
Abstract
Perinuclear retention of viral particles is a poorly understood phenomenon observed during many virus infections. In this study, we investigated whether perinuclear accumulation acts as a barrier to limit recombinant adeno-associated virus (rAAV) transduction. After nocodazole treatment to disrupt microtubules at microtubule-organization center (MT-MTOC) after virus entry, we observed higher rAAV transduction. To elucidate the role of MT-MTOC in rAAV infection and study its underlying mechanisms, we demonstrated that rAAV's perinuclear localization was retained by MT-MTOC with fluorescent analysis, and enhanced rAAV transduction from MT-MTOC disruption was dependent on the rAAV capsid's nuclear import signals. Interestingly, after knocking down RhoA or inhibiting its downstream effectors (ROCK and Actin), MT-MTOC disruption failed to increase rAAV transduction or nuclear entry. These data suggest that enhancement of rAAV transduction is the result of increased trafficking to the nucleus via the RhoA-ROCK-Actin pathway. Ten-fold higher rAAV transduction was also observed by disrupting MT-MTOC in brain, liver, and tumor in vivo. In summary, this study indicates that virus perinuclear accumulation at MT-MTOC is a barrier-limiting parameter for effective rAAV transduction and defines a novel defense mechanism by which host cells restrain viral invasion.
Introduction
C
The microtubule-organization center (MTOC) is a subcellular structure at perinuclear region, from which microtubules (MTs) are nucleated to form a radial filamentous network with minus ends anchored at MTOC and plus end reaching cell surface area. 2 Because of these features, MTs and MTOC, as a trafficking center, coordinate the transport of intracellular molecules and organelles between the perinuclear region and other areas of a cell. 2 For instance, intact MTs are required for the nuclear import of several transcription factors (e.g., p53, NF-kB, pRb), and on other hand, these structures are required to sequester other proteins (e.g., c-myc, MIZ-1, smad3) in the cytoplasm to block their nuclear entry. 3 –7 Numerous studies have also shown that cellular misfolded proteins are transported on MTs toward the MTOC region, where these misfolded proteins are then sequestered or degraded by proteasomes and lysosomes. 8,9
Strikingly, during the early stage of infection, many incoming viruses are also delivered to and retained at a perinuclear site after entering the cells. 10 –16 It has been suggested that this perinuclear site is co-localized with the MTOC region and, as observed for cellular proteins, that transport of these viruses along with MTs is facilitated by dynein motors. 9,17 Although this perinuclear retention of incoming virions seems to be a common phenomenon among many viruses, especially those that enter the nucleus, it remains unknown whether this accumulation is beneficial or inhibitory to viral infection. 9 Previous studies have shown that several viruses, including rotavirus, vaccine, and HSV, disrupt MTOC during early infection stage for reasons that have not been identified. 18 –22 Despite the lack of understanding of the role of such perinuclear retention on viral infection, these observations raise an intriguing hypothesis that MTOC region serves as a subcellular barrier to restrict access of the incoming virions to their replication site (nucleus in this study) during the early stage of viral infection.
On the other hand, during late infection stage, several viruses, such as HIV, HSV, poliovirus, and norovirus, have been suggested to exploit this perinuclear accumulation to concentrate newly synthesized viral components through MTs in order to facilitate late infection events, including virion replication, assembly, or egress. 23 –26 Classical virology assays usually study replication-competent viruses and measure the production of viral progeny as the readout, which is the sum of the total effects of all events in viral infection with these viruses. Given the potential involvement of MTs and MTOC throughout the entire viral infection, this method is unable to precisely dissect the perinuclear retention of incoming virions during early infection stage. Furthermore, viral replication and newly synthesized viral proteins will also confound virion distribution assays that use antibodies. 12,15
Recombinant viruses used as gene therapy vectors (designated as viral vectors) are purposely designed to exploit only the early infection steps, including delivering and expressing viral genomes, and are deficient in late infection events, including viral replication and assembly because of the deletion of critical viral proteins. 27 As a result, these viral vectors allow researchers to more precisely separate the early infection steps from the late events, offering a promising opportunity to specifically identify the mechanisms and roles of perinuclear retention on incoming virions. Adeno-associated virus (AAV) is a member in the family Parvoviridae and recombinant AAV (rAAV) is currently used as a gene therapy vector because of many desirable traits in gene delivery. 27 As a gene therapy vector, rAAV does not contain any viral DNA except for two inverted terminal repeats (ITR) flanking the exogenous transgene. 27 Therefore, rAAV only delivers and expresses transgenes in cells but is incapable of replicating and producing viral progeny. This attribute makes rAAV a useful model to specifically investigate virus–host interplay during the early stage of viral infection. Like many other viruses, its trafficking pathway typically starts with receptor-mediated endocytosis for cell entry, followed by cytoplasmic trafficking assisted by the endosomal routing system and MT network, and ends with nuclear entry and uncoating for successful transduction. 27 –29
In our previous study, using pharmacological agents, live-cell imaging, and flow cytometry analysis, we have demonstrated that rAAV2 exploits MTs for rapid cytoplasmic trafficking in endosomal compartments unidirectionally toward the perinuclear region and that rAAV2 transduction is reduced when the MT network is disrupted at early time point of rAAV infection. 30 In this study, we investigated the role of rAAV perinuclear retention on the effective transduction, and have further explored the detailed mechanism. A sensitive and reliable fluorescence imaging platform allowed us to examine viral trafficking in detail over time, which led us to the observation that the majority of viral particles finish cytoplasmic trafficking and localize at perinuclear region by 6–8 hr postinfection (p.i.). Manipulating host cells and viral particles using pharmacological interventions around 6–8 hr p.i. allowed us to efficiently investigate the underlying mechanism of perinuclear retention as well as the corresponding impact on viral infection.
Materials and Methods
rAAV2 production, purification, and labeling
HEK-293 cells were used to produce rAAV2 as described previously. 31 Briefly, cells were transfected with three plasmids: pXR2 (wt or with BR mutations), pXX680, and a plasmid containing the reporter transgene (GFP or Luciferase) flanked by two ITRs. At 60 hr after transfection, cells were collected and nuclei were isolated using hypotonic buffer and Kontes homogenizer. rAAV2 particles were recovered by resuspending the nuclear pellet in PBS with 0.5% deoxycholate (DOC) and then sonicating for 1 min. Highly pure virus was then retrieved as described previously. 32 Briefly, after DNase treatment, the virus suspension was subjected to one round of cesium chloride (CsCl) step gradient density fractionation and another round of fractionation using a continuous CsCl gradient. Determination of peak viral fractions, dialysis of virus, and measurement of viral titers by quantitative PCR (qPCR) were performed as previously described. The infectivity of AAV was determined to be approximately 1 transducing unit per 100 particles.
rAAV2 was covalently labeled with the Cy5 fluorophores as described in manufacturer's protocol with slight modification. Briefly, purified rAAV2 were incubated for 2 hr at 4°C in PBS with a 20-fold molar excess of Mono-NHS-Cy5 (GE Healthcare) over capsid protein units. Free dyes were removed from labeled viral particles by dialysis against PBS containing 5% sorbitol and viral solutions were stored at −80°C as small aliquots. Labeled viral titers were determined by both dot blot 31 and qPCR. Infectivity of the viral particles was determined by GFP reporter gene assay.
