
Abstract
Like most of the tools that enable modern life science research, the recent genome-editing revolution has its biological roots in the world of bacteria and archaea.
Introduction
M
The first CRISPR-cas locus was encountered in a K12-derived strain of E. coli in 1987, 6 and many more examples of these striking sequence arrangements were noted over the next 15–20 years. However, there were very few compelling hints of their functions until 2005, when three groups independently reported that many spacer sequences, which had previously been described as unique, in fact matched the sequences of fragments of bacteriophage genome and, at a lower frequency, other mobile elements. 7 –9 These three reports each proposed that the sequence matches reflect a defensive function for CRISPR, and one even noted a weak inverse correlation between phage killing and the number of spacers in the CRISPR locus. 7 The fundamental nature of CRISPR/Cas systems as sources of sequence-directed, adaptive immunity was conclusively revealed in 2007, along with the requirement for protein-coding cas genes to support this defense function. 10 By the end of 2008, phage immunity was shown to rely upon processed crRNAs, 11 and the ability of CRISPR systems to execute DNA interference was uncovered. 12 The latter three advances from 2007 to 2008—genetic interference, 10 essential RNA guides, 11 and DNA targeting 12 —enabled an initial glimpse of an engineered, RNA-directed, genome-targeting platform, and the “considerable functional utility” of such a crRNA-based DNA-targeting system in eukaryotes was first explicitly postulated. 12
In spite of these advances, several more years of mechanistic insights and advances would be required before this potential could be harnessed, and there were several steps along the way from the 2008 suggestion to today's realization. As became clear over time, one particular subset of CRISPR/Cas pathways—the “type II” systems 13 (see CRISPR/Cas Pathways and Mechanisms: Type II Systems) that use a protein called Cas9—was best suited for the development of such a platform. Critical steps included the demonstration that flanking sequence elements (protospacer adjacent motifs [PAMs]) are required in the target, 14,15 that type II interference proceeds via the introduction of double-strand breaks (DSBs) in the target DNA, 16 that Cas9 is the only type II Cas protein required for the interference function of existing spacers, 17 and that the tracrRNA (an additional small RNA besides crRNA) is an essential cofactor in type II pathways. 18 Each of these discoveries provided an essential piece of the puzzle that was dramatically completed in 2012, when the programmable, RNA-guided, DNA-cleaving capacity of an engineered Cas9 system was realized. 19,20 By early 2013, independently and within weeks of each other, several groups documented that the activity previously shown in bacterial cells and in vitro could also be observed in eukaryotic cells, resulting in genome editing. 21 –25 The ensuing two-and-a-half years have seen a virtual explosion of technical development and end-user adoption, 3,4 to the point that the CRISPR revolution is now being described as the most significant technological advance in the life sciences since the advent of the polymerase chain reaction approximately three decades earlier. 26 With the technological revolution in the laboratory already upon us, much attention is now turning toward its potential for engendering a comparable therapeutic revolution in the clinic.
As with RNA interference and other disruptive technologies, our ability to exploit CRISPR/Cas systems in experimental, biotechnological, agricultural, and clinical settings advances in strict parallel with our understanding of the underlying mechanisms. Here we review our current understanding of the natural CRISPR/Cas pathways that operate in bacteria and archaea, and briefly introduce the genome-editing applications that will be discussed in greater depth and detail throughout the rest of this issue of Human Gene Therapy.
Shared Features of CRISPR/Cas Systems
CRISPR/Cas systems are functionally, structurally, and mechanistically diverse. 1,2 Nearly all of them fit into one of three types (I–III), each of which is further subdivided into multiple subtypes. 5 Despite their diversity, CRISPR loci share a common underlying organization of short (usually ∼30–40 nt), direct repeats that are each separated from the other by the nonrepetitive, invader-derived spacers, which tend to be of comparable length (Fig. 1A). The array is commonly flanked by a “leader” sequence that, in most cases, includes a promoter that initiates unidirectional transcription through the CRISPR repeats and spacers. Type I and type III systems are found in both bacteria and archaea, whereas type II systems have been observed in bacteria only.
