
Abstract
The Sleeping Beauty (SB) transposon system can insert sequences into mammalian chromosomes, supporting long-term expression of both reporter and therapeutic genes. Hematopoietic progenitor cells (HPCs) are an ideal therapeutic gene transfer target as they are used in therapy for a variety of hematologic and metabolic conditions. As successful SB-mediated gene transfer into human CD34+ HPCs has been reported by several laboratories, we sought to extend these studies to the introduction of a therapeutic gene conferring resistance to methotrexate (MTX), potentially providing a chemoprotective effect after engraftment. SB-mediated transposition of hematopoietic progenitors, using a transposon encoding an L22Y variant dihydrofolate reductase fused to green fluorescent protein, conferred resistance to methotrexate and dipyridamole, a nucleoside transport inhibitor that tightens MTX selection conditions, as assessed by in vitro hematopoietic colony formation. Transposition of individual transgenes was confirmed by sequence analysis of transposon–chromosome junctions recovered by linear amplification-mediated PCR. These studies demonstrate the potential of SB-mediated transposition of HPCs for expression of drug resistance genes for selective and chemoprotective applications.
Introduction
T
In human cord blood-derived CD34+ progenitor cells, the SB system has been shown to mediate successful transposition and expression of reporter genes encoding enhanced green fluorescent protein (eGFP) 7,8 and Discosoma species red fluorescent protein (DsRed). 9 A hyperactive variant of SB transposase (SB100x), engineered by in vitro evolution of the transposase gene with targeted amino acid substitutions, was required to mediate transposition into hematopoietic stem and progenitor cells capable of multilineage, long-term engraftment. 7,9 The SB system was subsequently shown to mediate integration of the human β-globin gene into CD34+ human hematopoietic progenitor cells (HPCs). 10,11 SB-transposed CD34+ cells can be differentiated in vitro to T cells, B cells, natural killer (NK) cells, and myeloid cells, 9 and enucleated red blood cells. 10 Here we sought to extend previous studies with CD34+ cells to introduce a drug-selectable gene, conferring resistance to methotrexate (MTX).
Dihydrofolate reductase (DHFR) catalyzes the NADPH-dependent reduction of folate to dihydrofolate, and of dihydrofolate to tetrahydrofolate in mammalian cells. 12 Rapidly dividing cells require reduced folates as cofactors for enzymes involved in thymidylate synthesis, de novo purine biosynthesis, and glycine synthesis. MTX, a commonly used and clinically effective chemotherapeutic agent, is a potent competitive inhibitor of DHFR. 12,13 MTX has been successfully used to treat a number of malignancies such as acute lymphoblastic leukemia, non-Hodgkin's lymphoma, and osteosarcoma. 14 However, the therapeutic dose that can be administered is limited by toxicity in rapidly dividing cells of the bone marrow and gastrointestinal tissues. 15,16 Several amino acid substitutions in the DHFR enzyme have been described that confer resistance to MTX toxicity in normal drug-sensitive cells and tissues. 17 –19 For example, expression of murine DHFR with wild-type leucine substituted by tyrosine at position 22 (Tyr-22) results in a high level of resistance to MTX. 19,20 Expression of drug-resistant DHFR can mediate protection from antifolate toxicity after transplantation in mice using either transgenic hematopoietic stem cells (HSCs) 20 or HSCs transduced with DHFR-encoding retroviral or lentiviral vectors. 21,22 In vivo expansion of DHFR(Tyr-22)-transduced hematopoietic stem cells has been achieved after antifolate treatment, allowing for selective outgrowth. 23,24 These studies demonstrate the potential for protection of normal cells during administration of antifolate chemotherapy after DHFR gene transfer.
Here, we demonstrate that the nonviral SB transposon system can additionally modify human HPCs for expression of DHFR(Tyr-22), thus conferring resistance of hematopoietic colony-forming progenitors to high concentrations of methotrexate. These results have implications for the application of the nonviral SB system to drug resistance gene transfer for the purpose of protecting normal cells from the toxic side effects of chemotherapy as well achieving selective outgrowth of genetically engineered hematopoietic cells either in vitro or in vivo.