Cell culture, viral infection, and drug treatment
All cells (American Type Culture Collection) were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) with 10% FBS in 5% CO2 incubator. Cells were passaged every 2–3 days for up to 10 passages, when new aliquots of frozen cells were recovered from liquid nitrogen.
For pulse infection, cells were seeded on 12 mm glass coverslips at 24 hr before infection. The next day, after incubation in DMEM containing 20 mM HEPES at 4°C for 5 min, cells were inoculated with Cy5-rAAV2 (∼5,000 vgs/cell) or nonlabeled rAAV2 (∼1,000–5,000 vgs/cell) at 4°C for another 40 min. Cells were then washed with PBS to remove unbound virions and transferred to a 37°C incubator (regarded as 0 hr p.i.). Pharmacological drugs were added at 6–8 hr after pulse infection unless otherwise indicated. The concentrations of drugs were 30 μM nocodazole, 25 μM colchcine, 10 μM taxol, 10 μM rhizoxin, 10 μM maytansine, 2 μM MG132, 20 μM ALLN, 10 μM H1152, and 10 μM cytochalasin D. The drugs were maintained in the culture for approximately 3 hr unless otherwise indicated.
Flow cytometry and immunofluorescence
To evaluate viral transduction, flow cytometry analysis was used to measure the mean fluorescence intensity (MFI) of GFP expression. Since slight cell toxicities from the drug treatment were observed (data not shown), to exclude the potential effect of dead cells on the viral transduction measurement, we washed the cells in each well three times with PBS before harvesting for flow cytometry analysis, removing any floating or loosely attached cells. Either Trypan blue exclusion or 7-AAD exclusion assays was used to ensure cell viability over 95% for flow cytometry analysis.
For immunofluorescence, cells were washed with PBS and then fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature (RT). The cells were then permeabilized with 0.2% Triton X-100 in PBS for 5 min at RT. After blocking with immunofluorescence buffer (IFB) (5% normal goat serum in PBS containing 0.05% Tween-20) for 1 hr at RT, the cells were incubated with primary antibody to detect tubulin (Ab6161; rat monoclonal from Abcam Inc.), Golgi (Ab24586; mouse monoclonal from Abcam Inc.), and/or rAAV capsid (A20 mouse monoclonal antibody) diluted in 50% IFB overnight at 4°C. The cells were then incubated with secondary antibody, diluted 1:2,000 in 50% IFB (antimouse Alexa-Fluor 488; Molecular Probes) for 1 hr at RT. After six washes with PBS, coverslips were mounted cell side down on glass slides with mounting medium (prolong antifade gold with DAPI [4′,6′-diamidino-2-phenylindole]; Molecular Probes). After images were acquired using confocal microscopy, the existence of perinuclear accumulation in a cell in each image was examined by human eyes.
Nuclear isolation and viral genome quantification
Nuclei were isolated from cell fractionations as previously described, 31 with minor modifications allowing for viral infection. After incubation with rAAV and drugs, cells were washed three times with ice-cold PBS and harvested by centrifugation at 500 g for 10 min. The cell pellet was divided into two aliquots: one for direct viral genome extraction and the other for nuclei isolation. One aliquot was resuspended in hypotonic buffer and homogenized on ice using a Kontes homogenizer to the point where ∼90% of cells were broken with nuclei remaining intact. The homogenate was spun at 500 g for 10 min to separate the nuclei from the cytoplasmic components. The purity of nuclear fraction was assured by phase-contrast microscopy and immunoblotting using antitubulin and antilamin A/C antibodies, 33 and no cytoplasmic protein tubulin was detected in purified nucleus (data not shown).
rAAV genomes were recovered from both whole cells and nuclear fraction using DNeasy Blood and Tissue Kit (Qiagen Inc.) according to the manufacturer's protocol. The copy number of rAAV genomes was determined by qPCR on a LightCycler 480 using SYBR Green (Roche) as described for viral titering. 31
Animal studies
Housing and handling of the BALB/c mice and SCID mice used in the current study were carried out in compliance with National Institutes of Health guidelines and approved by the IACUC at the University of North Carolina–Chapel Hill. To grow xenografted breast cancer tumors, 106–107 MDA-231 breast cancer cells were injected into the mammary fat pad of SCID mice. When the tumor size was about 0.5 cm in diameter, the mice were used for intratumoral injection. Recombinant AAV2 vectors packaging luciferase or GFP transgenes were administered through the intravenous route (2 × 1010 vgs into tail vein) in a volume of 200 μl PBS or the intratumoral route (1 × 1010 vgs). DMSO or nocodazole (2 mg/kg) were administered via intraperitoneal or intratumoral injection at about 10–12 hr after viral injection. Bioluminescence from luciferase expression was visualized by using a Xenogen IVIS100 imaging system (Caliper Lifesciences) after intraperitoneal injection of luciferin substrate (120 mg/kg of body weight; Nanolight). Image acquisition and analysis were carried out using Living Image software. Quantitative data are based on values from 3 to 4 mice per group.
For vector infusions into mouse brain, animals were anesthetized with isoflurane and placed into a stereotaxic frame. Using a 32-gauge stainless steel injector and a Fisher Scientific infusion pump, mice received 1 μl per hemisphere (total of 5 × 108 vgs) into the striatum over 15 min. The injector was left in place for 3 min after infusion to allow diffusion from the site of injection.
To image viral trafficking in mouse tissues, Cy5-rAAV2 was administrated through intrabrain, intrahepatic, or intratumoral routes. DMSO or nocodazole were administrated through the intraperitoneal route at about 12 hr after viral inoculation. At 1 or 12 hr after vector injection or 4–6 hr after nocodazole injection (16–18 hr after viral injection), animals received an overdose of pentobarbital (100 mg/kg intraperitoneally) and were perfused transcardially with ice-cold 100 mM sodium PBS (pH 7.4), followed by 4% PFA in phosphate buffer (PB) (pH 7.4). After tissues were postfixed for 24 hr at 4°C in PFA, 34 10–20 μm coronal sections were cut using a cyrosection microtome and then the slides were directly sealed with mounting medium (Prolong Antifade Gold with DAPI [4′,6′- diamidino-2-phenylindole]; Molecular Probes).