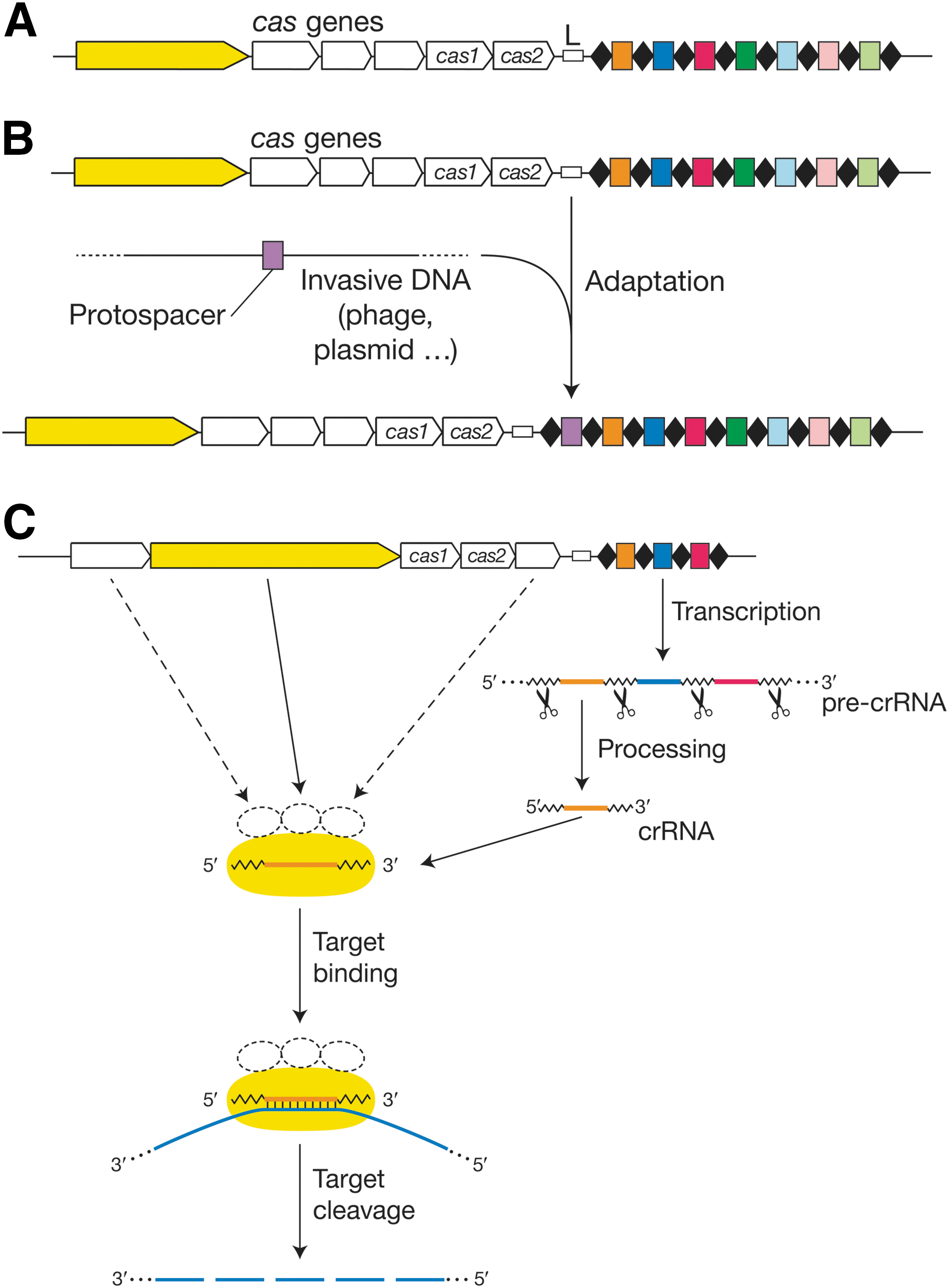
CRISPR-cas loci and fundamental features of all CRISPR immune systems.
CRISPR loci can only confer immunity in the presence of their cognate set of cas genes, which usually reside immediately adjacent to the CRISPR (Fig. 1A). The cas gene content is the most important determinant of that system's “type” designation, and each of the three primary types has its own, distinct, signature cas gene (Fig. 1A, in yellow) that is not found in either of the other two types. Only two cas genes, cas1 and cas2, are found in the cas loci of all three types.
In addition to these commonalities in organization, there are also three common molecular steps that drive CRISPR-mediated immunity: acquisition, expression, and interference. 1,2 Indeed, these three stages drive the molecular processes that originally defined CRISPR/Cas systems as providing DNA-encoded, 10 RNA-guided, 11 and complementary DNA- 12 or RNA-targeting 27 immunity, respectively (with the molecular target depending on the system; see Interference). Specifically, these three stages (1) allow CRISPR/Cas systems to build up immunity by acquisition of novel spacers derived from invasive genetic elements through sampling of foreign nucleic acid sequences 10 ; (2) generate small crRNAs through expression of CRISPR arrays and processing of these transcripts into mature guides that specify nucleic acid targets 11,28 ; and (3) drive interference against invasive elements through crRNA-Cas ribonucleoprotein complexes that specifically recognize and cleave complementary target sequences though endonucleolytic and/or exonucleolytic activities. 16,27,29 –33
Spacer acquisition
Since CRISPR-mediated adaptive immunity is DNA-encoded, the immunization process is driven by sampling of exogenous elements 10 that are copied and pasted into CRISPR arrays as novel spacers. These spacers provide immune memory that allows the adaptive immune system to store information about previous infections, and subsequently mount a response against similar invaders. 34 This process requires recognition of invasive elements, such as viruses and plasmids, by the Cas machinery; targeting of sequences that (for type I and II systems) are flanked by short, characteristic PAM elements; generation of a novel spacer sequence flanked by two CRISPR repeat sequences; and integration of a repeat-spacer unit at the leader end of the CRISPR locus array (Fig. 1B). Very little is known about spacer acquisition by the type III machinery; in contrast, several recent advances in type I and II model systems have shed light on the biochemical processes that drive copying and pasting of invasive sequences into CRISPR arrays, 35 –37 as well as biases in foreign nucleic acid sampling. 38 –43
Most spacer acquisition experiments have been carried out in Escherichia coli for type I systems 35 –38,41,43 –47 and in Streptococcus thermophilus for type II systems. 10,39,40,48 –51 Sequence features that are located immediately upstream of the first repeat are involved in locus orientation and integration of novel spacers, 34 and rely on a Cas1–Cas2–protospacer complex that drives cruciform structure-dependent integration of novel spacers at the junction between the leader sequence and the first CRISPR repeat. 34 –37 The polarized nature of the spacer acquisition process yields a genetic array that provides a record of iterative immunization events over time, and is useful for establishing phylogenetic linkage between strains that share ancestral spacers, as well as divergent paths between strains that have unique, recently acquired spacers. Though many different spacers may be acquired from invasive sequences, there are biased patterns of spacer acquisition that could be inherent to selection-driven differences in efficiencies, as well as “priming” of the immune system through existing spacer sequences that may provide partial resistance, 41,44,46,52 or that recruit the Cas machinery to partially complementary sequences. 