
Abstract
Key neuropathological hallmarks of Alzheimer's disease (AD) are extracellular amyloid plaques and intracellular accumulation of hyperphosphorylated Tau protein. The mechanisms underlying these neuropathological changes remain unclear. So far, research on AD therapy has had limited success in terms of symptomatic treatments although it has also had several failures for disease-modifying drugs. Gene transfer strategies to the brain have contributed to evaluate in animal models many interesting tracks, some of which should deserve clinical applications in AD patients in the future.
Introduction
A
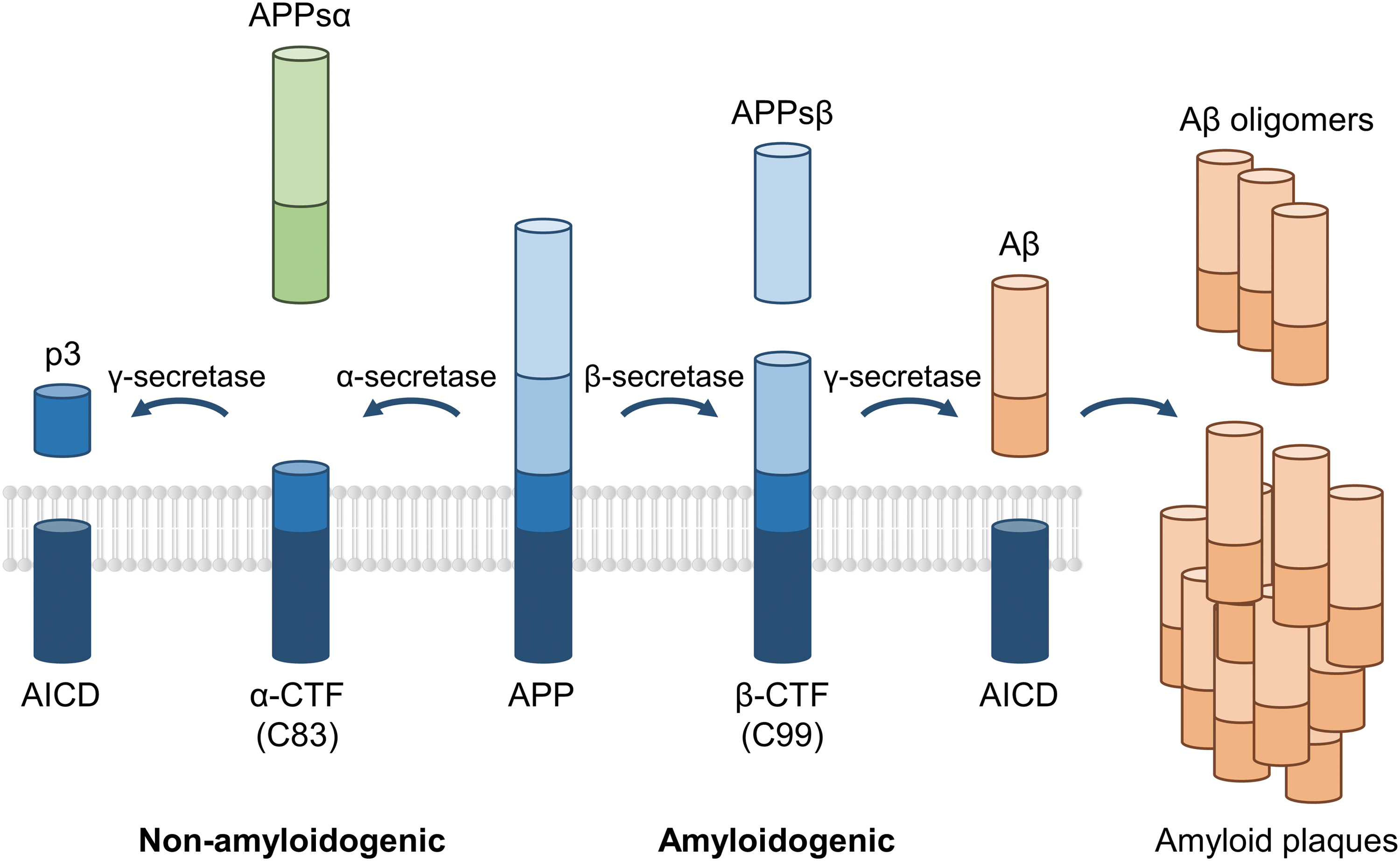
Scheme depicting the amyloid precursor protein (APP) processing. The APP can be proteolytically cleaved following two competing pathways. The nonamyloidogenic pathway involves the α-secretase cleaving the APP and giving rise to the soluble APPα (APPsα), which displays neurotrophic properties and the α carboxy-terminal fragment (α-CTF or C83). The last one is then processed by the γ-secretase liberating the p3 peptide and the APP intracellular domain (AICD) thought to function as a transcription factor. In the amyloidogenic pathway, APP is cleaved by the β-secretase inducing the release of the soluble APPβ (APPsβ) and the β carboxy-terminal fragment (β-CTF or C99) having cytotoxic effects. Finally, this peptide is processed by γ-secretase generating AICD and the amyloid β (Aβ), a soluble harmful peptide whose accumulation will form toxic oligomers and at later stages of the pathology, the amyloid plaques, hallmark of Alzheimer's disease patients' brains. Not drawn in proportion.