Quantitative 3D microscopy
Hela cells pulse infected with Cy5-rAAV2 were fixed with PFA and mounted to glass slides as described above. The distribution of viral particles in Hela cells was examined using a Zeiss LSM710 laser scanning confocal microscope equipped with a Plan-Apochromat 63×/NA 1.40 oil objective. Stacks of 20–30 focal planes were captured at 0.31 μm z-intervals through the depth of the cell. 3D images of the cells were reconstructed using the Z-stacks. All images were acquired at pixel dimensions of 0.13 μm × 0.13 μm × 0.31 μm (X, Y, Z) to fulfill the Nyquist sampling.
Deconvolution was performed by the AutoDeblur software (Media Cybernetics Inc.) using iterative and constrained algorithms as described previously. 32 The procedure started with a theoretical PSF derived from the actual settings of Zeiss710 confocal microscope, including NA of the microscope objective, refractive index of the medium, excitation wavelength, emission wavelength, confocal pinhole radius, pixel size, z-axis interval, and microscope type (i.e., wide field, confocal). A new adjusted adaptive PSF derived from the previous deconvolution round was used to generate the next adaptive PSF that fit the real imaging data better than the previous one (termed as one iteration or deconvolution round). Twelve rounds of iteration were used to deconvolve all the confocal images in this study.
All deconvolved image stacks were processed using the IMARIS software package (Bitplane AG, Zurich, Switzerland) for visualization and quantification as described previously. 32 Briefly, each deconvolved image stack was reconstructed using a volume-rendering module and smoothed by a 3D-median filter. Subsequently, an isosurface-rendering module was applied through thresholding by the fluorescence intensity that was slightly higher than background. For Cy5-rAAV2, the isosurface rendering was thresholded at the fluorescence intensity of 2000 a.u. (the upper boundary of background). For DAPI, isosurface rendering was thresholded at the fluorescence intensity of 3200 a.u. Parameters (volume, MFI, TFI) for these isosurface-coated Cy5-rAAV2 objects were extracted from the IMARIS program and analyzed to determine the localization of particles as described previously. 32
Negative staining and electron microscopy
Purified and dialyzed virus particles in 1× PBS were pipetted onto a glow-discharged copper grid. The grid was washed twice with water and then stained with 2% uranyl acetate. Electron microscopy images were taken with a LEO EM 910 transmission electron microscope at various magnifications.
Transient transfection of reporter plasmid and siRNA
For transfection of Hela cells with reporter plasmids, Hela cells were plated at a density of 4 × 105 cells/well and 24 hr later were transfected with TR-CMV-EGFP reporter plasmids using PEI. For each well, 500 ng DNA was mixed with 5 μl PEI (1 mg/ml) in 100 μl of serum-free DMEM and incubated at RT for 10 min before addition to cell medium. Culture medium was refreshed at 6 hr after transfection. At 16–20 hr after transfection, cells were treated with DMSO or other drugs as indicated. At about 20 hr after drug treatments, cells were harvested for flow cytometry analysis of GFP expression as indicated above. The control is the data without any treatment after transfection. The effect of treatments on transgene expression was calculated as 1× data of treatment/positive control.
Knockdown of RhoA was obtained by double transfection of siRNA (Dharmacom, Thermo Scientific) using Nucleofector (Lonza Inc.) performed with an Amaxa Electroporator according to the manufacturer's protocol. Knockdown efficiency was verified by Western blot using RhoA antibody (Abcam Inc.). Efficient knockdown was achieved two days after the first siRNA transfection. After verifying knockdown, approximately two days posttransfection, cells were processed with pulse infection and drug treatments as described above. Viral transduction was analyzed using flow cytometry.
Statistical analysis
Experiments were performed in triplicates and repeated at least three times independently, unless otherwise noted. For studies comparing quantification of luciferase or GFP expression in one treatment group to the corresponding control group, the Student t-test of the mean expression value was performed. For studies comparing quantification of luciferase or GFP expression in more than two treatment groups to the same control group, the one-way analysis of variance (ANOVA) was performed to test the global hypothesis that all mean values are the same across groups. If the global hypothesis is rejected, Dunnett's test was used to compare the mean of each treatment to the mean of control group simultaneously. Luciferase or GFP expression was presented as mean ± SD, and all tests were considered significant if p < 0.01, which was indicated by *.
Results
AAV retention in MOTC
AAV is a nonenveloped single-stranded virus and has successfully been used as gene delivery vehicle in clinical trials. To elucidate the role of MTOC in rAAV transduction, we first investigated the intracellular trafficking of rAAV2 over time using viral particles labeled with Cy5. With our optimized protocol, we were able to visualize a single viral particle, and the labeled virions remained as infectious as unlabeled particles.
32
Specifically, homogeneous labeling of virions was achieved by confocal microscopy of labeled particles on glass coverslips (Supplementary Fig. S1A, left; Supplementary Data are available online at
To study the intracellular trafficking of rAAV, we synchronized particle trafficking using “pulse infection,” in which cells are incubated with virions at 4°C for 40 min and unbound virions are then washed away before cells are transferred to 37°C for subsequent viral infection. After pulse infection, we studied the distribution of rAAV particles at several time points over 12 hr (Fig. 1A). During the first 2–4 hr p.i., the majority of viral particles were scattered in the cytoplasm and trafficking toward the perinuclear region. At 4 hr p.i., the majority of viral particles had finished their cytoplasmic trafficking and localized at the perinuclear region in approximately 30–40% of cells. At 8 hr p.i., perinuclear accumulation was observed in over 80% of cells and persisted for over 6 hr, suggesting retention of viral particles at this perinuclear site (arrows in Fig. 1A). To specifically investigate the role of the perinuclear retention, we focused on the time period between 6 and 8 hr after pulse infection, a time point at which, in majority of cells, most virions have finished cytoplasmic trafficking and remain at the perinuclear region.