45,53 A recent study showed that type I spacer acquisition can be replication-dependent, and that stalled replication forks yield DNA breaks that provide spacer acquisition hotspots. 38 This helps to promote spacer acquisition from plasmids or phages (which replicate especially prodigiously) as opposed to the host chromosome. Type II systems appear to be less inherently discriminatory against the initial acquisition of spacers from the host chromosome; instead, the ability of self-derived spacers to kill the cell because of chromosome interference leads to the immediate loss of self-derived spacers. 51
CrRNA biogenesis
Once immune markers have been integrated as spacers in CRISPR arrays, their sequence information must be rendered available to the effector machinery of the system to provide resistance against infections. This is achieved through the biogenesis of crRNA guides, each of which contains a unique sequence that drives the specific targeting of complementary nucleic acids.
11,18,54,55
First, the CRISPR repeat-spacer array is transcribed into a pre-crRNA that is processed into mature crRNAs by recognition and endonucleolytic cleavage of CRISPR repeat sequences or structures. The resulting crRNAs contain part or all of the spacer sequence, as well as part of one or both flanking CRISPR repeats.
1,2
The palindromic nature of most CRIS
Generally, CRISPR locus transcription is leader-dependent, constitutive to at least some extent so as to enable ongoing surveillance, but inducible by stress and phage exposure 56,57 to promote a rapid and efficient response to infection. Promoter elements have been identified in leader sequences, 43,50 as well as within CRISPR repeats, 58 that drive locus transcription and processing. Although the complete locus may be transcribed, studies have shown that there are biases in relative crRNA amounts across the locus, and that the first few spacers located at the leader end of the locus are often more abundant in the cell. 59 Intuitively, there is a greater evolutionary need to be resistant against invasive elements that were recently encountered, as opposed to those that were repelled in the more distant past. 60
Interference
Invasive element recognition and interference is based on sequence-specific binding and cleavage by the crRNA-Cas ribonucleoprotein complex. Mechanistically, each crRNA guide forms an interference complex with Cas proteins that drive endonucleolytic attacks on sequences that are complementary to the spacer-derived portion of the guide RNAs, as defined by Watson–Crick pairing between the crRNA and the target nucleic acid (Fig. 1C). Though most targets are dsDNA molecules, 12,16,19,20,29,33 some CRISPR/Cas systems have the ability to target complementary ssRNA. 27,29,31,32,61 –65 DNA-targeting CRISPR/Cas systems have evolved specific requirements for sequences adjacent to the protospacer (e.g., PAMs, 14,15 or crRNA-target noncomplementarity 66 ), whereas RNA-targeting modules have no such requirements. 27,62 Following initial endonucleolytic attack(s), the target may be further degraded by secondary nuclease activities that ensure further target destruction and eradication of the exogenous elements. 33,67 –70 Like most immune systems, CRISPR-mediated immunity (at least those aspects that function at the DNA level) must preclude self-targeting, in this case at the encoding CRISPR locus, and the solutions to that potential problem are type-specific (see next two sections).