Genetics and disease mechanism
Based on one's age at onset, AD is classified into early onset AD (EOAD, onset <65 years), accounting for 1–5% of all cases, and late onset AD (LOAD, onset >65 years), accounting for >95% of patients. EOAD is usually associated with a more rapid progression and a Mendelian pattern of inheritance. Three genes (APP, PSEN1, and PSEN2) that all encode proteins involved in APP metabolism and Aβ generation have been identified in the pathophysiology of EOAD, underlying the role of the amyloid pathway in its mechanism (Fig. 2). Mutations in these genes are mostly autosomal dominantly inherited and lead to Aβ aggregation and early onset disease. 6 –9
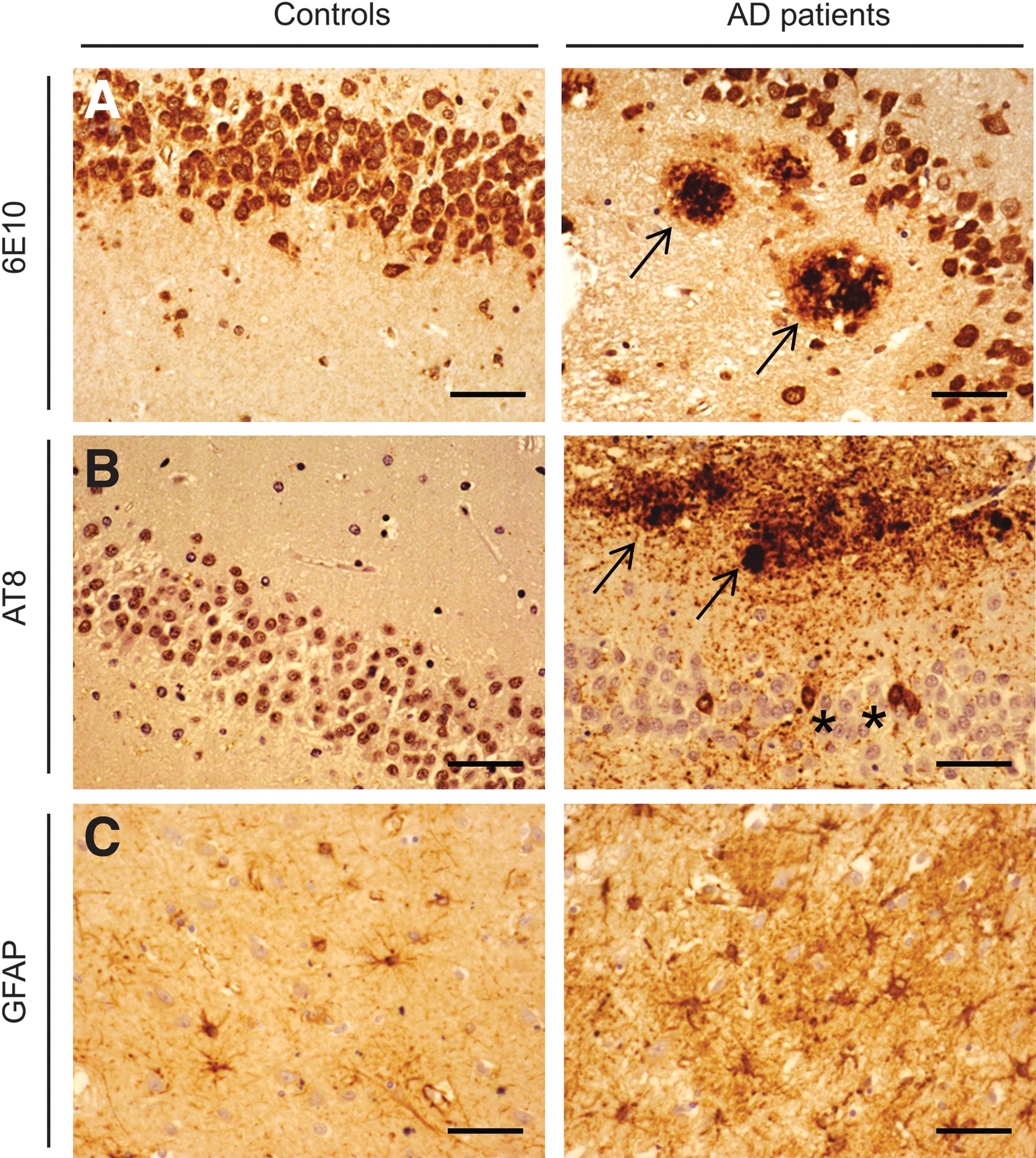
Alzheimer's disease (AD) neuropathology in the AD hippocampus.
Several genes are involved in LOAD. They increase disease risk in a non-Mendelian fashion. First-degree relatives of patients with LOAD have twice the risk of people without an AD-affected first-degree relative. The first identified gene and major genetic risk factor is the APOE ɛ4 allele. 10,11 APOE is a lipid-binding protein expressed in humans as three common isoforms coded for by three alleles, APOEɛ2, ɛ3, and ɛ4. A single APOE-ɛ4 allele is associated with a 2–3-fold increased risk, and two copies increase the risk to 5-fold or more. Each allele lowers the age at onset by 6–7 years. 8
