. Mononuclear cells were isolated by Ficoll density gradient centrifugation and staining with Fluor-conjugated antibodies of the appropriate cell markers. Labeled cells were analyzed by flow cytometric analysis. For the analysis of NSG-Hu-PBL engrafted mice, positive engraftment was first determined by the presence of human CD45+ cells, constituting the population of human leukocytes. CD45+ cells were further analyzed for CD3 expression, marking all human T cells. The CD45+CD3+ cell population was further analyzed for the presence of CD4+ and CD8+ T cells, determining the population of helper and cytotoxic T cells, respectively.
ADV injections and HIV challenge
Aliquots of ADV were thawed on ice and diluted in PBS to achieve the predetermined dose in 100 μl. Mice were anesthetized with isoflurane and 50 μl of vector was administered intramuscularly in each hind leg for single bNAb injections. For virus challenge experiments, mice were challenged intraperitoneally with HIV-1 as detailed elsewhere in text. In experiments involving multiple challenges with HIV, blood samples for assessing CD4+ T cell levels and plasma viral RNA loads were acquired from the mice on the day of challenge before virus challenge.
Quantification of mouse-generated bNAbs
Mice were retro-orbitally bled weekly and blood was allowed to clot at room temperature for 3 hr. Samples were spun at maximal speed in a tabletop microfuge for 2 min and serum was frozen in small aliquots at −80°C. For Western blots (WBs), 1 μl of NSG serum was diluted in PBS and mixed 1:1 with Laemmli sample buffer containing 2-mercaptoethanol and analyzed as described previously. For ELISAs, 1 μl of NSG mouse serum was diluted in 100 μl of blocking buffer for 2-fold serial dilutions and then analyzed as described previously. For TZMBL Env-pseudotyped HIV and infectious HIV neutralization luciferase assays, 2–5 μl of serum was diluted in 100 μl of complete DMEM for 2-fold serial dilutions and analyzed as described previously. For samples potentially containing infectious HIV, serum was incubated at 56°C for 30 min before being used in in vitro neutralization assays.
Viral load assessment
Plasma HIV RNA levels were determined with an Amplicor kit (Roche, Indianapolis, IN) according to the manufacturer's instructions. The limit of detection is 50 HIV RNA copies/ml.
Statistical analysis and mathematical calculations
ELISA standard curve plots were graphed and analyzed in GraphPad Prism. ELISA samples were measured in duplicate and only linear regression curves with coefficient of determination values of 0.95 and greater were used to calculate bNAb concentrations.
All neutralization assays were performed in duplicate and the data were graphed and analyzed with GraphPad Prism. Each point represents the mean of the indicated number of experiments; error bars denote standard deviations. Relative light unit (RLU) values were normalized to luciferase signal in the absence of recombinant bNAb, bNAb-containing supernatants, or bNAb-containing serum.
For NSG-Hu-PBL engraftment plots, experiments were performed in duplicate, using the indicated sample size. Error bars denote the standard error of the mean. Measurements of CD4+ T cell levels were performed in triplicate. Unless otherwise indicated, each point represents the mean of the indicated number of mice and error bars denote standard error. Statistically significant differences, when specified, were determined by analysis of variance (ANOVA) in GraphPad Prism. p<0.05 was considered statistically significant.
Results
FG ADV5 vectors encoding the heavy and light chain cDNAs (HC and LC, respectively) of the anti-gp120 bNAb VRC03 or PG16 (referred to as ADV-VRC03 and ADV-PG16, respectively) were constructed (Fig. 1a, top). Enhanced yellow fluorescent protein (eYFP) was also included to monitor transgene expression and viral spread in vitro. The coding sequences for the HC, LC, and eYFP were separated by distinct internal ribosomal entry site (IRES) elements to avoid directly repeated sequences that could trigger genome rearrangement during ADV vector amplification. 26 As controls, FG ADV5 vectors encoding only infrared red fluorescent protein (ADV-iRFP; Fig. 1a, middle) and eYFP (ADV-eYFP; Fig. 1a, bottom) were also generated. ADV VPs were purified from cultures of 293 cells transduced with the FG ADV5 vectors by cesium chloride gradient banding. Purified VP stocks of as high as 1013 total particles from up to 30 large (15-cm) 293 plates were routinely obtained and bNAb production was confirmed by Western blots (WBs) of culture supernatants (SNs) of 293 cells transduced with the ADV VPs. Although the stoichiometric ratio of the amounts of HC and LC appeared to vary in cell culture (Fig. 1b), they were expressed at approximately the same levels in animal sera (described below). Importantly, the expressed bNAbs were functional for viral entry inhibition in TZMBL luciferase virus neutralization assays using a single-cycle HIV-1 vector pseudotyped with R5- and X4-tropic envelopes derived from HIV-1 strains ADA, BaL, YU2, JRFL, NL4-3, and SF162 (Fig. 1c). They also potently neutralized infectious HIV-1 AD8, JRCSF, YU2, and BaL (Fig. 1d). Importantly, vesicular stomatitis virus envelope protein (VSVG)-pseudotyped HIV was not inhibited by ADV VP SNs, demonstrating that the inhibition was HIV-specific (data not shown).