Materials and Methods
Construction of transposons and transposase-encoding plasmids
Plasmid pCMV-SB100x, encoding hyperactive SB100x transposase expressed from the cytomegalovirus (CMV) immediate-early promoter,
7
was kindly provided by Z. Izsvak (Max Delbrück Center for Molecular Medicine, Berlin, Germany). The pKT2/mCAGGs-DHFR-GFP transposon plasmid was constructed from the previously described pDL2G plasmid,
22
containing the GGGGSGGGS glycine-serine linker between the mouse DHFR(Tyr-22) and eGFP domains of the fusion protein.
25
A fragment encoding DHFR(Tyr-22) and the [Gly4Ser]2 linker was amplified from the pDL2G template by PCR using sense oligonucleotide (5′-TA
Nucleofection of CD34+ cells
Umbilical cord blood (UCB) was obtained either from the New York Blood Center (New York, NY) under a protocol approved by the University of Minnesota (Minneapolis, MN) Institutional Review Board, or was purchased from the National Disease Research Interchange (Philadelphia, PA). CD34+ cells were purified by positive selection and cultured overnight in X-VIVO 10 serum-free medium with gentamicin (Lonza, Walkersville, MD), containing stem cell factor (SCF), thrombopoietin (TPO), interleukin (IL)-3, and Flt3 ligand (Flt3L) (30 ng/ml each; Bio-techne, Minneapolis, MN), as described previously. 28 CD34+ cells pooled from multiple cords were mixed with CD34− cells, supplemented with 10 μg of plasmid DNA consisting of a 3:1 ratio of transposon to transposase-encoding plasmid and/or or filler DNA, and nucleofected at a concentration of 1×106 cells/0.1 ml of human CD34+ cell Nucleofector solution (Lonza), using program U-08 and a Nucleofector I device. The transfected cells were immediately transferred to 24-well plates containing 37°C prewarmed medium.
In vitro culture of CD34+ cells
After 2–3 days in liquid culture, CD34+ cells were plated in methylcellulose medium containing human cytokines (HSC005; Bio-techne) in the presence or absence of 100 nM MTX (Hospira, Lake Forest, IL) and 5 μM dipyridamole (Sigma-Aldrich, St. Louis, MO). After 12–16 days at 37°C, 5% CO2, progenitor colonies containing >50 cells were counted with an inverted fluorescence microscope (Olympus IX71) (excitation filter, 485 nm; emission filter, 525 nm). Colonies were categorized as either erythroid (burst-forming unit-erythroid [BFU-E] or colony-forming unit-erythroid [CFU-E]) or granulocyte/monocyte morphology (CFU-G, CFU-M, or CFU-GM) 29 and scored as GFP positive or negative.
Linear amplification-mediated PCR
Individual progenitor-derived colonies were harvested from methylcellulose cultures and genomic DNA was purified with a Quick-gDNA MicroPrep kit (Zymo Research, Irvine, CA). Ten nanograms of genomic DNA (gDNA) was whole-genome amplified with an illustra GenomiPhi V2 DNA amplification kit (GE Healthcare, Piscataway, NJ), and 1 μg of amplified material was evaluated for transposon integrants by linear amplification-mediated PCR (LAM-PCR) as previously described, 30 using primers designed for recovery of SB transposon–chromosome junction sequences. 31 The recovered sequences were evaluated by nucleotide–nucleotide Basic Local Alignment Search Tool analysis 32 against the human genome, using the National Center for Biotechnology Information (NCBI, Bethesda, MD) database.