Among thousands of candidate-gene-based association studies aiming to identify additional susceptibility loci, the only gene that could be identified and consistently implicated in AD was the sortilin-related receptor (SORL1). SORL1 is involved in vesicle trafficking from the cell surface to the Golgi-ER. SORL1 directs APP to endocytic pathways for recycling and plays an important role in Aβ generation. Interestingly, SORL1 is also a receptor that binds lipoproteins, including APOE-containing particles. 12 –15 Recently, high-throughput genome-wide arrays allowed large hypothesis-free analysis of LOAD and identified more than 20 novel risk loci. 16 Most identified genes cluster in specific pathways: APP processing (SORL1, CASS4), Tau pathology (CASS4, FERMT2), immune response (HLA-DRB5/DRB1, INPP5D, MEF2C, CR1, CD33, TREM2), cell migration (PTK2B), endocytosis (SORL1, PICALM, BIN1, CD2AP, EPHA1), and lipid transport (APOE, ABCA7, CLU), strongly reinforcing the importance of these pathways in LOAD etiology. 12,17
Therapeutic strategies for AD
Research into AD therapy has succeeded in developing at least partly efficacious symptomatic relief, but has failed in terms of developing disease-modifying therapies. Drugs approved for AD treatment are limited to acetylcholinesterase inhibitors (donepezil, rivastagmine, galantamine) or NMDA receptor antagonists (memantine). 18 They potentially improve cognition, behavior, and general clinical state, but their efficacy is very limited.