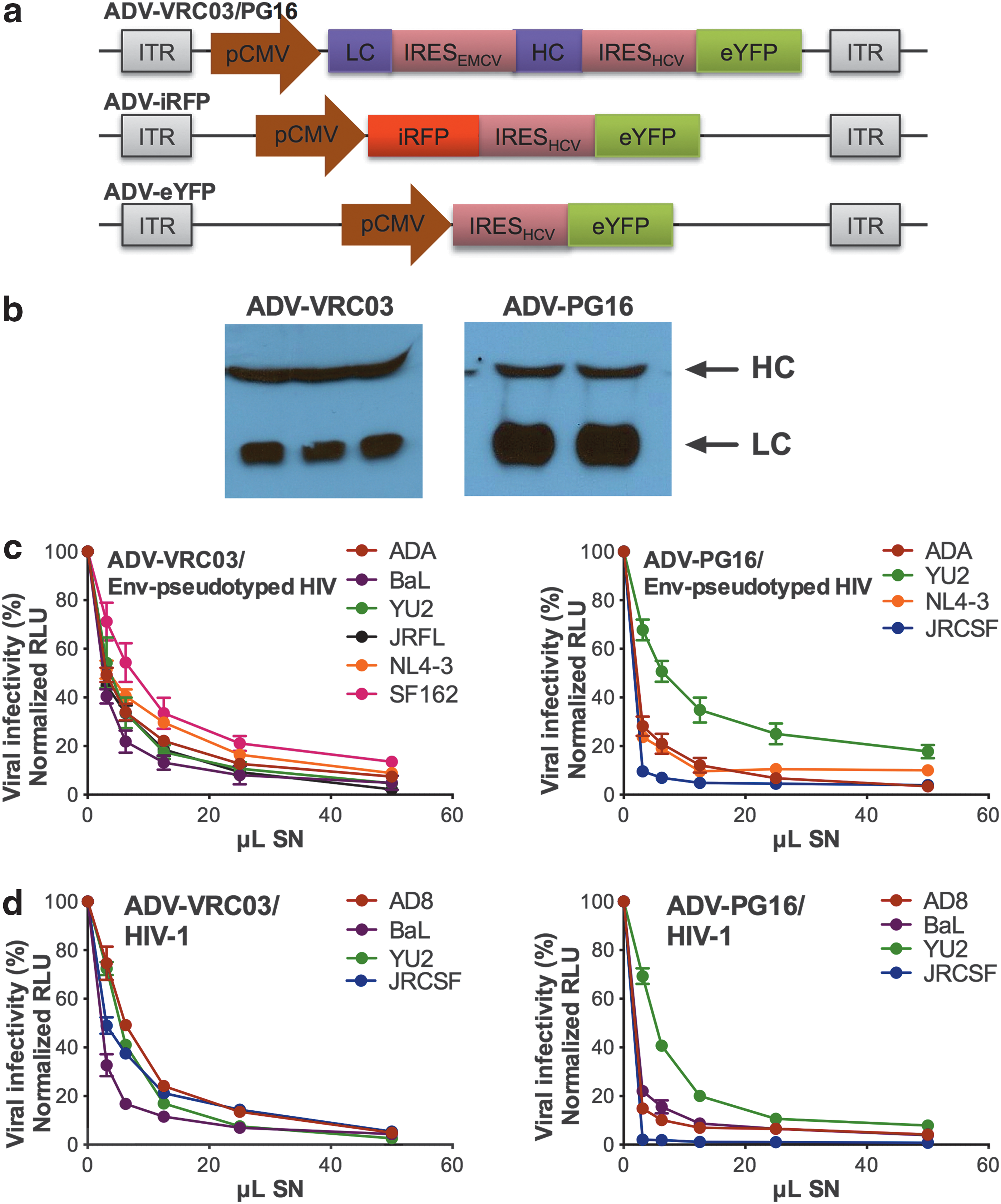
First-generation adenoviral serotype 5 (ADV5) vectors encoding broadly neutralizing antibodies (bNAbs) against HIV-1 gp120.