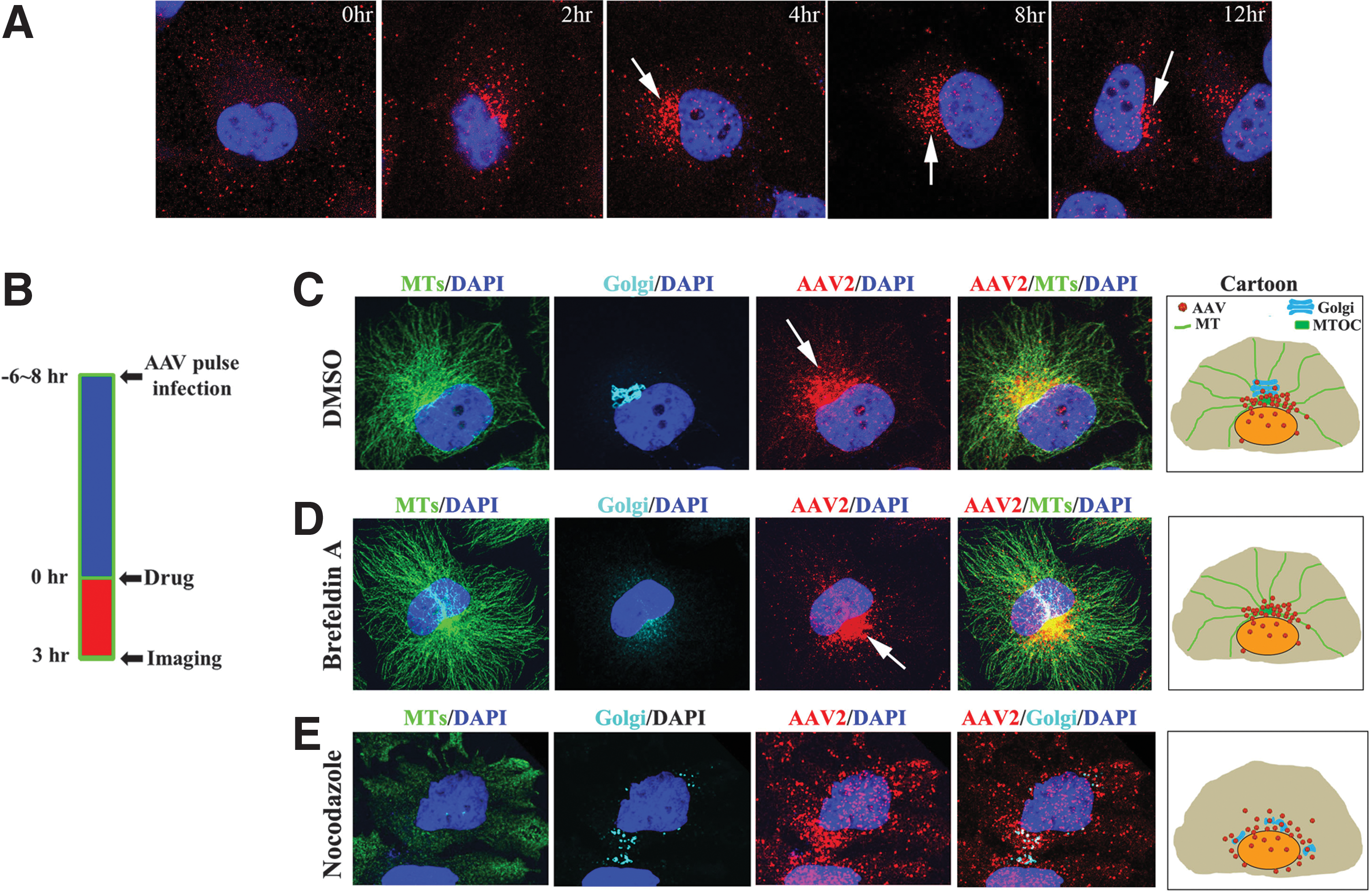
Perinuclear retention of rAAV by MT-MTOC. Hela cells were fixed to examine the distribution of Cy5-labeled viral particles at various time points after pulse infection. Nuclei were stained with DAPI (blue).
Previous studies have co-localized perinuclear rAAV particles to the Golgi apparatus, 36,37 and others have suggested that these virions may co-localize with MTOC. 38 As the perinuclear region is highly crowded and the resolution of classical fluorescent microscopy is only about 0.3 μm in lateral, images solely generated by such fluorescent microscopy alone could falsely co-localize viral particles with many cellular organelles at perinuclear region, namely, Golgi, MTOC, and ER. 38 To effectively discern which cellular structures at the perinuclear region co-localize with AAV particles and retain these virions, we used fluorescence microscopy and pharmacological interventions (Fig. 1B) to circumvent the above resolution limit issues. Specifically, if rAAV particles do co-localize to and are retained by a particular structure at the perinuclear region, we shall be able to predict that (1) the viral accumulation will be ablated upon the disruption of that structure by pharmacological agents, and (2) the majority of virions will still co-localize with that structure after release from the perinuclear region upon disruption of MTs and MTOC.
Using several antibodies, we demonstrated that rAAV particles can be co-stained with markers for Golgi apparatus and MT-MTOC (peri-MTOC MTs) at the perinuclear region (Fig. 1C). To distinguish whether virus localized to the Golgi or MT-MTOC, we used a panel of pharmacological reagents to disrupt these structures individually and visualized the localization of viral particles in each case. Upon Brefeldin-A treatment to specifically disrupt Golgi apparatus, perinuclear accumulation of AAV was not dispersed (Fig. 1D). In contrast, when MT-MTOC was fully disrupted by nocodazole, the perinuclear localization of AAV was ablated and few rAAV particles co-localized with dispersed Golgi cisternae (Fig. 1E). Although nocodazole treatment affects the Golgi apparatus as well, based on the result from treatment of Brefeldin A and nocodazole, these data suggest that rAAV is co-localized to and retained by MT-MTOC but not Golgi apparatus at the perinuclear region. This finding is consistent with previous studies that show that many other viruses traffic and localize to MTOC or MT-MTOC. 10,14,39,40
Disruption of MT-MTOC retention increases AAV transduction
To study whether the retention of AAV in MTOC affects rAAV transduction, we also used anti-MT drugs (nocodazol, colchicine, rhizoxin, and maytansine) to disrupt MTs as described in Fig. 2A. We infected 293 cells with AAV vector encoding GFP transgene. Eight hours later, drugs were added to the cells for 4 hr and washed out. The cells with GFP expression were analyzed by flow cytometry at 20 hr after nocodazole treatment (Fig. 2A). To exclude potential effects of the reagents on the second strand synthesis of viral DNA, self-complementary rAAV (rAAV2-CMV-GFPsc) was used in the transduction assay.

Disruption of MT-MTOC increases rAAV transduction.
Disruption of MTs by nocodazole treatment induced a 2–4-fold increase in viral transduction in multiple cell lines as measured by the expression of the transgene GFP (Fig. 2B). To rule out the possibility that the increased viral transduction was specific to nocodazole, a panel of anti-MT drugs (AMDs) was used (colchicine, rhizoxin, maytansine), all of which disrupt MTs. Similar increases in GFP expression were observed with all of the drugs (Fig. 2C). We also tested whether stabilization of MT-MTOC by taxol would reverse the effect of nocodazole, by comparing the viral transduction in cells treated with nocodazole alone or with nocodazole and taxol. We found that co-administration of taxol with nocodazole restored the enhanced viral transduction to the control level (Fig. 2D). Together, these results demonstrated that MT-MTOC disruption is able to increase rAAV transduction.