Among disease-modifying drugs that were evaluated, the most numerous trials concern passive immunosuppression. Results from animal studies have shown that anti-Aβ antibodies can prevent oligomer formation and reduce brain amyloid load with improvement in cognitive functions. Several monoclonal antibodies are being currently tested. 18 However, early clinical trials have been disappointing, leading to the initiation of clinical trials in which treatment is initiated in presymptomatic patients or in patients at a very early stage of the disease. 19 There remains a great unmet need to identify therapies with the potential to slow disease progression and improve cognitive function in AD.
Gene Therapy Strategies for AD
Acting directly on APP metabolism
Inhibiting secretase activity to decrease amyloid pathway
Anti-BACE1: Given the pivotal role BACE1 plays in Aβ production, it is an obvious therapeutic target. Small-molecule inhibitors of BACE1 have been in development, and many are now at various stages of clinical trials. Lowering BACE1 levels using lentiviral vectors expressing siRNAs reduced amyloid production and the neurodegenerative and behavioral deficits in AD mice. However, recent results showed unexpected and undesirable effects of BACE1 inhibition on synaptic function and cognition. In addition to the well-documented neurotoxic effects of Aβ, there is evidence that Aβ has neuroprotective properties and positive role on mechanisms underlying cognition at low or physiologic levels, raising the possibility that inhibiting BACE1 in wild-type animals may reduce endogenous Aβ production to such levels that its neurotrophic and synaptotrophic properties are lost. 20 –22
Inhibiting gamma secretase activity
γ-Secretase is a membrane protease carrying out cleavage of more than 100 single transmembrane-spanning proteins, including APP, Notch, and N-cadherin. Presenilin subunit is the catalytic component: mutations in the presenelin gene are a major cause of early onset familial AD (FAD) 8 because they lead to an increase in the production of the highly amyloidogenic Aβ42 isoform. Drugs aimed at γ-secretase are considered to be promising therapeutic targets for AD. However, inhibition of γ-secretase can cause severe adverse events, particularly because of the blockage of other pathways particularly the Notch signaling process. 23,24
Increasing amyloid degrading enzymes: ECE, IDE, and NEP
Several proteases, including neprilysin (NEP), endothelin converting enzyme (ECE), and insulin degrading enzyme (IDE), have been shown to cleave Aβ. 25 AAV5-ECE-1 administration was evaluated in APP/PS1 transgenic mice. Strong expression was obtained in areas surrounding the injection sites, allowing Aβ and plaques decrease in the anterior cortex and hippocampus. 26
Another study compared AAV-induced increased expression of NEP and IDE. AAV vectors expressed either native forms of NEP (NEP-n) or IDE (IDE-n), or engineered secreted forms of NEP (NEP-s) or IDE (IDE-s). In a six-week study, total Aβ and plaques were decreased in animals receiving the NEP-n and NEP-s but not for IDE-n or IDE-s in either the hippocampus or cortex. Thus, NEP, but not IDE, may be a good candidate for AD gene therapy. 27
Delivering anti-amyloid antibodies
Monoclonal antibodies or polyclonal immunoglobulins targeting Aβ have been used to promote its clearance. Results from animal studies have shown that anti-Aβ antibodies can prevent oligomer formation and reduce brain amyloid load with improvement in cognitive functions. Several studies are ongoing to evaluate the tolerance and the efficacy of these strategies in human patients. 19,28
A gene therapy strategy was also used based on the delivery of AAV encoding anti-Aβ single chain antibody (scFv) injected into the corticohippocampal regions of AD mouse models. One year after injection, expression of scFv was detectable in the neurons of the hippocampus. Amyloid deposits were strongly decreased at the injection sites with no sign of neurotoxicity. 29
