
Abstract
Over the last few years, huge progress has been made with regard to the understanding of molecular mechanisms underlying the pathogenesis of neurodegenerative diseases of the eye. Such knowledge has led to the development of gene therapy approaches to treat these devastating disorders. Challenges regarding the efficacy and efficiency of therapeutic gene delivery have driven the development of novel therapeutic approaches, which continue to evolve the field of ocular gene therapy. In this review article, we will discuss the evolution of preclinical and clinical strategies that have improved gene therapy in the eye, showing that treatment of vision loss has a bright future.
Introduction
V
As a gene therapy target, the retina is a particularly well-suited organ for therapeutic interventions. The retina is a small tissue, highly compartmentalized, immune-privileged, and easily accessible. Optical transparency of the eye enables safe evaluation of reporter gene expression and therapeutic effects by noninvasive methods, 2 such as electroretinography (ERG), funduscopy, and optical coherence tomography (OCT). These favorable factors, along with a thorough knowledge of the molecular pathogenesis of many retinal diseases, the development and characterization of animal models that mimic human diseases, and the advances in gene delivery tools, have fueled a rapid development of multiple gene therapy strategies for several forms of retinopathies. This review is focused on emerging strategies that use gene therapy to combat vision loss, particularly for the treatment of retinal diseases caused by mutations that directly affect the photoreceptors.
Gene Replacement Therapy for LCA2: The First Success of Ocular Gene Therapy
The most successful example of ocular gene therapy was the gene replacement therapy for RPE65, Leber's congenital amaurosis 2 (LCA2), an early onset form of autosomal recessive retinal degeneration caused by mutations in the RPE65 (RPE-specific 65 kDa protein) gene. RPE65 encodes an isomerase expressed mainly in the RPE that is critical for recycling the visual chromophore involved in the visual cycle. 3 Mutations in RPE65 result in defective visual pigment formation (both rhodopsin and cone opsin), 4 hence severely affecting photoreceptor function and vision. 5 Large amounts of opsin apoprotein in photoreceptors, as well as accumulation of toxic retinyl esters in the RPE, are thought to promote the progressive death of photoreceptors. Clinical phenotype analyses revealed that the degenerative component of RPE65-LCA2 starts at an early age in patients with a functional loss that is much larger than expected for the amount of cells retained. 6 It is this phenotype that provided a very good starting point for a gene-based intervention for this disorder.
Several murine and canine models of LCA2 have shown marked functional benefits with gene therapy. 6 In particular, results obtained in the Rpe65 −/− Briard dog yielded deep excitement in the field because of its more human-like eye anatomy and immune system. The first study was carried out by subretinal delivery of recombinant adeno-associated virus (AAV) 2 vectors expressing the wild-type canine Rpe65 cDNA under the control of the ubiquitous chicken β-actin (CBA) promoter. 7 This study revealed a dramatic improvement in photoreceptor function and vision in treated dogs. Subsequent dog studies extended the use of other AAV serotypes, including AAV1, AAV4, and AAV5, and different promoters. 8 –18 Improvement of vision persisted for over 11 years after a single injection of the vector. 16 In addition, successful restoration of both cone and rod function was achieved in 20 out of 22 treated eyes at more advance stages of the disease (dogs over 2 years of age). 12,14,16,18
Based on these preclinical studies, four separate phase I–II clinical trials were initiated, which yielded promising results after subretinal administration of AAV2-hRPE65 vectors (NCT00481546, NCT00516377, NCT00643747, NCT00749957; Supplementary Table S1; Supplementary Data are available online at