Increased transduction upon MT-MTOC disruption is independent of promoter activity, cell cycle arrest, and viral degradation
Next, we explored the underlying mechanism associated with the increase in viral transduction induced by MT-MTOC disruption. We envisioned three possible mechanisms for the increase in transgene expression: (1) MT-MTOC disruption leads to the activation of the promoter on the viral transgene cassette; (2) MT-MTOC disruption results in cell cycle arrest that, in turn, leads to increased viral transduction; (3) MT-MTOC disruption affects intracellular viral processing or trafficking. First, we employed in vitro plasmid transfection to examine the potential effect of MT-MTOC disruption on promoter activity. Our flow cytometry data demonstrated that MT-MTOC disruption by AMDs did not affect the reporter gene (GFP) expression from the viral transgene cassette, which was directly transfected into the cells using PEI (Fig. 3A). This excludes the possibility of AMD treatment affecting promoter activity (mechanism 1). To test the second possible mechanism, we measured the effects of AMD treatment on viral transduction in cells whose cell cycle was already arrested at S-phase by double thymidine block, a step preceding the M-phase block caused by AMDs. Our data showed that MT-MTOC disruption also induced an enhancement of viral transduction in these prearrested cells, suggesting that the increase in viral transduction was independent of cell cycle arrest induced by disruption of MT-MTOC (mechanism 2; Fig. 3B). Additionally, it was previously reported that intracellular degradation of rAAV limits viral transduction and its blockage using proteasome inhibitors resulted in dramatic increase in viral transduction. 41 It was also previously proposed that MT-MTOC facilitates the degradation of intracellular proteins by transporting these proteins to and retaining proteasome machineries at the perinuclear region. As a result, we tested if MT-MTOC disruption has reduced the rAAV degradation. Data from quantitative PCR demonstrate that the amount of rAAV measured as genome copy number (vgs/cell) was not significantly changed upon MT-MTOC disruption, suggesting that AMD treatment did not affect the viral degradation (Fig. 3C). This notion was further supported by our data from pharmacological intervention studies that showed that proteasome inhibitor can further enhance viral transduction if co-administrated with AMDs (Fig. 3D). Therefore, we have excluded the hypotheses (1) that MT-MTOC disruption directly increases transgene expression, (2) that MT-MTOC disruption acts to increase transduction through cell cycle arrest, and (3) that MT-MTOC disruption increases rAAV transduction by decreasing the degradation of rAAV virions.

MT-MTOC disruption does not affect promoter activity and viral degradation.
Interestingly, previous studies, including ours, demonstrated that disruption of MTs during rAAV entry has a negative effect on transduction because of impaired cytoplasmic trafficking. 30 The data from previous study and our current findings suggest that (1) MTs are not required for later steps of transduction after rAAV trafficking to perinuclear region because MT disruption did not inhibit rAAV transduction after that time, and (2) though MTs play a central role in trafficking of the virus to the perinuclear region, at later time points these structures act to retain virions and thus limit transduction. It is unclear how rAAV accumulates at MTOC and whether any motors are involved in the process; further studies are warranted.
Nuclear entry of rAAV is increased upon the disruption of MT-MTOC
Based on the above findings, the most likely mechanism for elevated viral transduction upon MT-MTOC disruption occurs at the level of trafficking. It has been previously demonstrated that nuclear entry is an essential trafficking event for rAAV's infectious pathway. 42,43 Moreover, nuclear entry is the most probable event subsequent to and being limited by perinuclear retention in rAAV's infectious trafficking pathway. We hypothesized that the retention of viral particles at the MTOC region may limit the subsequent nuclear trafficking of the virus. To test this hypothesis, we visualized viral trafficking steps with and without Nocodozole treatment by fluorescent confocal microscopy and quantitatively evaluated the distribution of viral particles at different time points after administration of nocodazole at 6–8 hr p.i. (Fig. 4A). At about 1–1.5 hr after MT-MTOC disruption (∼8–9 hr p.i.), the perinuclear retention of rAAV particles was ablated in most cells examined (Fig. 4C) and the viral particles spread around the outside of nucleus (Fig. 4B). Later, ∼3–4 hr after MT-MTOC disruption (∼10–11 hr p.i.), an approximate 2-fold increase of viral particles was observed in the nucleus as shown by the representative images from confocal microscopy (Fig. 4B, C) and quantitative data from 3D microscopy (Fig. 4B-f/i/l, and 4D). Increased viral entry into the nucleus was also supported by quantitative PCR analysis of the viral genome copy number in the nuclear fractions of treated and untreated cells (Fig. 4E). Together, these results suggest that disruption of MT-MTOC at 6–8 hr p.i. leads to an increase in the fraction of viruses in nuclear proximity and may explain the increased transduction we have observed (Fig. 2).

Release of rAAV and increased nuclear entry upon MT-MTOC disruption.
Three major basic regions (BRs) on the capsid have been shown to function as nuclear localization signals (NLS) with varying strength. 42,43 rAAV mutants with deletions at these regions can still accumulate at the perinuclear region but are unable to enter nucleus. 37 Our prior study has demonstrated that pronounced Golgi localization is observed with the BR1 mutant, and the BR2 and BR3 capsid mutants appear to have diffuse localization throughout the cell in addition to localizing to the Golgi. 37 To test whether NLS are required for the increased nuclear entry and viral transduction, we examined the effects of MT-MTOC disruption on these BR deletion mutants. In both DMSO- and nocodazole -treated cells, BR deletions resulted in the reduced viral transduction (Fig. 4F). Importantly, nocodazole treatment was unable to restore the impaired transduction of these BR mutants, indicating that AMD-induced enhancement of viral transduction and nuclear entry requires the capsid NLSs. These data suggest that MT-MTOC disruption releases rAAV to continue down its normal nuclear entry pathway and does not cause rAAV's nuclear entry pathway to change.