Increasing physiological APP pathway
The proteolytic processing of APP can be achieved following two competing pathways: the amyloidogenic and the nonamyloidogenic pathways 4 (Fig. 1). The last one not only prevents the production of amyloid toxic forms, but also enables the release of the soluble APPsα. APP has important physiological functions, many of which are thought to be mediated via the secretion of the APPsα. 30 In vivo the administration of APPsα enhances memory performance and long-term potentiation (LTP) in rodents, and displays crucial physiological properties for synaptic plasticity and hippocampal function. 31 In APP-KO mice, APPsα knock-in completely rescued spatial learning. 32
The idea emerged that, beside the well-established neurotoxic and synaptotoxic properties of β-CTF and soluble and oligomeric Aβ, loss of the neuroprotective APPsα also contributes considerably to the development of AD. APPsα levels are decreased in the cerebrospinal fluid of AD patients, in both genetic and sporadic forms, which is correlated with poor memory performance. 33,34 APPsα was also shown to inhibit tau phosphorylation through GSK3β modulation. 35 This suggests that overexpression of APPsα could be of great interest to alleviate AD-related symptoms. One way to do this would consist of overexpressing the α-secretase (ADAM10). However, ADAM10 was shown to have many substrates and its upregulation was implicated in tumorigenesis, thereby precluding a human clinical trial. 36 APPsα was overexpressed by means of AAV virus in hippocampal neurons of an aged AD mouse model (APP/PS1). 37 Two months after gene therapy, APPsα partially rescued spatial memory defects, restored synaptic plasticity and spine density, and decreased soluble Aβ and amyloid plaques. This was associated with microglial activation and phagocytosis (increase of IDE and TREM2) around amyloid plaques.
Increase neurotroprotection
Nerve growth factor
Nervous system growth factors prevent neuronal death in various correlative animal models of AD. 38 Specifically, nerve growth factor (NGF) stimulates and prevents the death of the function of basal forebrain cholinergic neurons that undergo early and prominent degeneration in AD. 39
Degeneration of cholinergic neurons is an early and prominent contributor to cell loss and cognitive decline in AD. Indeed, NGF levels in the basal forebrain region decline in AD. Previous studies in animal models of AD have shown that NGF can stimulate cholinergic neurons that are necessary for maintenance of cognitive function, and undergo atrophy in early AD and prevent their death. 40 However, growth factors can cause off-target adverse effects, necessitating a targeted delivery strategy to control their localization and spread in the brain. 41
In a first phase I trial, NGF was delivered through transplantation of autologous fibroblasts transduced with a Moloney leukemia viral vector to express human NGF into the basal forebrain region containing cholinergic cell bodies that send their projections throughout the cortex and hippocampus. The clinical findings of the phase I ex vivo trial suggested possible beneficial effects over a 2-year observation period compared with pretreatment rates of cognitive decline. A second phase I clinical trial included 10 patients who received AAV2-NGF into the basal forebrain region. An escalation dose protocol was used (1.2 × 1010 to 1.2 × 1011 vector particles 42 ). The brains of all 8 patients in the first phase I trial were examined. All patients exhibited a trophic response to NGF in the form of axonal sprouting toward the NGF source. Cholinergic neuronal hypertrophy occurred on the NGF-treated side. Activation of cellular signaling and functional markers was present in the 2 patients who underwent AAV2-NGF gene transfer. An overall lower rate of cognitive decline and increased cortical glucose uptake were reported; two individuals had subcortical hemorrhage during implantation. No other adverse pathological effects related to NGF were observed.