CRISPR/Cas Pathways and Mechanisms: Type I and Type III Systems
Two of the CRISPR/Cas families—types I and III—operate through large multiprotein complexes 2,71 –73 that load their cognate crRNAs and execute interference. There are a few other similarities between them, most notably the involvement of a Cas6 family member as the pre-crRNA processing enzyme 11,54,55 (Fig. 2, purple) (with a Cas5 ortholog occasionally substituting), 74,75 and the role of multiple subunits of a Cas7 ortholog 76 as the “backbone” of the complex (Fig. 2, green). 2,71 –73 These and other similarities between type I and type III systems have been interpreted as signs of common molecular ancestry. 5
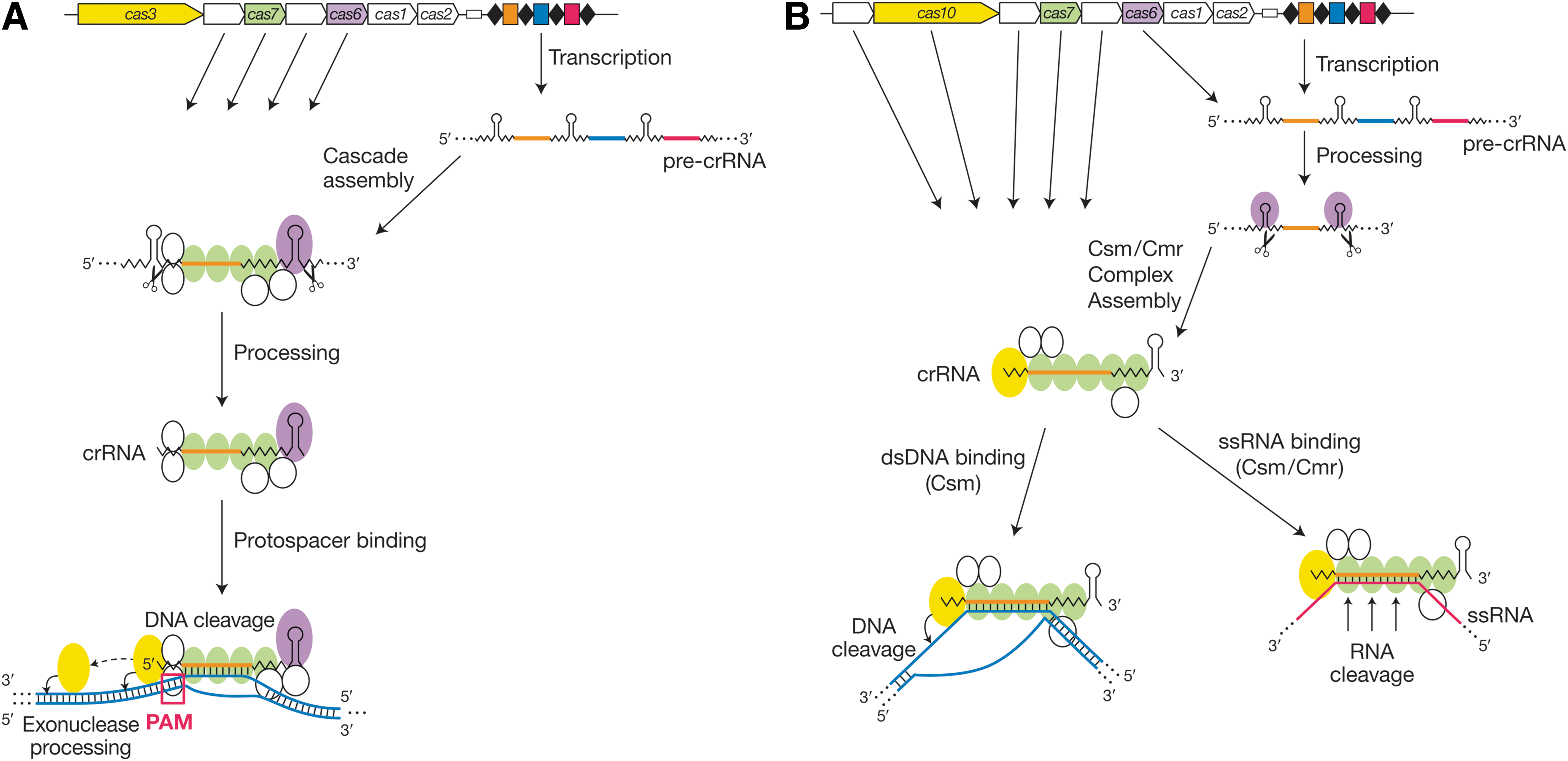
The type I and type III CRISPR interference pathways.
The type I-E machinery from E. coli is among the best-characterized CRISPR/Cas systems and is frequently invoked as a prototype. The type I assembly (“Cascade”) includes the Cas6 family member as an integral subunit, and the pre-crRNA is processed within the complex, 11 with a liberated, mature crRNA remaining bound following processing (Fig. 2A). 77 The complex then searches for its target site, probably by first recognizing its PAM and then testing for complementarity in the adjacent “seed” region (protospacer nts 1–5, 7, and 8). 53,78 –80 Type I systems, with their reliance on PAM elements during targeting, avoid recognizing the CRISPR locus itself by virtue of the absence of a PAM consensus from the CRISPR repeats. R-loop formation within long dsDNA-embedded protospacers requires negative supercoiling, presumably to facilitate NTP-independent unwinding. 33 The successfully established Cascade–protospacer complex is then bound by the type I signature protein, Cas3, which carries an HD nuclease domain as well as an NTP-dependent unwindase/translocase domain. The HD domain makes an initial nick near the PAM, and Cas3 then disengages from Cascade and uses its NTPase domain to translocate along the DNA, introducing additional nicks along the way (Fig. 2A, bottom). 33
The structure of crRNA-loaded E. coli Cascade has been solved at near-atomic resolution, both without 81,82 and with 83 a bound DNA target strand. The target is recognized by Watson–Crick base pairing, though with every sixth base of the protospacer splayed out and unpaired. This allows an overall geometry to the interaction that is non-double-helical, with repeated half-helical turns of duplex interrupted by unpaired bases that allow the DNA to cross back over the guide without wrapping around it. Distinct binding kinetics and structural signatures can be discerned at cognate versus near-cognate protospacers, 53 which is significant because the former trigger interference, whereas the latter can induce a mode of new spacer acquisition known as “primed” adaptation. 41,44,45,52 In this mode, the prior existence of a partial or complete match between a CRISPR spacer and a target protospacer promotes the acquisition of new spacers, allowing the system to update its immune specificity even after mutational evasion by the target. Primed adaptation, which requires Cascade and Cas3 as well as Cas1 and Cas2 (Fig. 1B), differs from naïve adaptation, which requires Cas1 and Cas2 only. 43
Type III systems are divided into two subtypes, III-A and III-B. Both of them employ a signature protein called Cas10, which is the largest subunit within the Csm (type III-A) and Cmr (type III-B) complexes (Fig. 2B). Both also employ a Cas6 ortholog for crRNA processing, 54,84 though in these cases the processing enzyme is not usually a stable component of the corresponding effector complex. In 2008, the type III-A system from Staphylococcus epidermidis was the first to be shown to operate at the DNA level, 12 whereas in 2009, the type III-B system from Pyrococcus furiosus was the first RNA-degrading system to be documented. 27 In neither subtype does target recognition and interference require PAM elements, 12,27,62,66 unlike the type I and II systems. In 2014, two observations initially deepened the mysteries behind the apparently disparate target specificities of the type III-A and III-B machineries. First, unexpectedly, the S. epidermidis type III-A DNA-targeting interference pathway was shown to depend upon ongoing transcription across the protospacer. 