A sandwich ELISA was performed to determine the IC50 values of bNAb-containing supernatants. For VRC03, our measured IC50 values of 36 and 13 ng/ml, against YU2- and JRFL Env-pseudotyped HIV, respectively, were in good agreement with the reported IC50 values of 37 and 9 ng/ml. 19 For PG16, inhibition of JRCSF Env-pseudotyped HIV and infectious JRCSF was particularly potent with IC50 values of 8 and 2 ng/ml, respectively, in agreement with the low reported IC50 value against this strain (1 ng/ml). 20
ADV transgene expression in mice was analyzed in two ways. First, transgene expression at the site of intramuscular injection was monitored by whole-body imaging of immunodeficient NOD/SCIDIL2rγ−/− (NSG) mice administered 3×1011 ADV-iRFP VPs (Fig. 2a). As expected, iRFP expression was greatest at or near the site of injection in the right hind leg. For the humanized mouse model, we used the NSG-Hu-PBL model of HIV infection,
21
wherein adult NSG mice are reconstituted with human peripheral blood mononuclear cells (PBMCs). The NSG background ensures severe immunodeficiency that maximizes engraftment and xeno-activated T cells constitute the dominant human cell type in this model, supporting rapid HIV infection and CD4+ T cell depletion.
9,10,21
bNAb-encoding ADV VPs (9×1011) administered intramuscularly into NSG mice produced high levels of VRC03 and PG16, and qualitative assessment of mouse sera in WBs revealed HC and LC amounts of approximately similar stoichiometry (Fig. 2b, top and bottom). Intramuscular administration of as much as 1×1012 ADV VPs did not impact engraftment of most donor PBMCs tested. The percentages of peripheral CD4+ T cells as well as CD4+/CD8+ T cell ratios in ADV-treated NSG-Hu-PBL mice were similar to those of mock-treated NSG-Hu-PBL mice (Supplementary Fig. S1a; Supplementary Data are available online at

Kinetics of bNAb production after intramuscular injection of ADV-bNAb.
Having verified high levels of circulating bNAb, ADV-VRC03-injected Hu-PBL mice were challenged with the R5-tropic HIV isolate YU2 (Fig. 3a). Prechallenge serum VRC03 titers were confirmed by neutralization assays with JRFL and YU2 envelope-pseudotyped HIV on day 10 postengraftment (Fig. 3b). On day 35 postengraftment, mice were challenged intraperitoneally with 1×105 focus-forming units (FFU) of YU2. ADV-treated mice demonstrated similar levels of PBMC engraftment as the mock-treated group at the time of YU2 challenge (Supplementary Fig. S1b), and sera collected on day 7 postchallenge retained the same potency of neutralization against infectious YU2 as prechallenge sera (Fig. 3c). Despite the fact that the challenge was well over 1 month after ADV-VRC03 administration, YU2-induced peripheral CD4+ T cell depletion was prevented (Fig. 3d), and this correlated with undetectable plasma HIV RNA loads (<50 copies/ml) in ADV-VRC03-treated mice, in contrast to mock-treated control mice that displayed severe peripheral CD4+ T cell depletion and high HIV RNA loads (∼107 copies/ml; Fig. 3e).