A phase II multicenter, sham-surgery-controlled trial of NGF in AD is ongoing in 49 patients with mild to moderate AD based on a single administration of AAV-NGF vector that encodes the gene for NGF (CERE-110) or an appropriate sham (placebo) surgery control treatment. Data are expected soon. 43
Brain-derived neurotrophic factor
Brain-derived neurotrophic factor (BDNF) is expressed in multiple cortical regions, including the entorhinal cortex and hippocampus. 44 BDNF levels decline in AD. 45 Administration of BDNF using a lentiviral vector (under the control of the CAG promoter) to the entorhinal cortex in an AD mouse model (APP transgenic mouse line J20) improved learning and memory, enhanced expression of the synaptic protein synaptophysin, 38 and prevented neuronal loss with early life BDNF treatment. The lentiviral-mediated BDNF gene was delivered into the entorhinal cortices of mice at age 2 months, and mice were examined 5 months later. BDNF revealed neuroprotective properties. Interestingly, this beneficial effect was not accompanied by a decrease of amyloid plaques.
Glial cell-derived neurotrophic factor
Glial cell-derived neurotrophic factor (GDNF) is emerging as a potent neurotrophic factor with therapeutic potential against a range of neurodegenerative conditions, including AD. Lentiviral vectors were used to overexpress the GDNF gene in hippocampal astrocytes of 3xTg-AD mice in vivo. After 6 months of GDNF overexpression, 10-month-old 3xTg-AD mice showed preserved learning and memory. GDNF therapy did not significantly reduce amyloid and tau pathology, but upregulated the expression of BDNF and induced neuroprotection. 46
IGF1 and IGF2
Insulin-like growth factor 2 (IGF2) plays a critical role in memory consolidation in rats and mice, and IGF2 expression decreases in the hippocampus of patients with AD. AAV-IGF2 administration in the hippocampus of aged wild-type mice enhances memory and promotes dendritic spine formation. AAV-IGF2 or AAV-IGF1 injection into the hippocampus of APP Tg2576 mice rescues behavioral deficits, promotes dendritic spine formation, and restores synaptic transmission. IGF2, but not IGF1, injection allows significant reduction in amyloid levels. Results demonstrate that IGF2/IGF2R is involved in the extracellular Aβ degradation mechanism, and suggest that IGF2R may act as an Aβ scavenger.
47
Another study showed that intracerebroventricular infusion of IGF2 in APP/PS1 mice that express the green fluorescent protein in cholinergic neurons
Nrf2 antioxidant pathway activation
Oxidative injury is thought to be central in the pathogenesis of AD. Binding of the transcription factor nuclear factor E2-related factor 2 (NRF2) to the antioxidant response element (ARE) enhancer sequence is an endogenous defense system against oxidative stress, triggering the simultaneous expression of numerous protective enzymes and scavengers.
A lentiviral vector was used to deliver NRF2 bilaterally into the hippocampus of 9-month-old transgenic AD mice (APP/PS1 mice). Reduction in spatial learning deficits of aged APP/PS1 mice was achieved. Six months after injection, NRF2 gene transfer was associated with a reduction in astrocytic, but not microglial activation, and induction of NRF2 target gene heme oxygenase 1 in hippocampal neurons. 49
Boosting autophagy-mediated pathways
Autophagy, the major cellular pathway for degradation of long-lived proteins and protein turnover, has been implicated in AD. Evidence suggests that increasing the levels of autopagy-related proteins may have potential for therapy. Lentiviral-mediated overexpression of beclin-1 in the hippocampus and cortex of APP transgenic mice reduced both intracellular Aβ as well as extracellular β-pleated Aβ depostis. 50 Thus, restoring beclin-1 and enhancing autophagy may be a novel approach to treat AD.