Although the pioneering RPE65 trials went far beyond the primary expectations, they uncovered a number of unforeseen challenges, mainly in the magnitude and the longevity of the therapeutic benefits. Contrary to the remarkable disease rescue obtained in the Rpe65 −/− dog, none of the treated RPE65-LCA eyes in any clinical trials have shown improvements in retinal function measurable by full-field ERG. 6 The reason for this species-specific difference remains unknown, but is likely related to the extent of disease progression in patients at the time of treatment. Unlike early-onset degeneration in patients with LCA2, the RPE65-mutant dogs exhibit no evidence of photoreceptor degeneration for up to 1.5 years of age and show good preservation of photoreceptors in the peripheral retina at 5–7 years of age (57% of rods and 85% of total cones). 6,14,15 Severe photoreceptor degeneration in patient retinas at the time of treatment may have limited the success of the gene therapy strategies. Nonetheless, a recent study investigating efficiency of AAV2-hRPE65 delivery in older Rpe65-mutant dogs showed no correlation between improvement in ERG, the age of the dogs at the time of treatment, and the number of surviving photoreceptors. These results suggest that additional factors could influence the degree of rescue associated with RPE65 treatment. 14 Health of the remaining RPE and photoreceptor cells may be a factor to consider. One might also predict that disease progression negatively impacts the mean transduction efficiency (i.e., the percentage/density of total RPE cells transduced and the level of RPE65 expression in transduced cells), thereby reducing the magnitude of rescue. In the animal studies, the effect of RPE65 replacement is highly dose dependent, 17 with lower doses associated with improvement in visual-guided behavior without detectable rescue by ERG recordings. 14,17 Inefficient transduction is in agreement with reports of incomplete restoration of dark adaptation in patients after gene therapy, 17,22,27 indicating that partial restoration of the visual cycle may be insufficient to meet the demand of the surviving photoreceptors. Consistent with that, Bainbridge et al. found that RPE65 RNA level in the human eye is 2.5 times greater than that in the dog eye, which suggests that the demand for RPE65 by the human retina is higher. 17
A recent long-term assessment of photoreceptor preservation in treated RPE65-LCA2 patients also questioned the longevity of the therapeutic effects. Two studies (NCT00481546 and NCT00643747) demonstrated that, despite visual improvement, photoreceptor death remained unchanged and followed the expected natural history of the disease. 16,17,22 Disappointingly, retinal degeneration was associated with a progressive contraction of the areas of improved vision over a period of 5–6 years after intervention 22 and with a sharp decline of retinal sensitivity by 3 years postinjection in a subset of treated patients, 17 indicating a possible loss of the functionally rescued cells. However, loss of a therapeutic effect at 2–4 years posttreatment has not yet been reported by Spark Therapeutics, potentially reflecting differences in vector design or manufacturing. Nonetheless, the loss of the therapeutic effect observed by Jacobson et al. 22 and Bainbridge et al. 17 has important implications for the evaluation of any retinal gene therapy, and several hypotheses have been formulated to explain it. Cideciyan et al. proposed that gene replacement therapy does not slow the degenerative component of the disease if the intervention is initiated after a threshold of accumulative molecular changes has been reached in RPE and/or photoreceptors. 16 Alternatively, the continuous changes that occur in the tissue not exposed to the vector could have overcome the small number of stably rescued RPE/photoreceptors in these patients. 28 Should results of the phase III clinical trial confirm the lack of long-term efficacy, appreciating the mechanism will be important for managing expectations about the benefits of gene therapy for each patient, as well as for designing strategies to improve the durability of the treatment. If a “point of no return” exists, alleviating stress in cells before treatment may be a solution to extend the efficacy of the gene therapy. Otherwise, it may be necessary to increase the number of transduced cells and also protect rescued photoreceptors against secondary degeneration. Such strategies are being developed for use in other retinal diseases 28,29 and could be adapted to the RPE65 deficiency.
Recent Progress in Clinical Applications of Retinal Gene Replacement Therapy
Gene replacement therapy for Mer proto-oncogene tyrosine kinase (MERTK)- and Rab-escort protein 1 (REP1)-associated inherited retinal diseases
Pioneering results of RPE65-LCA2 trials laid the groundwork for the initiation of trials for different forms of inherited retinopathies caused by mutations in another RPE-specific gene (MERTK retinitis pigmentosa) and in a gene required for both RPE and photoreceptor survival (REP1 choroideremia).