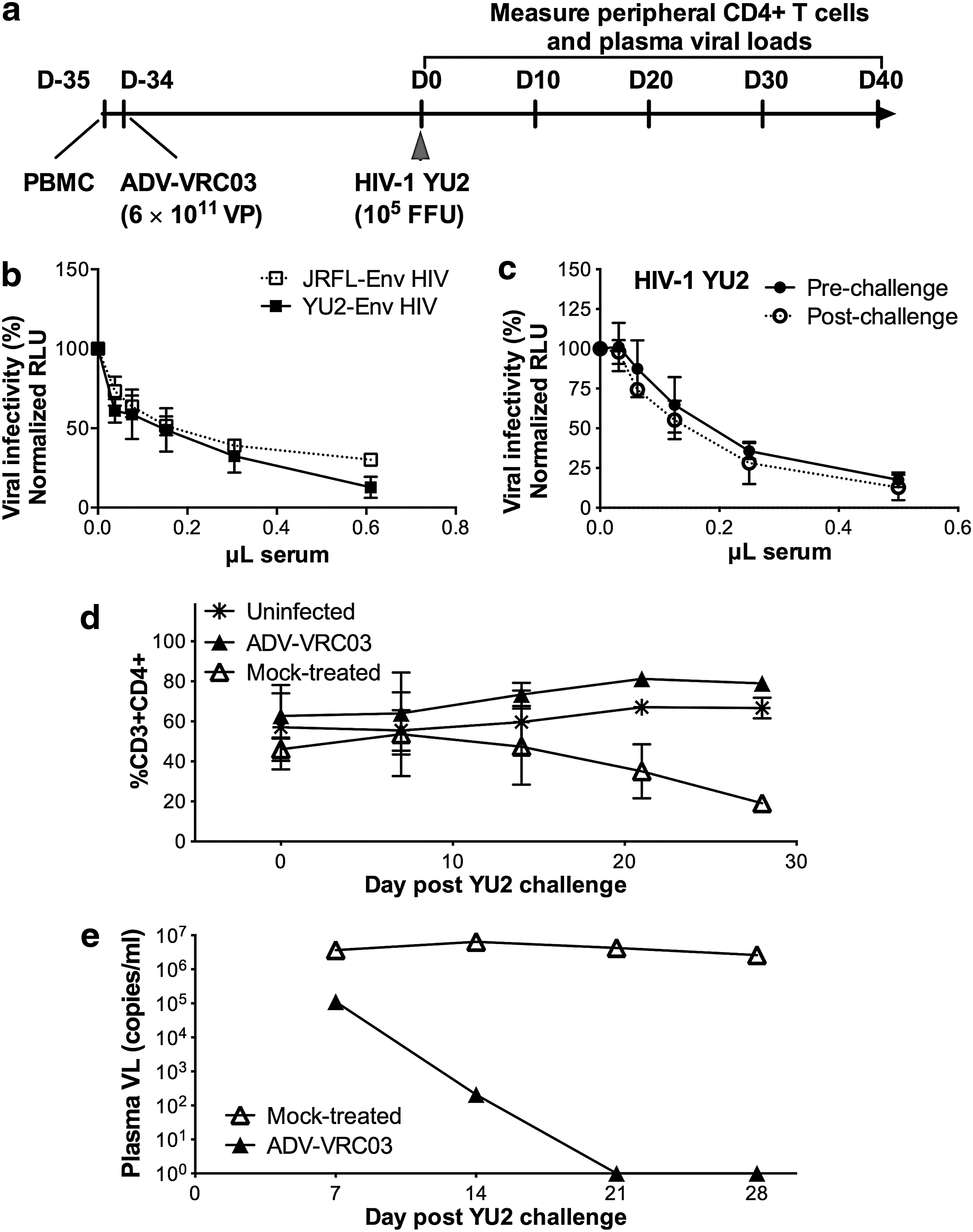
A single intramuscular administration of ADV-VRC03 prevents YU2 acquisition.
We next investigated whether intramuscular injection of ADV-PG16 could protect against repeat HIV-1 acquisition (Fig. 4a). In these experiments, we used the R5-tropic JRCSF isolate, given the high in vitro potency of PG16 against this virus. In addition, in our experience this viral strain results in a rapid depletion of peripheral CD4+ T cells, allowing a study incorporating multiple challenges with JRCSF during the ∼2- to 3-month time period of Hu-PBL engraftment in ADV-injected mice that are protected by bNAb expression. For these experiments, Hu-PBL mice were intramuscularly administered either ADV-PG16 or ADV-eYFP as control (3×1011 VPs) or left untreated (denoted as mock-treated). Circulating levels of PG16 were confirmed prechallenge in neutralization assays using ADA and JRCSF Env-pseudotyped HIV (Fig. 4b). Levels of human leukocyte engraftment between mouse cohorts were similar at the time of challenge (Supplementary Fig. S1c), and mice received an intraperitoneal challenge dose of 3×104 FFU of JRCSF on day 13 alone or also on day 28 after ADV treatment. All ADV-PG16-treated animals maintained peripheral CD4+ T cell levels similar to mock-challenged mice whether they received a single or a double challenge dose of JRCSF, whereas CD4+ T cell levels rapidly declined in control mice after the first and only viral challenge inoculum (Fig. 4c and d, left). Importantly, ADV-PG16-treated mice had no detectable plasma HIV RNA loads 15 days postchallenge, in contrast to the ∼107 viral RNA copies/ml observed in control Hu-PBL mice that were treated with ADV-eYFP or mock-treated (Fig. 4c and d, right).