rAAV is imported into the nucleus via the RhoA-ROCK-Actin pathway upon MT-MTOC disruption
The above findings suggested that MT-MTOC disruption itself allows only the release of rAAV particles from the perinuclear region. The subsequent increase in nuclear entry is achieved by a mechanism that requires the NLS on viral capsid. It is well established that MT disruption can lead to RhoA-ROCK mediated rearrangement of actin filaments (AFs), which in turn may affect actin-mediated intracellular trafficking. 44,45 Perinuclear AFs have been suggested to be involved in nuclear import/export of intracellular materials via association with the nuclear envelope or perhaps nuclear pore complexes. 10,46 –55 In this study, we tested whether the AFs regulated by the activation of RhoA-ROCK upon MT disruption contributed to the increased rAAV nuclear entry and transduction. First, we transiently transfected siRNA to knockdown the expression of endogenous RhoA as demonstrated by immunoblotting for the RhoA protein (Fig. 5A). Interestingly, treatment with nocodazole induced an increase in viral transduction only in cells transfected with the control (scrambled) siRNA and had minimal effect in RhoA knockdown cells (Fig. 5B). This observation suggests that enhanced AAV transduction via MTOC disruption requires RhoA activation. H1152 is a potent inhibitor of ROCK, a downstream effector of RhoA, and induces actin filament alterations. Cytochalasin D (CytoD) is an inhibitor of AFs that induces the de-polymerization of these filaments. 56 Both H1152 and CytoD were found to ablate the transduction enhancement induced by MT-MTOC disruption with nocodazole; therefore, the enhancement of transduction required that ROCK not be inhibited and that AFs be intact (Fig. 5C). The data suggest that the RhoA-ROCK-Actin pathway is required for the increased viral transduction upon MT-MTOC disruption. Visualization of viral particles by fluorescence microscopy revealed that nocodazole -induced viral dispersion from the perinuclear region was unaffected by co-administration with H1152 or CytoD (Fig. 5D). Our results from 3D quantitative microscopy further demonstrated that co-administration of either H1152 or CytoD blocked the increase in viral nuclear entry only after MT-MTOC disruption (Fig. 5E). Additionally, administration of H1152 or CytoD alone at 6–8 hr p.i. did not affect the transduction and nuclear entry of rAAV (Fig. 5C, E).

Disruption of MT-MTOC increases viral transduction and nuclear entry through an RhoA-ROCK-Actin pathway.
Together, the above data suggest a two-step working mechanism for the increased rAAV transduction observed upon MT-MTOC disruption: (1) viral particles are released from the perinuclear region and spread around the outside of the nucleus upon MT-MTOC disruption, and (2) some of these viral particles are then transported into the nucleus via the RhoA-ROCK-Actin pathway.
Perinuclear retention of rAAV and nocodazole-mediated enhancement on viral transduction also occur in vivo
In cell culture, we observed a bottleneck in viral transduction at the stage of accumulation and retention of rAAV at MTOC region. rAAV is currently widely used as a gene therapy vector in many clinical trials to tackle a variety of human diseases. 27 The effort to improve its efficiency in gene delivery has been compromised by the lack of knowledge about rAAV trafficking in in vivo settings. With the sensitive imaging technique used in this study, we examined whether perinuclear retention of rAAV also acted as a bottleneck in vivo. To examine this hypothesis, we injected Cy5-rAAV into mouse brain, liver, or xenografted tumor, and determined the viral distribution at different time points after viral infection. At an early stage (∼1 hr) after viral inoculation, viral particles were scattered across cells in these tissues, as is clearly observed in the brain image (Fig. 6A). Because of the relatively high background fluorescence observed in both liver and xenografted tumor, we were not able to visualize single viral particles in these tissues as observed in the brain; instead, we were able to visualize particles only at the sites of viral injection, where a higher concentration of labeled virions was present (arrowheads in Fig. 6A). At 6–12 hr p.i., a large number of viral particles were observed to accumulate at the perinuclear region, consistent with our in vitro studies (Fig. 6A). Approximately 60–80% of cells in these tissues had perinuclear accumulation by 12 hr after infection (arrows in Fig. 6A, B).
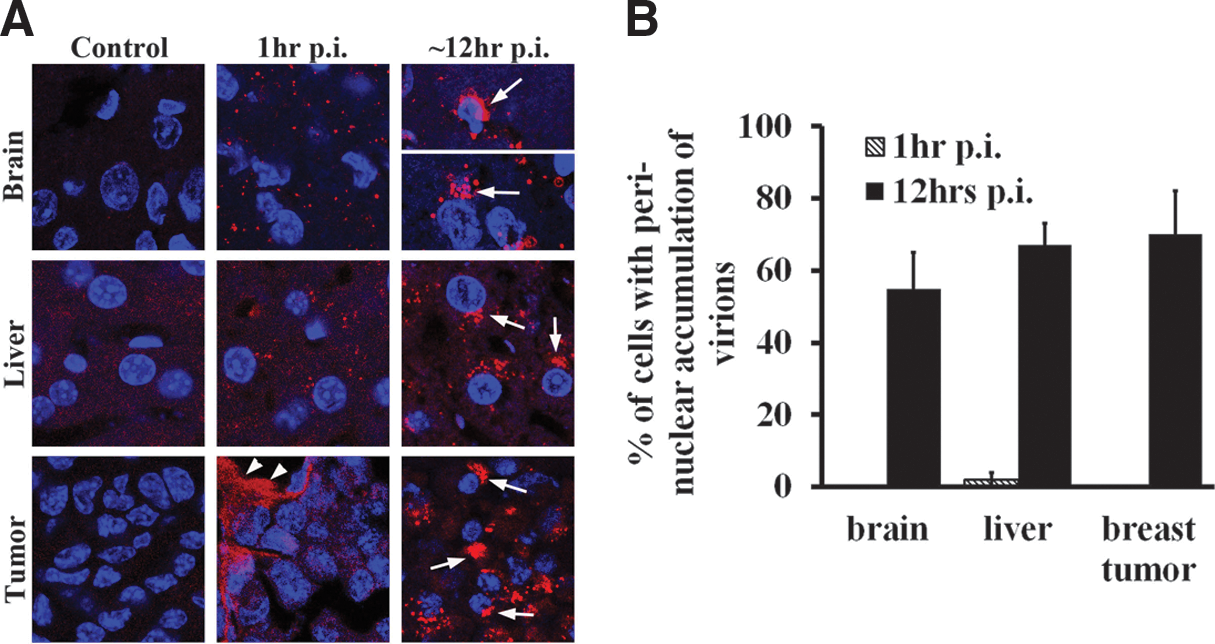
Perinuclear accumulation of rAAV in mouse tissues. Mouse tissues (brain [cerebrum], liver, xenografted breast tumor) were removed at 1 or 12 hr after injection of Cy5-rAAV and sectioned using a cryo-microtome. Nuclei are stained with DAPI (blue). The viral particles were visualized by confocal microscopy.
Furthermore, we tested whether AMD treatment could disrupt this perinuclear retention and increase viral transduction in mouse as was observed in our in vitro studies. Approximately 10 hr after viral inoculation, nocodazole was administrated to the animal through intraperitoneal or intratumoral injection. At 4–6 hr after nocodazole injection (∼14–16 hr p.i.), perinuclear retention (arrows in Fig. 7A) was observed only in DMSO-treated animals and was ablated in the cells of both liver and xenografted breast tumor in nocodazole -treated animals (Fig. 7A). Using GFP as a reporter gene packaged by rAAV, a significantly stronger GFP fluorescent signal was observed in the nocodazole-treated breast tumors (Fig. 7B). Moreover, by measuring luciferase activity using the IVIS imaging system, we observed a roughly 10-fold increase of viral transduction in both liver and xenografted tumor of nocodazole-treated mice (Fig. 7C, D). Together, these data suggest that rAAV takes a similar subcellular trafficking route in animal tissues to that in vitro and is retained at the perinuclear region, a trafficking bottleneck for viral transduction.