85 Shortly thereafter, Csm complexes from S. thermophilus and Thermus thermophilus were convincingly shown to harbor RNA cleavage activity 31,32 with 6 nt periodicity, analogous to activities shown for Cmr complexes. 27,61 –65,86 These observations have since been dramatically unified by the development and analysis of a combined transcription/interference system based upon the Csm machinery from S. epidermidis. 29 This allowed the direct detection not only of transcript degradation, but also of transcription-dependent DNA cleavage that depends upon specific Cas10 residues that are not required for RNA targeting. Conversely, the RNA hydrolysis activities of the Csm and Cmr complexes are catalyzed not by Cas10 but by the Csm3 29,31,32 and Cmr4 61,86 –88 subunits, respectively. Thus, the type III-A pathway has both DNA- and RNA-targeting capabilities (Fig. 2B, bottom, left and right branches, respectively), and it has been proposed that the well-established type III-B RNA-targeting activity is also accompanied by a DNA-targeting mode. 29,89,90
As with types I and II, when type III CRISPR/Cas systems engage in DNA interference, they must distinguish between self versus nonself to avoid attacking the crRNA-complementary DNA within the CRISPR locus. The lack of a PAM requirement in type III necessitates a distinct mode of discrimination. In the Csm pathway, differential crRNA repeat pairing between the host chromosomal sequence (in which the spacer DNA is flanked by a CRISPR repeat), as opposed to the lack of crRNA repeat pairing with the bona fide target DNA sequence, 29,66 allows the system to make the necessary self versus nonself distinctions and avoid autoimmunity. Intriguingly, DNA cleavage occurs very close to the sites of differential crRNA/DNA base pairing. 29 Almost nothing is known about spacer acquisition in type III systems. Adaptation mechanisms for these PAM-independent and RNA-targeting pathways are intriguing topics for future research.
CRISPR/Cas Pathways and Mechanisms: Type II Systems
Type II CRISPR/Cas systems constitute an outlier branch within prokaryotic adaptive immune systems, given their peculiar genetic underpinning and corresponding biochemical mode of action. Specifically, the multiprotein complexes driving crRNA biogenesis and interference in type I and type III systems are replaced by a single protein, Cas9, which is involved in all three fundamental steps of CRISPR biology. Furthermore, the crRNA biogenesis step relies on ancillary elements that are unique to type II systems. This has warranted the split of the three major types of CRISPR/Cas systems into two distinct classes, with type II systems serving as the model for the second class. Intriguingly, type II systems are uniquely present in bacteria, and are the least frequently observed CRISPR/Cas type.
Within type II systems, three distinct subtypes have been identified, II-A, II-B, and II-C, based on the occurrence and sequences of associated cas genes. 5,91 Specifically, in addition to the aforementioned universal cas1 and cas2 genes, the type II signature gene is cas9 (Fig. 3), which encodes the Cas9 DNA endonuclease that drives acquisition, 51,92 crRNA accumulation, 18,58,93 and interference. 16,17,19,20 Furthermore, in type II-A systems, csn2 encodes a protein involved in novel spacer acquisition. 10,51,92 In type II-B systems, cas4 replaces csn2, and neither is present in type II-C systems. Congruence in phylogenetic relationships between CRISPR repeats, cas1, cas2, and cas9 sequences have consistently identified three distinct type II subtypes. 13,94 The Cas9 signature protein varies in size between these various groups, with II-C subtypes usually bearing shorter orthologs of Cas9.

The type II CRISPR interference pathway. Pre-crRNA is usually driven from the leader region. A separate noncoding RNA called the tracrRNA is transcribed separately and anneals to the pre-crRNA repeats. Both strands of the resulting duplex are cleaved by the host factor RNase III, and the resulting crRNA also undergoes 5′-terminal trimming by unknown nucleases. The RNAs engage a single Cas protein, Cas9, and guide it to its target DNA via Watson–Crick base pairing with one of the two strands. Cas9 then cleaves both strands of the bound DNA.