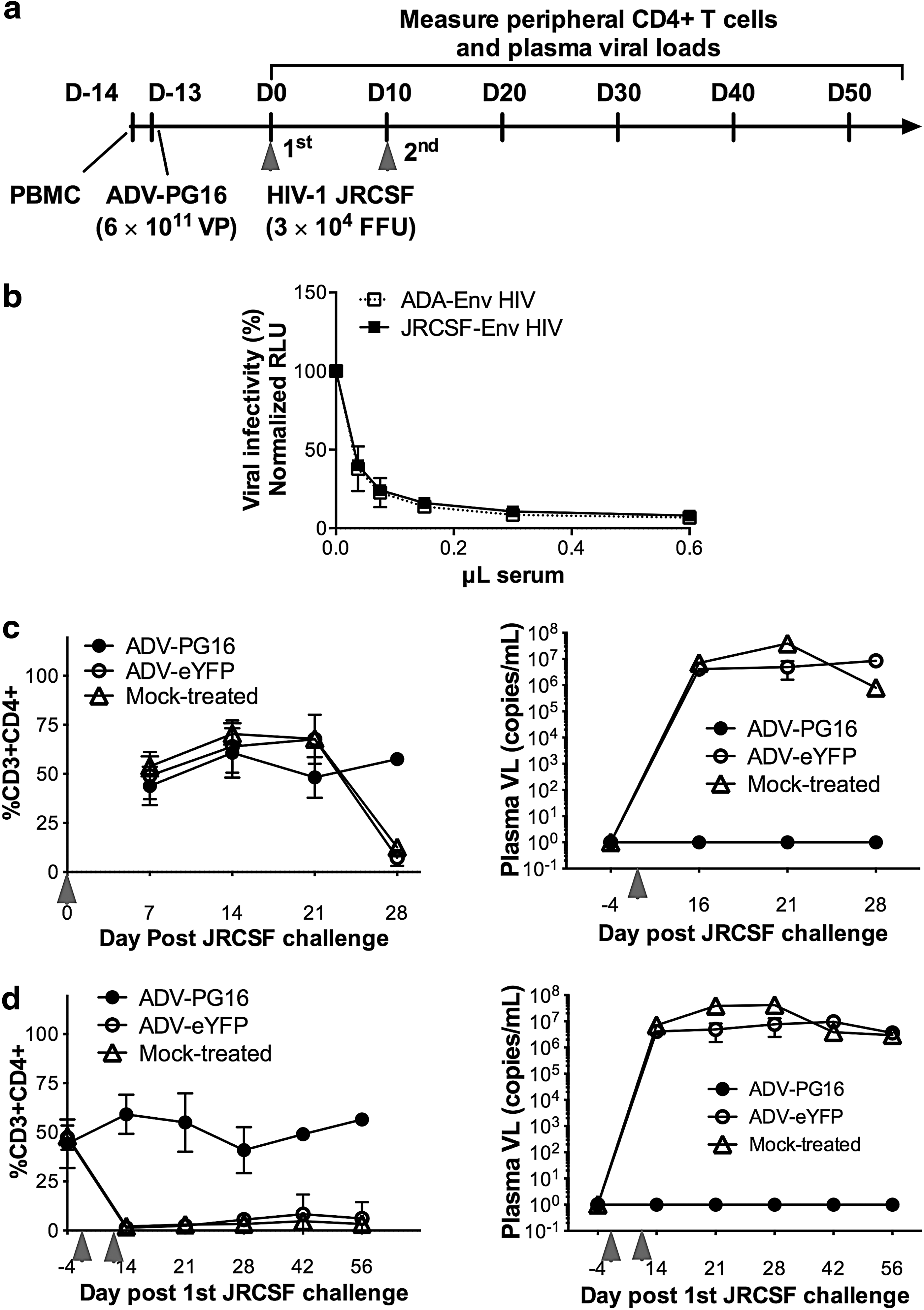
A single ADV-PG16 administration prevents virus acquisition from two consecutive JRCSF challenges.