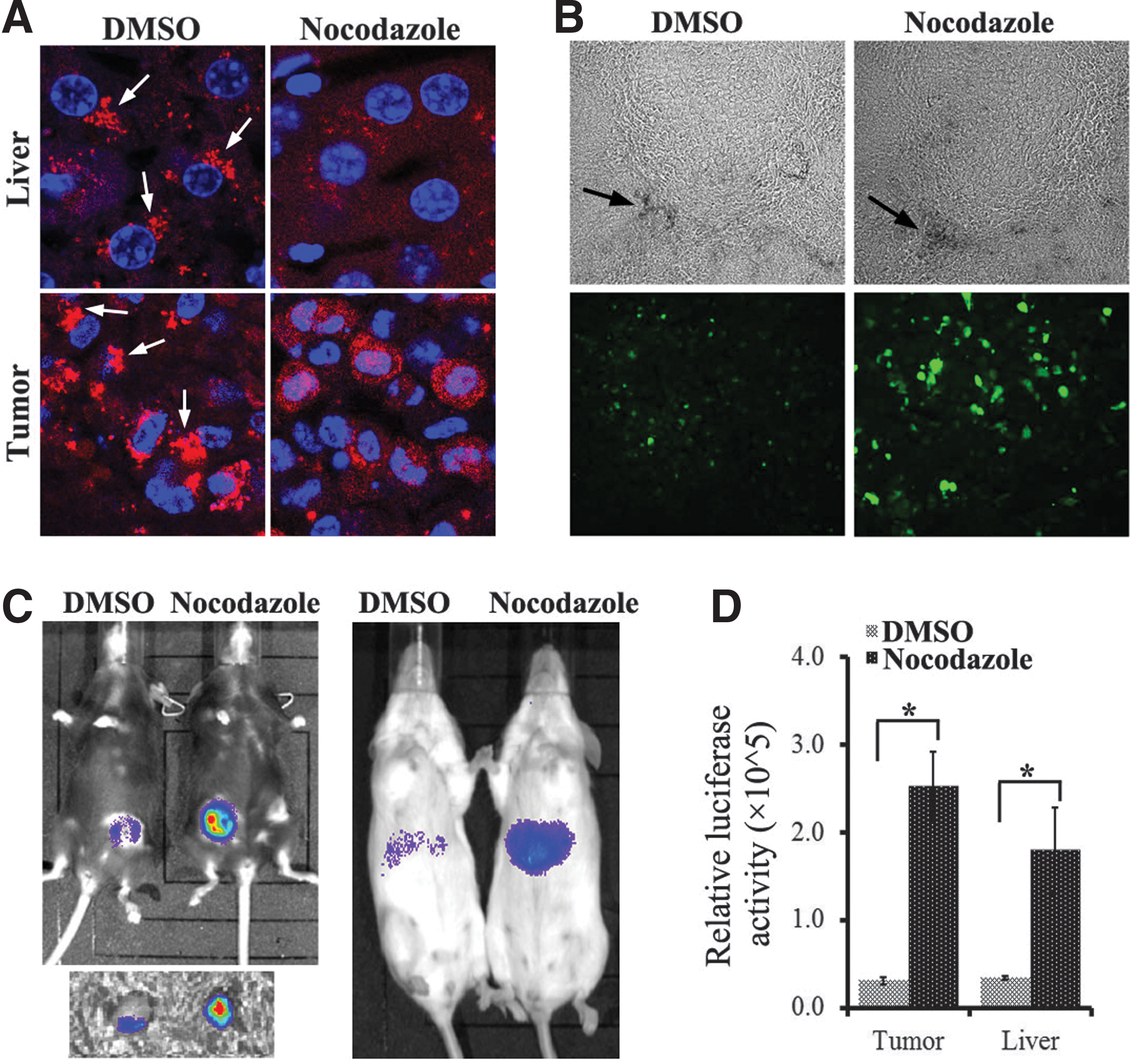
Disruption of perinuclear accumulation of rAAV particles with nocodazole treatment increases transduction in vivo.
Discussion
Viral accumulation at the perinuclear region is commonly observed during the infection of many viruses (e.g., Ad, HSV, HIV, influenza, parvoviruses). 11,13 –15,39 However, it remains unclear regarding whether and how this accumulation affects viral infection. Two plausible hypotheses 9 are (1) viruses exploit the cellular protein-trafficking machineries to approach the replication site or to concentrate viral components for viral replication and later virion assembly, thus supporting viral infection, and (2) perinuclear accumulation represents an innate cellular response that recognizes virus components as foreign or misfolded proteins and targets them for sequestration and degradation at the perinuclear region, thus interfering with viral infection. Our present study has supported the hypothesis that perinuclear accumulation is a cellular defense mechanism to sequester virions in order to limit the infection of rAAV.
Both Golgi and MTOC are perinuclear structures that coincidentally co-localize at the perinuclear region with many viruses. 10,11,13 –16 However, it remained unclear which cellular structure maintains perinuclear accumulation of rAAV. Earlier studies have suggested that the perinuclear viral particles co-localize to the Golgi apparatus. 36,37 Another study suggests that these virions may co-localize with MTOC. 38 In this study, to examine which structure was involved, we used confocal fluorescence microscopy and pharmacological intervention to circumvent the difficulties in studying the interactions between virions and cellular structures at the highly crowded perinuclear region because of the resolution limits of classical fluorescence microscopy (∼0.3 μm in lateral). Our results suggest that MT-MTOC but not the Golgi co-localizes with and retains the rAAV particles at perinuclear region (Fig. 1). Consistent with our findings, many other viruses, including Ad, HSV, and HIV, have also been shown to move on MTs to the MTOC region and associated with MT-MTOC. 10,14,39,40,57 –62 Furthermore, Fig. 1E shows that only a small portion of viral particles co-localized with the dispersed Golgi cisternae, which are released from highly packed MTOC region upon MT-MTOC disruption. This suggests that fewer particles physically localize in the Golgi apparatus. Consistent with previous reports, we did find that disruption of the Golgi apparatus at an earlier time during infection could impair rAAV transduction (data not shown). These data indicate that the Golgi does play a role in viral infection, probably through modification of viral capsid critical for viral infection, but is dispensable for perinuclear retention of incoming virions. Further investigation of the exact role of the Golgi apparatus on the infection of rAAV may advance our current knowledge of virology.