The crRNA biogenesis process in type II CRISPR/Cas systems is unique, as it requires processing by RNase III, in conjunction with trans-encoded CRISPR RNA (tracrRNA) and in addition to the aforementioned Cas machinery and CRISPR repeat-spacer array (Fig. 3). Specifically, the tracrRNA includes an antirepeat segment that is partially complementary to the crRNA sequence, and it forms a dual-RNA complex that allows processing of the pre-crRNA into mature crRNA. 18,93 The resulting mature crRNA–tracrRNA–Cas9 complex contains a short crRNA that is composed of ∼20–24 nt derived from the 3′ end of the CRISPR spacer, as well as ∼20–24 nt derived from the 5′ end of the CRISPR repeat (Fig. 4A). The first pre-crRNA processing step occurs within the CRISPR repeat sequence, to define the 3′ edge of the crRNA. A subsequent 5′ trimming step (executed by unknown nucleases) occurs within the CRISPR spacer sequence, to define the 5′ edge of the crRNA. Cas9 is essential for crRNA accumulation in cells, 18,58 but it is not yet proven whether this reflects an active role for Cas9 in processing, 93 postprocessing crRNA stabilization, or (perhaps most likely) both.
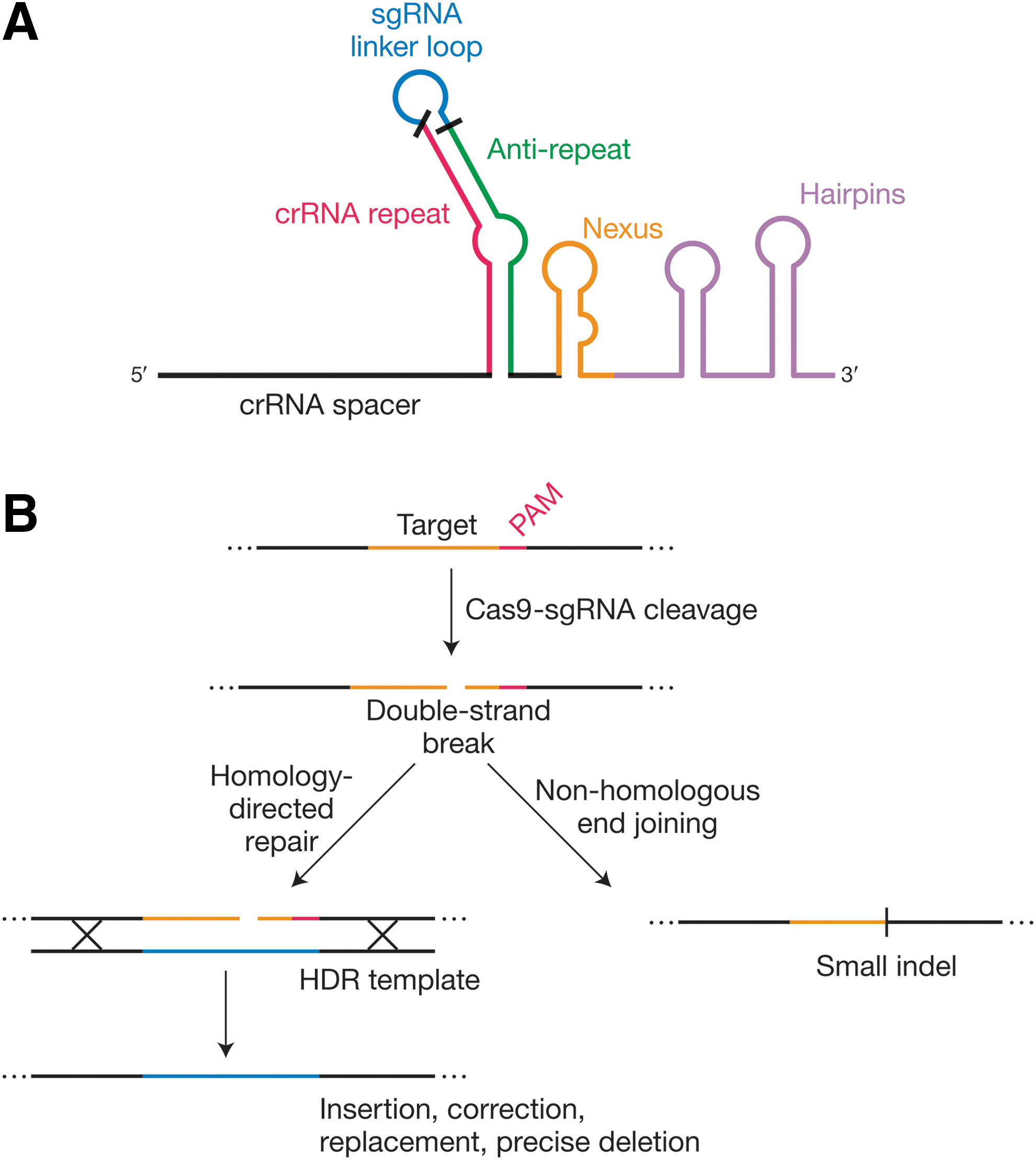
Genome editing by engineered Cas9 systems.