Results
Sleeping Beauty transposons were constructed to express murine DHFR(Tyr-22) using the mini-CAGGs promoter, 26 containing the CMV early enhancer, chicken β-actin promoter, and a truncated β-globin intron sequence (Fig. 1). Because a minimum of 1 million cells is ideal for the nucleofection procedure, we routinely mixed purified CD34+ cells with CD34− cells (25–50% CD34+) and nucleofected at a 3:1 ratio of transposon/transposase-encoding plasmids in trans. 9 After 3 days of liquid culture with human cytokines, CD34+ cells exhibited selective uptake of nucleofected DNA, expansion, and expression of eGFP, without any difference in eGFP levels when transposase was either present or absent (Fig. 2). eGFP was expressed by the CD34+CD38low/– population (Fig. 2B), containing less mature, pluripotent hematopoietic progenitors that can reconstitute a functional human immune system in immunodeficient mice. 33,34 Cells were plated in semisolid methylcellulose medium containing human cytokines and methotrexate for selective outgrowth of DHFR-transposed, drug-resistant progenitors into erythroid (BFU-E, CFU-E) and myeloid (CFU-GM, CFU-G, CFU-M) colonies. Dipyridamole (DP), a nucleoside transport inhibitor, was included in the medium to prevent uptake of exogenous purine nucleosides and thymidine that can compromise MTX selection. 