MERTK is involved in the engulfment of outer segment debris by the RPE. Patients with MERTK deficiency exhibit severe dysfunction of both rods and cones, associated with an early-onset and progressive degeneration of photoreceptors. In the first clinical safety study for MERTK retinitis pigmentosa (NCT01482195), 30 six patients received a submacular injection of AAV2-VMD2-hMERTK, in which the MERTK cDNA was expressed under the control of the RPE-specific VMD2 (vitelliform macular dystrophy) promoter. No complications attributed to the vector were observed, and three patients displayed improved visual acuity in the treated eye within the first week/month postintervention. However, the improvement declined to baseline 2 years posttreatment in two of the patients. Contrary to the LCA2 gene therapy studies, this functional decline was not associated with major changes in retinal thickness, as assessed by OCT. However, variability in OCT measurements in these patients with nystagmus was very large. It is possible that the quality of treatment, including the number of cells treated and expression levels of MERTK, may have been too low to promote long-term benefits.
Recently, promising results from a multicenter phase I/II study of AAV2-mediated gene replacement therapy for choroideremia (CHM) were reported (NCT01461213), 31,32 strengthening the use of gene therapy for retinal diseases. CMH is a severe X-linked recessive disorder leading to blindness through progressive degeneration of the choroid, RPE, and photoreceptors caused by the loss of function of the Rab escort protein 1 (REP1). CHM can be identified in childhood; however, it differs from RPE65-LCA2 in that most patients retain 20/20 vision because of fairly well-preserved central retinal cones until the fifth decade of life. The main objective is to preserve this area from further degeneration, which will require long-term follow-up to detect the outcome of the gene therapy. Importantly, no retinal thinning or loss of visual acuity was observed despite the surgically induced retinal detachment, establishing a favorable safety profile for this gene transfer protocol targeting the fovea. In addition, at 6 months posttreatment, significant improvement in retinal sensitivity was noted in the five patients who received the highest vector dose, as well as large gains in visual acuity in the two patients in whom visual acuity was already reduced at baseline. Preservation or gains in visual acuity were sustained until at least 3.5 years after treatment, while, over the same period, visual acuity in the control noninjected eyes decreased progressively. 32 It will be interesting to see if photoreceptor degeneration is prevented in these patients in a long-term follow-up examination. A 30-patient phase II study (NCT02341807) has started to further assess the functional and anatomical outcomes. Additionally, a phase I/II clinical trial using AAV5, a serotype with increased tropism for photoreceptors as compared with AAV2, is planned (Horama/Nantes Hospital, France). This approach may maximize the efficacy of the therapy as it was demonstrated that rod photoreceptors could degenerate independently from the RPE. 33,34
Additional clinical trials of retinal gene replacement therapy
To date, eight clinical trials testing gene replacement for four other retinal diseases are in progress (Supplementary Table S1). All of them build on the existing AAV2 platform, with the exception of two trials using the equine infectious anemia virus (EIAV)-based lentiviral vectors for the treatment of Stargardt disease (NCT01367444) and Usher type 1B syndrome (NCT01505062). These diseases are caused by mutations in the ABCA4 and MYO7A genes respectively, which are too large to be packaged in an AAV. 35 Lentivirus, which has a higher cargo capacity than AAV, has therefore been chosen as an alternative to AAV.