The crRNA–tracrRNA–Cas9 complex drives complementary target dsDNA binding in a PAM-dependent manner. 95 As with the type I systems, the absence of a PAM consensus from type II CRISPR repeats prevents the targeting of the CRISPR locus itself. Once Cas9 recognizes the PAM, flanking DNA is interrogated for complementarity to the loaded RNA guide sequence. 79,95 Subsequently, cleavage of the target nucleic acid occurs by dual nicking involving RuvC and HNH motifs that each cleave one strand of the target DNA to generate a blunt DSB precisely near the PAM-proximal end of the proto-spacer within an R-loop structure (Fig. 3, bottom). 16,17,19,20 The precision of the blunt cleavage relies in part on a ruler-anchor mechanism defined by the flanking PAM, 96 and occurs within the “seed” sequence, three base pairs upstream of the PAM-proximal end of the protospacer. 16,19,20
Type II systems are arguably the most studied and best characterized CRISPR/Cas systems, with model systems derived from Streptococcus thermophilus (Sth), Streptococcus pyogenes (Spy), and Neisseria meningitidis (Nme) as prototypes. Early work in Sth first showed adaptive immunity 10 ; characterized the PAM 14,97 ; demonstrated endonucleolytic cleavage of target DNA 16 ; provided proof of concept for engineering, reprogramming, and heterologous transfer of CRISPR targeting 17 ; and established a requirement for the HNH and RuvC active-site residues for Cas9's interference function. 17,19 The tracrRNA, 18 the role of RNase III, 18 the requirement of the tracrRNA for dsDNA cleavage, 20 and the ability to fuse the dual RNAs into a single-guide RNA 20 were first shown in Spy, contributing to the widely popular use of SpyCas9 for genome-editing applications. With regard to type II-C systems, the Nme model has been used to show an alternative crRNA biogenesis pathway, which relies on promoters embedded within CRISPR repeats, an alternative transcription direction pattern, and an ability to function even in the absence of RNase III pre-crRNA processing. 58 These features may be applicable across multiple or even all type II-C systems.
More recently, structural studies have provided insights into the biochemical underpinning of guide RNA–Cas9–target DNA interactions, 72,79,96,98 –100 and set the stage for engineering of Cas9 variants with increased affinity, specificity, efficiency, and overall functionality. The Cas9 protein is not only the signature element for type II CRISPR/Cas systems, but is actually the cornerstone of all three steps of CRISPR biology in these systems, including acquisition of novel spacers, 51,92 accumulation of crRNAs, 18,58 and interference. 10,16,17,19,20 This illustrates the broad functional potential of this protein, and reflects the multiple domains that it carries, including those that direct guide RNA binding, target (PAM) DNA binding, dual nicking, and likely more.
CRISPR-Based Genome Editing
In the years leading up to the discovery of the CRISPR pathway 10 and its core features, 1,2 considerable excitement arose about the potential of zinc-finger nucleases (ZFNs) and then TAL endonucleases (TALENs) to enable locus-specific, user-targetable genome editing. 101 Tremendous advances were made on both fronts, but the challenges, costs, inconsistencies, inefficiencies, and modest throughput inherent in these “hard-wired” technologies, which generally require the design, expression, and validation of an entirely new pair of polypeptides for every locus to be targeted, limited their adoption. It was in this context that the potential of a natural, 10 crRNA-guided 11 DNA-targeting 12,16 system, with an invariant Cas protein “hardware” that could be programmed (and re-programmed) by a swappable crRNA “software,” was first recognized and postulated 12 and then, over the next several years, developed, 19,20 further foreshadowed, 102 and applied. 21 –25,103 ZFNs and TALENs are still in use and are even in advanced clinical trials, but the simplicity, efficacy, and economy of CRISPR/Cas9 have vaulted it to most-favored-technique status for most user-definable genome-editing and DNA-binding applications. The revolutionary nature of CRISPR/Cas9 genome engineering is now clear, 26,104 as described in recent comprehensive reviews 3,4 and throughout this issue.