35 –37 Nucleofection in the presence or absence of pCMV-SB100x plasmid did not alter the morphology of colony-forming cells (CFCs); approximately 300 erythroid and 135 granulocyte/monocyte colonies per 1000 CD34+ cells were enumerated. Colonies scoring GFP positive by fluorescence microscopy were observed only when pCMV-SB100x plasmid was conucleofected with either DHFR–GFP fusion or GFP transposon DNA before plating (Fig. 3A). Only half of the drug-resistant colonies scored positive for GFP (Fig. 3B), likely due to the high background fluorescence of methylcellulose. Growth of drug-resistant progenitor colonies was observed only when cells were nucleofected with transposon encoding DHFR(Tyr-22)-eGFP along with pCMV-SB100x plasmid (Figs. 3B and 4). CFC colonies that were eGFP+ ranged from uniform bright expression of eGFP to dim eGFP expression (Fig. 4). This variability in expression has been previously reported in CD34+ cell-derived colonies, 38 and likely is due to position effect variegation.

Transposons used in this study. Maps of pKT2/mCAGGs-DHFR(Tyr-22)-eGFP, pKT2/mCAGGs-eGFP, and hyperactive pCMV-SB100x are shown. eGFP, enhanced green fluorescent protein gene; DHFR, dihydrofolate reductase; mCAGGs, mini-CAGGs; pA, poly(A); CMV, cytomegalovirus promoter. Color images available online at

CD34+ cells selectively express GFP 2 or 3 days after nucleofection of a mixed population of CD34-positive and -negative cells.
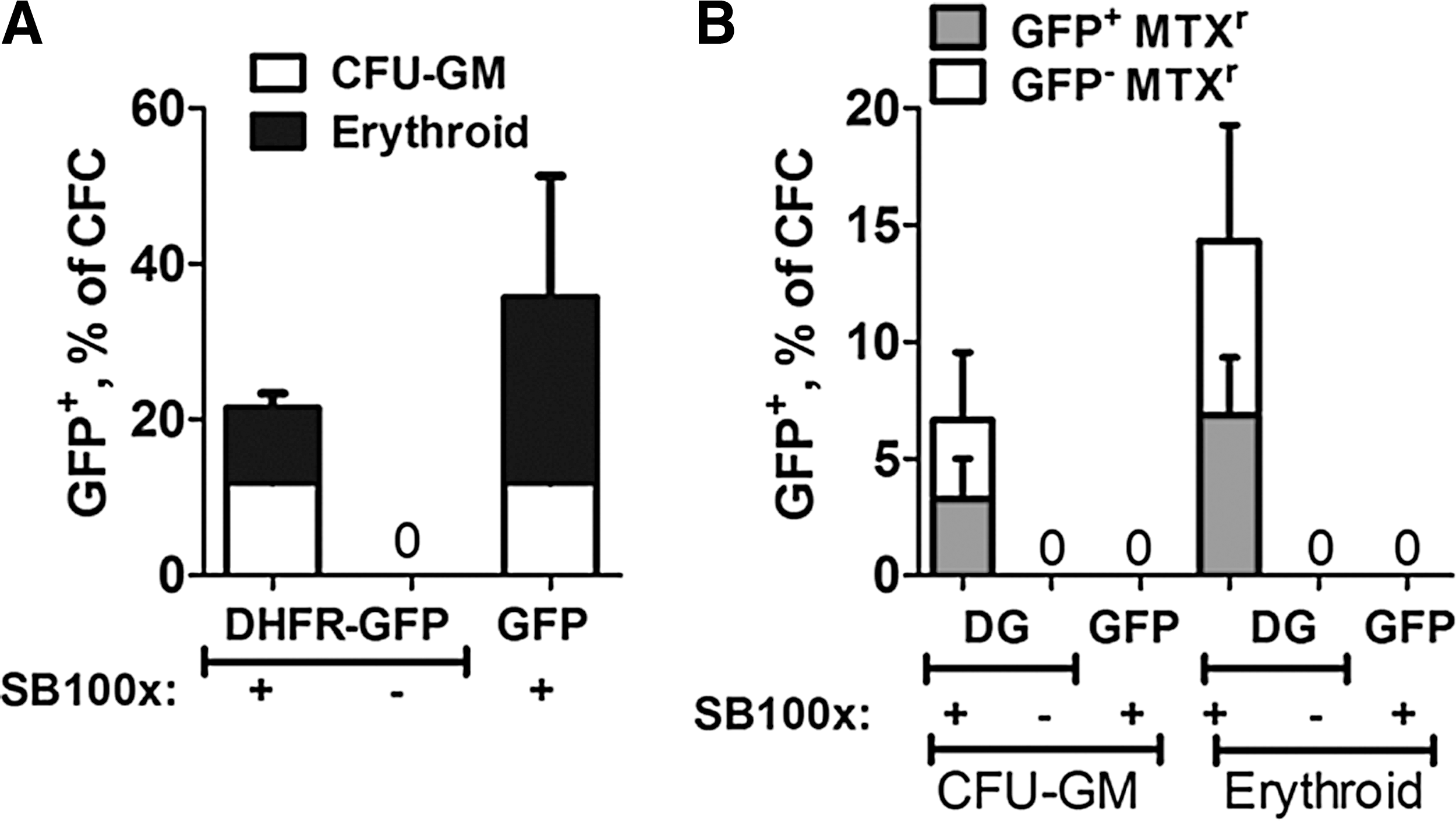