ABCA4 is localized in the outer segments of photoreceptors and acts as an important membrane transporter for the recycling of the visual chromophore. ABCA4 loss of function is associated with accumulation of toxic products in RPE cells, followed by severe RPE and macular photoreceptor death. A proof-of-concept study in the Abca4 −/− mice demonstrated beneficial effects after subretinal injection of EIAV-Abca4 at postnatal day (PN)4–PN5. 36 MYO7A is expressed in cochlear hair cells of the inner ear as well as in retinal photoreceptors and the RPE, 37 where it plays a role in multiple cellular processes, including endocytosis and cellular transport. Although the amount of MYO7A in photoreceptors is lower than that in the RPE, photoreceptors are affected before RPE cells in patients with Usher1B, indicating that photoreceptors are important cells to target in this disease as well. The ability of LV-MYO7A to restore RPE abnormalities has been shown in the Myo7a −/− mouse after subretinal injection of the vector at birth. 38,39 However, a major concern of lentiviral vectors for clinical use is its relative inability to transduce postmitotic photoreceptors. Physical barriers, which are not present in the newborn rodent retina, have been hypothesized to dramatically limit access of the large lentivirus particles to adult photoreceptors. 40 At this point the results of these two clinical trials remain unknown. Nonetheless, these results will provide valuable information not only regarding the efficacy of lentivirus in the retina, but also to determine whether lentivirus may serve as a safe alternative vector for RPE diseases in which high level of transgene expression is required.
Preclinical Advances in Gene-Specific Therapy for Photoreceptor Diseases
One of the major challenges over the next 20 years will be to initiate treatment of many retinal disorders in which the disease-causing mutation is primarily expressed in photoreceptors. Compared with RPE-associated diseases, primary photoreceptor dystrophies have been considered as more difficult to treat. This is because photoreceptors are directly impaired by the genetic mutation and, second, because the efficiency of photoreceptor transduction is relatively lower compared with that of RPE cells. 41 AAV2 and lentiviral vectors, though excellent vectors for transducing the RPE, were found to be inefficient in transducing photoreceptor cells. 42,43 However, several novel AAV serotypes have been identified and characterized. Among these, AAV5, 7, 8, 9, and rh10, as well as vectors with modified capsid proteins, show remarkable improvement in terms of photoreceptor transduction efficiency when compared with AAV2 (for review, see refs. 41,44 ). The main question that remains to be answered now is whether the use of these serotypes can offer clinical benefits. To illustrate advances in gene-specific therapies for recessive photoreceptor diseases, we will describe two approaches in more detail: (1) the promising application of gene therapy for the treatment of stationary disorders, in particular cone disorders, and (2) the novel approaches for the treatment of progressive retinal dystrophies initiated by defects that are either in rods or in both rods and cones.
Gene replacement therapy in animal models of stationary photoreceptor disorders
Despite the fact that stationary photoreceptor diseases are relatively rare, they are ideal translational models for the development of gene replacement therapies targeting photoreceptors. Stationary disorders are associated with congenital retinal dysfunction and can thus be diagnosed early. Their slowly progressive degenerative nature presents therefore a wide window of opportunity for intervention and effects of the therapy can be rapidly assessed through the restoration of retinal function.