We next evaluated whether a single intramuscular ADV injection could protect against multiple intraperitoneal challenges with JRCSF. Mice were challenged intraperitoneally six times with 3×104 FFU of JRCSF on a weekly basis, starting on day 16 (D16) after ADV treatment (Fig. 5a). As assessed by peripheral CD4+ T cell levels, a single ADV-PG16 injection was fully protective against all six HIV challenges (Fig. 5b) whereas control mouse cohorts infected with JRCSF had profound CD4+ T cell loss after the first and only virus challenge inoculum. Accordingly, control mouse groups also displayed high plasma HIV RNA loads, representative of active viral replication, which declined rapidly once target CD4+ T cells were fully depleted. HIV RNA load measurements in ADV-PG16-treated mice, however, remained high through the six challenges when serum samples were tested on a weekly basis (Fig. 5c). Furthermore, the plasma viral RNA loads decayed to below detection limits approximately 2 weeks after the last viral challenge. Given that the postchallenge sera from these ADV-PG16-treated mice continued to display excellent neutralization in vitro (data not shown), we reasoned that these viral RNA loads were likely due to delayed clearance of viral RNA originating from the repeated inoculation of the virus stock that was used specifically for this experiment. We confirmed this by inoculating 3×104 FFU of heat-killed JRCSF on a weekly basis, as described previously, into Hu-PBL mice (Supplementary Fig. S2a and b). We again observed high plasma VLs and, as expected, there was no reduction in CD4+ T cell levels. Note that this viral RNA decayed at approximately 0.5 log weekly, consistent with the results of Fig. 5c. As further proof of the extensive protective ability of ADV-PG16, we rechallenged with JRCSF for a seventh time, 3 weeks after the sixth challenge. Again, CD4+ T cell levels remained unaffected, and HIV RNA loads were detectable 1 week later. It is thus likely that ∼2 weeks is required to clear existing viremia from each challenge dose (Fig. 5b and c).

A single ADV-PG16 administration prevents virus acquisition from multiple and repeated challenge with JRCSF.
Collectively, our data demonstrate that FG ADV vectors are a viable means by which to deliver anti-gp160 bNAb genes, and that a single intramuscular injection yields bNAb expression levels high and persistent enough to protect against multiple repeated challenges with HIV-1.
Discussion
Herein, we demonstrate the usefulness of FG ADV5 vectors that encode full-length bNAbs against HIV gp120 for HIV prophylaxis in a humanized mouse model. A single intramuscular injection of bNAb-encoding ADV vector was sufficient to prevent virus acquisition and peripheral CD4+ T cell depletion after single or repeated intraperitoneal challenge with a high dose of infectious HIV-1. Although ADV-bNAb-treated mice challenged multiple times with HIV maintained peripheral CD4+ T cell levels, they initially displayed HIV RNA loads that fell to undetectable levels in the absence of repeated virus challenge, suggesting an absence of productive infection.
This study follows a steady line of antibody-based approaches targeting HIV Env, but is novel in the use of an ADV5 vector. Since the identification of the first bNAb against gp160 in 1992, 27 there has been much interest in the use of these antibodies, either prophylactically or therapeutically. Until more recently, such efforts have met with a lack of success, likely for several reasons. First, previous approaches often involved the elicitation of bNAbs against HIV by antigen immunization. bNAbs against HIV are now well known to possess unusual primary sequence, structural, and binding properties, making them difficult to elicit with immunogens. 5 –7,28 Second, methodological approaches for high-throughput screening and recovery of HC and LC genes from individual patient B cells did not exist until more recently, and the previously identified bNAbs were not potent enough to prevent viral escape, even when delivered in combination. 29 –32 Compared with the current crop of bNAbs, first-generation bNAbs have much reduced binding affinities and higher IC50 values, likely insufficient to be used prophylactically or therapeutically in humans.