Long-term expression of GFP and methotrexate (MTX) resistance by hematopoietic progenitor colonies after transposition with transposon encoding GFP or DHFR(Tyr-22)-eGFP fusion gene and SB100x.
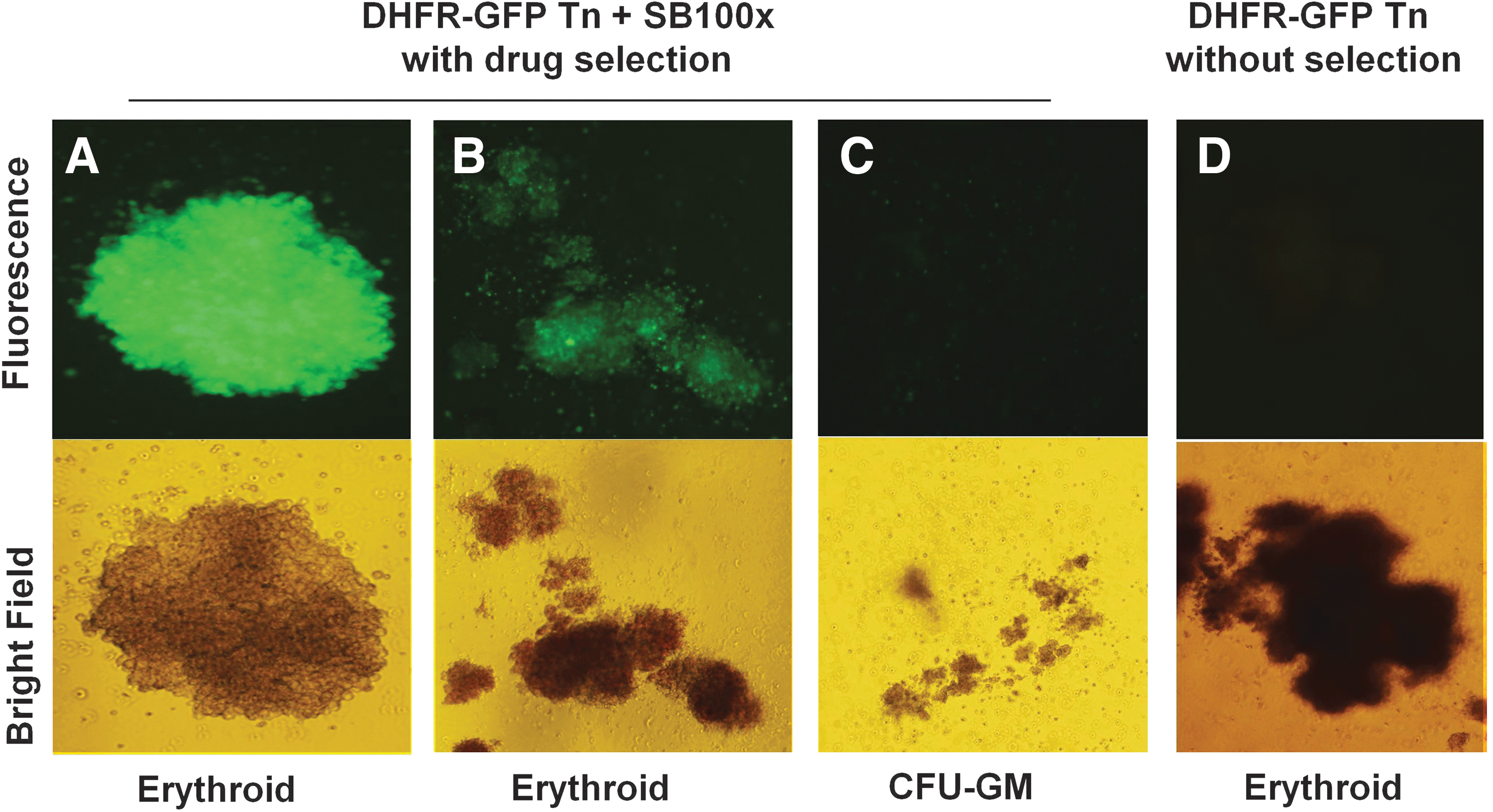
Enhanced GFP expression by drug-resistant hematopoietic progenitor colonies ranged from bright
For molecular analysis of SB-mediated transposition, chromosome–transposon junction fragments were recovered from individual colonies by linear amplification-mediated PCR (LAM-PCR) after culture for 12–14 days, when episomal plasmid was no longer likely to be present. Eleven transposon integrants were identified in the genome and contained the signature TA dinucleotide positioned between chromosomal and SB inverted repeat sequences (Table 1). In this small sample set, 5 of the 11 integrants were intergenic, and 6 of the 11 were found within the introns of genes. These data verify that the formation of MTX-resistant, eGFP+ hematopoietic colonies was molecularly associated with SB-mediated transposition and transgene expression.
Molecular Analysis of Chromosomal Integrants in Colony-Forming Cells
CFC, colony-forming cell; CFU-GM, colony-forming unit-granulocyte/monocyte; IR/DR, indirect repeat/direct repeat; N/A, not applicable.
Erythroid (BFU-E or CFU-E) or granulocyte/monocyte morphology (CFU-GM).
The sequences from the right indirect repeat/direct repeat (IR/DR).
pKT2/mCAGGs-DHFR-eGFP transposon, CFCs cultured under selection conditions.
pKT2/mCAGGs-eGFP transposon, CFCs cultured without selection.