Our results, together with previous studies, strongly suggest that perinuclear retention by MTOC during early infection serves as a cellular barrier limiting viral transportation to the replication sites and leads to decreased viral transduction. During early infection stage, in which a virus delivers its genome to the target site for replication, accumulation of incoming virions at MTOC region is observed for most viruses that exploit MTs for earlier cytoplasmic trafficking. 10 –15,39 Consistent to these findings, we demonstrated that retention of rAAV by MT-MTOC limits viral transduction, and that disruption of MT-MTOC during infection led to an increase in rAAV transduction (Fig. 2E). Strikingly, some viruses may have evolved to overcome the MTOC retention by breaking MTs during their infection. 18 –22 For instance, HSV dismantle MT-MTOC after entering the cells as one of the functions of viral protein ICP0 and deletion of ICP0 seems to limit viral infection. 21
Our current study raises an interesting question for future exploration, which is whether and how the endosomal vesicles at MTOC region may contribute to the rAAV transduction enhancement observed upon MT-MTOC disruption. A reasonable hypothesis is that at 6–8 hr p.i., some viral particles observed at the MTOC region still remain inside the MT-localized endosomes/lysosomes, 32,63 whereas others have escaped the endosomes and directly associated with MTs. Direct interaction between MTs and the rAAV capsid has been demonstrated previously. 64 Both endosomal and escaped viral particles would be released from the MTOC region upon MT-MTOC disruption. Using rAAV mutants with defects in the phospholipase (PLA) domain that helps rAAV for endosome escape, 37 we determined that MT-MTOC disruption was unable to rescue the impaired viral transduction of these mutants (data not shown), which suggests that MT-MTOC disruption does not promote the escape of rAAV from endosomes. Further experiments should be performed to test whether nocodazole-mediated RhoA activation is not involved in endosome escape in the MTOC region with PLA mutant. Furthermore, increased viral particles reaching the nucleus upon disruption of MT-MTOC are most likely the subset of virions that have escaped from endosomes and are directly associated with MTs. Those virions that failed to escape endosomes are not eligible for nuclear entry regardless of the integrity of MT-MTOC. This notion is consistent with our observation that the proportion of nuclear particles increases from only about 30% to 50%, possibly because about 30–40% of virions are still inside the endo/lysosomes at 8 hr p.i. 32 In the future, it will be interesting to investigate the potential role of endo/lysosomal vesicles on viral retention at the perinuclear region, which would add valuable information to the mechanism of this perinuclear phenomenon. Additionally, the relationship of free rAAV particles in the cytoplasma with MT should be elucidated.
Actin filaments (AFs) and small Rho GTPases have been shown to be involved in viral infection, including cell entry, cytoplasmic trafficking, and nuclear entry. 36,56,65 –67 Recently, perinuclear and nuclear AFs were shown to associate with the nuclear envelope and nuclear pore complex, and were suggested to be involved in nuclear transport of macromolecules and viral particles. 46 –50,66 rAAV has been suggested to utilize AFs to enter nucleus during an earlier stage of infection when MTs were disrupted. 30 Furthermore, the disruption of MTs leads to a RhoA-ROCK-mediated rearrangement of AFs. 44,45 Consistent with these studies, our data show that AFs may facilitate the increase in nuclear entry through the RhoA-ROCK pathway upon MT-MTOC disruption (Fig. 5). Interruption of this pathway in the presence of intact MTs does not affect the level of viral transduction. This finding indicates that, at this time point, nuclear import of rAAV by AFs occurs only upon the disruption of MT-MTOC. It is possible that the AAV capsid has a much higher binding affinity for MTs than for AFs, and therefore remains associated with MT-MTOC. Moreover, the requirement of RhoA and ROCK activity indicates that MT disruption-induced alterations of AFs are necessary for the increase in viral nuclear entry and transduction. One possibility is that activated RhoA-ROCK results in more AFs connecting to the nuclear pore complex, which in turn transports more rAAV particles into the nucleus. Further study of this mechanism will add more detail to current knowledge of viral nuclear entry as well as the biology of cellular cytoskeletons.
Although tremendous progress has been made in understanding the trafficking of rAAV virions in cell culture, little is known about the trafficking pattern of this virus in vivo. This has limited one's ability to improve the efficiency of viral transduction for gene delivery in its natural environment. In this study, we examined the trafficking of rAAV in mouse tissues, including brain, liver, and xenografted tumors. In each of these tissues, the viral particles accumulate at a perinuclear site, resembling our in vitro observations (Fig. 6). Treatment with nocodazole was able to disrupt the perinuclear retention of rAAV and increase the viral transduction in all tissues tested (Fig. 7). Although further investigations are needed to refine the details of viral trafficking in these tissues, to our best knowledge, this is the first study to verify an in vitro trafficking pathway in in vivo settings. Additionally, the data showing that nocodazole treatment can enhance rAAV transduction in breast cancer cells indicate that there may be a synergistic effect between chemotherapeutic drugs, such as AMDs, and rAAV-mediated gene therapy in treating cancer patients. In this study, we studied the enhancement effect of MT-MTOC disruption after virus entry on rAAV2 transduction. Since different serotypes of AAV may use different mechanism for intracellular trafficking, it should be warranted whether the MT disruption also has an enhanced effect on transduction from other serotypes of AAV after cell entry.
In summary, our current study examined the mechanism of perinuclear retention of the parvovirus AAV and the corresponding effects on viral infection. Our data demonstrated that perinuclear retention by MT-MTOC serves as a subcellular barrier and limits rAAV infection, specifically at the step of nuclear entry, providing a novel host defense mechanism to sequester incoming virions. These studies may shed light on mechanisms of viral pathogenesis, especially under circumstances of MT disassembly during cell division and that of MTOC malfunction caused by misfolded-protein diseases or treatment with chemotherapy drugs. Moreover, further understanding of the determinants on the virus responsible for perinuclear retention may also lead to novel strategies for the enhancement of rAAV vector for gene delivery.
Footnotes
Acknowledgments
This research was funded by NIH Grants R01DK084033 (to C.L. and R.J.S.) and P01HL112761, R01AI072176, and R01AR064369 (NIH to R.J.S.). We thank Marc Weinberg for technical assistance, and members of the UNC Gene Therapy Center, particularly Thomas Lentz, Jayme Warischalk, and Matt Hirsch, for productive discussions. We greatly appreciate Swati Yadav for calculating virus titers by qPCR. We also thank the members in the Microscopy Services Laboratory, especially Robert Bagnell, Steven Ray, and Victoria Madden, for providing resources.
Author Disclosure
R.J.S. is the founder and a shareholder at Asklepios BioPharmaceutical. He receives research support through the University of North Carolina (UNC) from Asklepios BioPharmaceutical. He holds patents that have been licensed by UNC to Asklepios Biopharmaceutical, for which he receives royalties. He has consulted for Baxter Healthcare and has received payment for speaking.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.