With the advent of the more potent, newer generation bNAbs, there has been a resurgence of antibody-based approaches, and studies thus far have yielded promising results. bNAb administration was reported to reduce viral rebound from latently infected cells after discontinuation of antiretroviral therapy and inhibited the replication of reservoir-derived virus. 33,34 Interestingly, passive immunotherapy with earlier generation bNAbs also delayed viral rebound in human subjects after HAART interruption. 35 This is significant given that HAART is unable to eradicate latent virus. Passive infusions of tri- and pentamix bNAbs targeting distinct nonoverlapping epitopes were able to lower HIV RNA loads and prevent viral escape in HIV-infected humanized mice. 36 Similarly, passive infusions of one or more bNAbs resulted in complete virological suppression in simian/human immunodeficiency virus (SHIV)-infected macaques, with no evidence of viral escape. 37,38 Viral rebound in both the humanized mice and macaques occurred only after decline in serum bNAb levels. 36 –38 Compared with the delivery of recombinant bNAbs, vectored bNAbs may circumvent the need for frequent reinjections by enabling sustained, high levels of transgene expression. Although bNAbs can be vectored in AAV, 9,10 AAV has a limited carrying capacity 12 that would necessitate multiple vectors and injections if combinations of bNAbs were to be administered. In contrast, the present work uses FG ADV5 vectors, which have a large-enough carrying capacity to allow the encoding of multiple full-length bNAbs on a single vector, if necessary, or the addition of regulatory or other cis-acting sequences. 14,15
One concern associated with ADV vectors is a limited duration of transgene expression. Other reports document ADV-vectored antibody expression ranging from 1 to 4 weeks in mice. 39 –41 In our study, a single intramuscular injection evoked high-level serum bNAb titers for >6 weeks. Although we did not monitor the kinetics of transgene expression for the entire period in vivo, ADV vector-injected mice were completely protected even at 6 weeks, when bNAb levels were significantly lower than peak levels at 2 weeks after vector administration. The decline in bNAb production was unlikely to have been caused by immune clearance as nontransplanted NSG mice displayed the same dynamics. The comparatively shorter duration of ADV vector-delivered transgene expression relative to AAV-vectored transgenes has been documented previously, 12,41 but nonetheless this limitation may be outweighed by the unique advantages of ADV vectors. First, in addition to being able to encode combinations of bNAbs, which has been shown to prevent or delay viral escape, the larger carrying capacity of ADV vectors should support the delivery of the newly engineered bispecific and multivalent bNAbs, which have been reported to be 100–200 times more potent than conventional gp160-targeting bNAbs. 42,43 By encoding even more highly potent bNAbs, viremia may be fully suppressed for a longer period of time, reducing the chances of virological resistance or escape. Second, clinical safety is a major concern with any viral vector, and immunogenic effects have been observed, albeit rarely, for both ADV 44 and AAV. 45 If donor immunity to specific ADV serotypes preexists, ADV capsids based on alternative serotypes or chimeric ADV5 vectors incorporating proteins from other serotypes are readily available, and have been shown to circumvent ADV5-related immunogenicity. 46,47 To reduce or ameliorate treatment or vector-related toxicity, inducible ADV vectors allowing for the encoded gene(s) to be regulated in vivo may be engineered. For AAV, separate vectors would be required if an inducible system is desired. Third, ADV vectors are more readily and easily amplified and purified in comparison with AAV, making them more cost-effective when manufactured at scale for clinical use. 11,13 Finally, should long-term transgene expression be desired, helper-dependent or “gutted” ADV (HDADV) vectors could be used. Not only do HDADV vectors have a carrying capacity of more than 30 kb, 14,15 but nonhuman primate studies have demonstrated extremely long-lived expression, equivalent to that of AAV-based vectors. 48 –51
In summary, the new anti-HIV bNAbs have unlocked novel therapeutic and prophylactic approaches to HIV-1. Herein, we explored the possibility that such bNAbs can be delivered via well-characterized FG ADV5 vectors. Although our studies examined prophylactic efficacy in Hu-PBL mice, an acute model for HIV-1 infection, our data demonstrate the feasibility of a treatment approach with ADV5-vectored bNAb for potent and complete suppression of HIV-1 replication in other humanized mouse models. Future investigations may explore the use of more recent and potent bNAbs, regulatable vectors, and combinatorial use in HDADV vectors, in conjunction with HAART in large animal models.
Footnotes
Acknowledgment
The authors thank Drs. John Mascola (Vaccine Research Center, NIAID, NIH, Bethesda, MD) and Heinrich Gottlinger (Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA) for their kind gifts of expression plasmids encoding VRC03 and PG16, respectively. The authors also thank the contributors at the AIDS Reagent Repository for the use of cell lines, antibodies, and HIV plasmids. This work was supported by NIH-NIDA (F31 DA033925-01). R.E.S. is an NIDA HIV Avant Garde awardee.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.