Achromatopsia (ACHM) is an autosomal recessive disease associated with severe cone dysfunction, caused by a loss of function of some cone-specific proteins. To date, four small 45 –48 and three large animal models 29,49,50 of ACHM have been successfully treated using AAV-mediated gene replacement therapy (Supplementary Table S2). The first report of efficient gene therapy for ACHM was obtained in the cpfl3 (cone photoreceptor functional loss 3) mouse model of GNAT2 (guanine nucleotide binding G-protein) deficiency. 45 This mouse model retains ∼25% of the normal cone-mediated ERG response up to 4 weeks of age with no detectable responses by 9 months of age. In contrast, cone structural integrity is maintained for at least 14 weeks. Subretinal injection of AAV5 encoding the mouse Gnat2 under the control of the human red green cone opsin promoter (AAV5-PR2.1-mGnat2) at PN23-PN29 in cpfl3 mice restored cone ERG responses and visual-guided behavior to levels indistinguishable from age-matched controls in 80% of treated eyes, for at least 7 months. Interestingly, when the cpfl3 mice were treated at later stages of the disease (>9 months of age), the degree of rescue was variable, with only one eye showing cone ERG responses within the normal range. 45
Consistent with this, Carvalho et al. demonstrated that an optimal therapeutic window is present in the Cngb3 −/− (cone-specific cyclic nucleotide gated channel subunit b3) mouse model of ACHM, 48 in which cone degeneration is comparable to that of the cpfl3 mice. While subretinal injection of AAV8-mCAR-hCNGB3, expressing CNGB3 under the control of the mouse cone arrestin promoter at PN15, resulted in long-term restoration of cone-mediated ERG responses up to 90% of wild-type levels, treatment at PN90 and PN180 resulted in functional rescue at 70–80% and 60–70% of wild-type levels, respectively. Restoration of visual acuity was not possible after PN90. Komaromy et al. also showed restoration of vision in two canine models of CNGB3-ACHM using AAV5, marking an important milestone in the photoreceptor gene therapy field. 49 However, the magnitude and longevity of the therapy in dogs was also age dependent. 29,49 Younger dogs (<6 months) treated with AAV5-PR2.1-cCngb3 displayed the most sustained therapeutic effects, with a restoration of cone function up to 5–10% of wild-type levels, and a restoration of visual-guided behavior in bright light. Therapeutic effects were maintained for at least 33 months postinjection in two dogs. In contrast, only two out of seven dogs treated with less efficient vectors showed any sustained response, whereas the others had either a transient or no functional cone response. At older ages, only 1 dog out of 11 showed any sustained response. The authors hypothesized that the failure might result from the improper reassembly of the components of the phototransduction cascade when retinas were treated at more advanced stages of the disease. Combination of gene therapy with administration of the ciliary neurotrophic factor (CNTF), which promotes outer segment deconstruction and reconstruction, increased the efficacy and durability of gene therapy in older dogs. 29 CNTF alone also transiently restored cone function, 29,51 an effect not reproduced in five patients with CNGB3-ACHM. 52
Taken together these results indicate that the health of photoreceptor cells at the time of intervention and the associated efficacy in transgene expression could be limiting factors for a functional rescue after gene therapy in ACHM. Nonetheless, accurate selection of patients, choice of area for treatment, and selection of an optimal vector system may realistically allow for a favorable outcome in humans. Based on the encouraging results using animal models, Applied Genetic Technologies Corporation is conducting a phase I–II clinical trial to evaluate the potential of an optimized vector system 53 (AAV2tYF-PR1.7-codon-optimized hCNGB3 [Supplementary Table S1] for the treatment of CNGB3-ACHM).
X-linked retinoschisis (XLRS) is characterized by compromised retinal integrity and a subsequent slow loss of central photoreceptors. Juvenile XLRS is caused by loss-of-function mutations in the RS1 gene (retinoschisin), which encodes for a protein primarily expressed and secreted from photoreceptors and bipolar cells. The function of RS1 is unknown, but the protein is thought to play a role in cell adhesion and in the organization of the photoreceptor-bipolar cell synapse. 54 Numerous studies have shown that delivery of the RS1 gene to the photoreceptors, by subretinal (AAV5-mOP-hRS1; mouse opsin promoter 55 ) and intravitreal injections (AAV2-CMV-Rs1, 56 AAV8-RS1-hRS1, 57 AAV8-RS/IRBP-hRS1; under the control of RS/interphotoreceptor binding protein promoter 58 and AAV7m8-RHO-hRS1; rhodopsin promoter 59 ) can lead to long-term structural and functional preservation. Nonetheless, gene transfer at advanced stages of the disease (>7 months of age) showed no improvements of ERG responses. 60 Applied Genetic Technologies Corporation recently initiated a phase I/II clinical trial to evaluate the safety and efficacy of intravitreal injection of AAV2tYF-CBA-hRS1 in patients with XLRS (NCT02599922; Supplementary Table S1). AAV8-RS/IRBP-hRS1 also entered phase I/II (NCT02317887; Supplementary Table S1).