Targeting inflammatory pathway
Interleukin-4
Increasing data demonstrate the role of inflammation in AD. Anti-inflammatory cytokine signaling may play an emerging role as neurotransmitters, neuromodulators, and neurohormones in the brain. IL-4 has been characterized as a potential modulator of neuronal activities in the brain. IL-4 receptors are expressed in the hippocampus, and downregulation of IL-4 causes aging-related deficits of hippocampal LTP. 51 Moreover, IL-4 stimulation of human macrophages or microglia enhances Aβ degradation. AAV-mediated expression of the mouse IL-4 gene in APP/PS1 mice attenuates AD. Vector encoding IL-4 injection into the hippocampus resulted in sustained expression of IL-4, reduced astro/microgliosis, Aβ oligomerization and deposition, enhanced neurogenesis, improved spatial learning, and promoted phosphorylation of N-methyl-D-aspartate receptor subunit 2B. 52
Interleukin-10
Anti-inflammatory cytokines, such as IL-10, can have significant therapeutic potentials in AD. AAV-mediated neuronal expression of the mouse IL-10 gene ameliorates cognitive dysfunction in APP/PS1 mice. Sustained expression of IL-10 reduced astro/microgliosis, enhanced neurogenesis, and improved spatial learning. 53
Triggering receptor expressed on myeloid cells 2
The triggering receptor expressed on myeloid cells 2 (TREM2) gene was a recently identified susceptibility gene for AD, as its low-frequency variants increase the risk of AD similar to APOE ɛ4 allele. The TREM2 transmembrane protein is a receptor expressed on microglia that stimulates phagocytosis and suppresses inflammation. TREM2 is trafficked to the cell surface, where it binds with several ligands. Homozygous mutations in TREM2 are associated with autosomal recessive forms of dementia. 12
Overexpression of TREM2 using a lentiviral injection in the brain ameliorated AD-related neuropathology (Aβ deposition, neuroinflammation, and neuronal loss) in APP/PS1 mice and improved spatial cognitive functions. Upregulation of TREM2 could serve as a compensatory response to Aβ and protect against AD progression by modeling microglia functions. 54
Modulating genes related to LIPID metabolism
Targeting APOE, the major susceptibility gene for AD
APOE is a regulator of lipoprotein metabolism in the central nervous system, and APOE plays several important roles such as cholesterol transport, neuroplasticity, and inflammation. APOE binds to Aβ and influences its clearance and Aβ aggregation. 11
Direct intracerebral administration of lentiviral vectors expressing the three common human APOE isoforms differentially alters hippocampal Aβ and amyloid burden in the PDAPP mouse model of AD. Expression of APOE ɛ2 in the absence or even in the presence of mouse APOE after direct intracerebral administration of lentiviral vector expressing APOE ɛ2 in the PDAPP mouse model markedly reduced hippocampal Aβ burden. In contrast, expression of APOE ɛ4 in the absence of mouse APOE increased hippocampal Aβ42 levels and amyloid accumulation. 55
Recently, a gene transfer approach to target the cortex of amyloid plaque-bearing transgenic mice with APOE was used by injection into the lateral ventricles of AD mice an AAV vector expressing the various human APOE alleles to transduce the ependymal layer. Human APOE proteins diffused into the cerebrospinal fluid and interstitial fluid (ISF). Human APOE isoforms affected the concentrations of soluble oligomeric Aβ in the ISF, the pace of Aβ fibrillization and deposition, and the extent of peri-plaque neurotoxic effects. Increase in soluble Aβ, an exacerbation of synaptic loss, and an increased number of dystrophic neurites around each deposit were observed in AD mice receiving APOE ɛ4, whereas a relative protective effect was observed with APOE ɛ2. These results suggest that therapeutic approaches aimed at decreasing APOE ɛ4, or increasing APOE ɛ2, may be beneficial in AD. 56
Decreased plaque number was also observed when overexpressing APOE ɛ2 in astrocytes, the cells that physiologically produce APOE (Fol et al., unpublished results).