Discussion
We demonstrate that the nonviral SB transposon system can effectively deliver a pharmacologically relevant therapeutic drug resistance gene to human hematopoietic progenitor cells. Drug-resistant variants of DHFR have been expressed in human umbilical cord blood CD34+ cells after gammaretroviral vector transduction. 39,40 However, in clinical trials to treat X-linked severe combined immunodeficiency by transduction of hematopoietic cells with gammaretroviral vectors, genotoxicity was observed. 41 Although deep sequencing of SB integrants in CD34+ cells has not been reported, analysis of other human cells (HeLa cells, primary T cells, etc.) has shown that SB-mediated integration in human cells is closer to random integration than that mediated by lentiviral vectors. One of the advantages of SB for gene therapy is the lack of preferential integration at transcriptional start sites. 42 –44 Lentiviral (HIV-1-derived) vectors tend to integrate within gene sequences of CD34+ cells, whereas gammaretroviral vectors (based on Moloney murine leukemia virus) tend to integrate near areas of actively transcribed regulatory elements in CD34+ cells. 45 This supports the use of nonviral gene therapy for genetic modification of hematopoietic progenitor cells as it is less prone to insertional mutagenesis.
SB has been the most extensively tested transposon system for preclinical gene therapy studies and is currently being used in clinical trials to engineer T cells for treatment of patients with B-lineage lymphoid malignancies. 46 Since it was initially described, several variants of SB transposase exhibiting increased activity have been generated, first by targeted mutagenesis to create SB11, 47 and later a hyperactive variant, SB100x, was engineered and has been a key component in achieving stable reporter gene expression in human CD34+ cells. 7,9
There are two possible applications for drug-resistant DHFR gene transfer in HSCs. First, DHFR gene transfer in HSCs followed by transplantation and engraftment would permit administration of normally toxic doses of antifolate chemotherapy. 48 –50 Expression of drug-resistant DHFR in hematopoietic cells has also been found to confer a protective effect to normally drug-sensitive gastrointestinal tissues. 20 Second, drug-resistant DHFR could be coexpressed along with a nonselectable gene, followed by exposure to MTX, allowing enrichment of human hematopoietic progenitors or lymphoid cells expressing both genes, thereby permitting an in vivo selective growth advantage. 51,52 Such an approach would overcome the lower efficiency of SB-mediated gene transfer compared with viral vectors.
Our data demonstrate that the SB transposon system can be used to confer methotrexate resistance after drug-resistant DHFR gene transfer in CD34+ human myeloid progenitors. The next challenge is to use the SB transposon system to integrate therapeutic genes into hematopoietic stem cells with multilineage differentiation potential, capable of engraftment and long-term reconstitution in recipient animals. Others have demonstrated detectable expression of fluorescent reporter genes after transplantation of SB-engineered CD34+ cells into immunodeficient mice. 9 However, the frequency of stable SB-mediated gene transfer, reported as a mean of 7% in the bone marrow of immunodeficient mice, 9 is lower than that achieved using retroviral or lentiviral vectors, reported as 20–25% of peripheral blood cells in human patients. 53 One possible reason for the low efficiency of gene transfer using the SB system is the toxicity of nucleofection, with only 50% of cells surviving the procedure. 10 This could be overcome by using mRNA encoding SB transposase along with DNA encoding the transposon 54 or by modification of nucleofection conditions. Coexpression of drug-resistant DHFR along with a therapeutic gene would allow for selective outgrowth of transposed cells and enrichment of cells expressing the nonselectable therapeutic gene either in vitro or in vivo.
Footnotes
Acknowledgments
The authors thank Cindy Eide (University of Minnesota, Pediatric Department) for technical assistance and Colin Sweeney (NIAID, NIH) for his protocol, adapting the LAM-PCR method to SB transposons.
Author Disclosure
This work was supported by grant R44 HL076908 from the National Institutes of Health (NHLBI) to Discovery Genomics, Inc. (DGI) (R.S.M.). R.S.M. is a founder (with stock) of DGI. K.A.H., E.R.O., and R.S.M. are current employees of DGI, including stock options.