At its heart, the basic CRISPR/Cas9 genome engineering technology reflects its natural function: the use of an RNA guide to identify a complementary dsDNA target sequence (next to a PAM), leading to DNA cleavage at that site (Fig. 4B). In eukaryotic cells, however, the most frequently desired consequence is not the wholesale degradation of the cleaved DNA molecule, but rather the repair of the Cas9-induced break, either by nonhomologous end joining (NHEJ) or homology-directed repair (HDR) 21 –25 (Fig. 4B, right and left branches, respectively). The former often leaves behind small insertions or deletions (indels) that can disrupt the reading frames of protein-coding genes, leading to loss of gene function. Larger deletions 105,106 and even inversions 107 can be achieved by the simultaneous induction of multiple breaks via multiplexed guides. HDR, in contrast, results in the replacement of the targeted sequence with another, usually through the purposeful provision of a repair template designed by the user. Accordingly, HDR can be used to revert unwanted mutations, generate new alleles, insert or fuse useful domains, and introduce transgenes. 3,4 The platform's utility goes much further: mutational inactivation of Cas9's RuvC or HNH domain converts it into an RNA-guided nickase, and inactivation of both domains converts it into an RNA-guided DNA binding (but not cleaving) platform. In the latter case, the fusion or tethering of effector domains can be used to evoke a wide range of locus-specific outcomes (transcriptional activation or repression, chromatin modification, forced looping, and many others) as well as experimental benefits such as fluorescence visualization or affinity tagging for physical isolation. 3,4
The CRISPR/Cas genome engineering system that is best developed and most frequently applied to date is, by far, SpyCas9. 3,4 However, the continued development of additional Cas9 proteins that are orthogonal (i.e., are guided by nondistributive and incompatible guides) 108 –110 allows for the concurrent use of multiple systems for distinct combinations of these purposes. Targeting scope and deliverability will be expanded and modulated as shorter Cas9 proteins 109,111,112 associated with diverse PAM sequences, and perhaps additional functional distinctions, are characterized. Regarding the RNA guides, separately transcribed crRNAs and tracrRNAs are effective in genome editing, 22 but the system can be simplified further thanks to the development of sgRNA technology 20 (Fig. 4A), in which the native RNase III–crRNA–tracrRNA–Cas9 four-component system is engineered into an sgRNA–Cas9 two-component system. This represented a key tipping point in the technology's development, as reflected in the currently pervasive use of sgRNAs rather than dual guides. Our ability to optimize these systems is benefitting from our ever-increasing understanding of the functional anatomy of Cas9 and sgRNA domains. 98 –100,108 Finally, the potential for off-target effects is broadly acknowledged (and sometimes overstated) 113 ; several routes toward improving the target specificity of Cas9 have already been reported, 114 –119 and more are undoubtedly in the pipeline.
Now that the genome-editing capabilities of CRISPR/Cas9 platforms are so well established in academic and industrial laboratories, one of the most difficult and important directions will be to develop it for clinical treatment of inherited and infectious diseases, as well as other promising applications such as cancer immunotherapy.
Perspective and Conclusions
Although the CRISPR craze has yielded tremendous scientific progress and critical technological advances, it is important to keep in mind that the sgRNA–Cas9 technology is only 3 years old, and that notwithstanding current progress and momentum, we are yet to fully unleash the potential of these tools. Indeed, the frenetic and ever-increasing pace of Cas9-oriented scientific publications illustrates the ease with which this revolutionary technology can be both implemented and further engineered to carry out a vast array of applications across disciplines and in a wide diversity of organisms and cell types. The CRISPR craze has reached beyond the realms of the scientific literature into the global media, with noteworthy coverage in both print and electronic outlets. Beyond academic research and the media, the most significant impact of CRISPR may well turn out to be in industry, with unprecedented levels of interest and investment from multiple distinct business segments, including pharmaceuticals and biotech, as well as covering the food supply chain from agriculture to livestock to other food products. With the brightest lights shining on translational medicine in general, and gene therapy in particular, the recent advances and current momentum have established a very promising basis and opened up a strategic path toward the clinic. Early industrial successes at Danisco/DuPont and elsewhere illustrate the speed with which CRISPR can be commercially exploited, and is a reminder of the full span of development between 2005 and 2013. 120
Nevertheless, gaps and issues are being and remain to be addressed, including technical, legal, and ethical dimensions. With regard to technical challenges, the community is focused on the practicalities of delivering the genome-editing machinery to target tissues, assessing and minimizing off-targeting by Cas9, rationalizing guide design for optimal targeting and cleavage efficiency, expanding the Cas9 and target sequence space, and rebalancing DNA repair mechanisms toward HDR for surgical genome editing and the replacement of undesirable alleles. As for legal concerns, the tremendous economic potential has already yielded a complex and fascinating intellectual property situation that is unfolding before us, and will be instrumental in allowing key players to license foundational patents and enjoy freedom to operate in developing this technology into viable, valuable, and promising commercial products. Lastly, with regard to ethical concerns, as with any powerful technology, the limiting factor lies in the users rather than the technology. It is important to bear in mind that, historically, scientists, regulating bodies, and industrial groups have frequently succeeded in cautiously assessing real and perceived concerns for the full spectrum of stakeholders. We are optimistic that the CRISPR and gene therapy communities, together with policy makers, will establish principles and guidelines under which this powerful technology may be best exploited and implemented. This is too concerning and visible not to be properly addressed.
Lastly, as we evaluate the state of the CRISPR field, it is important to note that the bacterial origins of the CRISPR-fueled genome-editing revolution are a reminder of the wealth of biological gold that lies buried within microbial genomes. Discoveries in this area are ongoing (e.g., the DNA-guided DNA-cleaving roles of some bacterial Argonaute proteins, 121,122 the increased repurposing of the type I and type III CRISPR/Cas systems, 62,86,123 –125 and others) and will continue to expand the molecular genetic toolbox. Looking forward, we are confident that the current body of work and the present momentum are just the beginning of an even longer and more transformative CRISPR era that will forever change both laboratory and clinical practice.
Footnotes
Author Disclosure
E.J.S. is cofounder, shareholder, and advisor of Intellia Therapeutics, and is an inventor on a pending patent on Nme Cas9 technology. R.B. is cofounder, shareholder, and advisor of Intellia Therapeutics, and is director, shareholder, and advisor of Caribou Biosciences. R.B. is also an inventor on several patents related to CRISPR/Cas systems and their various uses.