Inhibiting Acyl-CoA cholesterol acyltransferase enzyme
Acyl-CoA cholesterol acyltransferase (ACAT) enzyme catalyzes the formation of cholesteryl esters and mediates storage of cholesterol and cellular cholesterol homeostasis. 57 AAVs expressing artificial microRNA (miRNA) sequence targeting Acat1 were used to test whether a specific genetic knockdown of Acat1 in the mouse brain, administered at 10 months after the onset of the disease, could benefit AD. Analysis at 12 months showed that vector administration was well tolerated and allowed to decrease the levels of brain Aβ and full-length APP to levels comparable to full genetic ablation of Acat1. 58
Activating cholesterol 24 hydroxylase enzyme
In AD, a mechanistic link between cholesterol metabolism in the brain and progression of AD has been clearly reported and altered cholesterol metabolism seems to play a pivotal role in the formation of amyloid plaques and in tau pathologies. 59 In the brain, cholesterol is synthesized in situ but cannot be degraded or cross the blood–brain barrier (BBB). The major exportable form of brain cholesterol is 24-hydroxycholesterol (24S-OHC) generated by the neuronal cholesterol 24-hydroxylase enzyme (CYP46A1). Because there is currently no molecule able to increase CYP46A1 activity and cross the BBB, overexpression of CYP46A1 by administering an AAV vector carrying the CYP46A1 cDNA in the brain seems to be an effective strategy. The therapeutic effect of AAV5-mediated CYP46A1 overexpression on rodent models of AD with amyloid pathology (APP23 and APP/PS1 mice) has been reported, by reducing the number of amyloid plaques (Fig. 3) and by improving spatial memory. 60 The same strategy was successful in THY-Tau22 mice, in which CYP46A1 overexpression corrected cognitive deficits, impaired long-term depression, and spine defects. 61 In conclusion, restoring CYP46A1 expression improves behavioral and clinical performances in vivo in both amyloid and Tau mouse models. Importantly, hippocampal shRNA-mediated inhibition of Cyp46a1 induced an AD-like phenotype in normal mice characterized by an increase in neuronal cholesterol and Aβ peptides followed by cognitive deficits, apoptotic neuronal death, and hippocampal atrophy. In addition, inhibition of the CYP46A1 gene aggravated the phenotype in APP23 mice. 62 These data strongly suggest CYP46A1 as a relevant target to modulate AD progression, thus opening new avenues for treatment.

Overexpression of CYP46A1 in the hippocampus of AD mice decreases amyloid plaques and astrogliosis.
Conclusions and Future Perspectives
Research on AD therapy based on disease-modifying drugs has so far revealed limited achievements in terms of symptomatic treatments. Gene transfer strategies targeting specific regions of the brain have contributed to evaluate several potential tracks in AD animal models, some of which should deserve to be looked as potential candidates for clinical applications in AD.
As for most clinical conditions, the success of the development of new disease-modifying therapies strongly relies on critical issues, among which are the detailed knowledge of the natural history of the disease; its clinical, radiological, and biological endpoints; the selection of the patient population (EOAD, LOAD, genetic background); and the definition of valuable outcome predictors of disease evolution, but also biomarkers of treatment functionality (mechanism of action). This is particularly crucial for new innovative biotherapy strategies in which the number of patients included in phase I and phase II trials will be low. Gene therapy has a place in the battle against AD, given that it is designed to focus on one specific target and on specific delivery to affected brain regions. Moreover, it has the capacity to bring strong insight into the beneficial effects of modulating pathways to the brain that will help to better understand the physiopathology of the disease and contribute to stop its progression.
Footnotes
Acknowledgments
This work was supported by NeurATRIS: A Translational Research Infrastructure for Biotherapies in Neurosciences, the Fondation France Alzheimer, the ANR-10-MALZ-0103 CholAD, the ERA-Net Neuron (01EW1305A), and the Fondation pour la Recherche Médicale, Bioingénierie pour la Santé 2014 “Projet DBS20140930765.”
Author Disclosure
N.C. is a founder and owner of founder equity of BrainVectis Therapeutics. The other authors declare that they have no competing interests.