Gene replacement therapy in models of progressive photoreceptor disorders
Progressive photoreceptor degenerations are the most common causes of complete blindness in humans. When inherited, they are primarily caused by mutations in genes expressed in rods only (retinitis pigmentosa [RP]), or in both rods and cones (LCA, RP, and cone–rod dystrophies). 61 The fact that loss of cones is associated with the most devastating aspect in these diseases points to the need to preserve cone function and survival as the primary therapeutic outcome. However, as cone death is invariably linked to the death of rods, 62 the therapy should be able in most cases to also robustly protect rod photoreceptors from degeneration.
Mutations in the PDE6β gene, which encodes the β subunit of the rod phosphodiesterase (PDE6) enzyme, are associated with one of the most common and aggressive forms of recessive rod-initiated RP. In the absence of PDE6β, PDE6 activity is severely impaired and high levels of intracellular cGMP and Ca2+ accumulate, leading to rod death. Rod dysfunction with early-onset degeneration is collectively seen in all animal models of PDE6β-RP, including the rd1, 63 –66 rd10, 67,68 and Pde6β-H620Q 69 mice, as well as the PDE6β-deficient rod cone dysplasia (rcd1) 70,71 and cone rod dystrophy 1 (crd1) dogs. 72 In all of these models, rod loss is always followed by a mutation-independent loss of cones.
Gene therapy approaches to delay rod death were first employed in the rd1 mouse, in which the majority of rods are lost by 3 weeks of age. Initial attempts to treat photoreceptor degeneration in these mice were made using adenoviral, 73,74 lentiviral, 69,75 and AAV2 vectors. 76 These gene delivery tools result in overall poor photoreceptor transduction, and these studies revealed minimal structural and functional ERG rescue. Using more efficient gene delivery tools such as AAV8-RHO-hPDE6β and AAV9-RHO-hPDE6β, long-term (13 months) functional and structural improvements were obtained after subretinal injection of rd1 mice at PN9, after removal of a confounding mutation in the Gpr179 gene. 77
Benefits of the levels and kinetics of transgene expression were also evident in the slightly slower degenerating rd10 mouse model, where injection of an AAV8-mPde6β (but not AAV5-mPde6β) vector at PN14 resulted in a robust functional rescue. 78 In comparison, subretinal injection of AAV5-CMV-hPDE6β or AAV8-CMV-hPDE6β in the same animal model at PN2, when rod differentiation is incomplete and rod transduction is inefficient (Petit L. and Punzo C., unpublished data), had only minimal therapeutic effects. 79 Another study showed that AAV8(Y773F), a vector that results in high levels of transgene expression within 2–3 days in nearly 100% of rods, 80 successfully altered the course of the disease. 81 In rcd1 dogs, injections of AAV5 and AAV8 vectors expressing cPde6β under the control of the rhodopsin kinase (RK) promoter (AAV5-hRK1-cPde6β and AAV8-hRK1-cPde6β) resulted in similar levels of functional and morphological rescue after subretinal delivery at PN20, 82 for at least 40 months posttreatment. 83 Intervention before the onset of photoreceptor degeneration (PN25) and a relatively slower progression of the disease when compared with the mouse models may have likely allowed for therapeutic transgene expression in a high proportion of photoreceptors in time, independent of the serotype used. Importance of the quality of treatment is in agreement with results obtained by the group of S. Tsang, who tested whether lack of sustained benefit in the PDE6β-deficient mouse after gene therapy is (1) because of insufficient transduction efficiency and/or (2) because the disease is too advanced at the time of treatment. Using a Cre-inducible Cre-loxP rescue allele, they demonstrated that photoreceptor degeneration in Pde6β-H620Q/LoxP is halted if DNA recombination is initiated at early, mid, and late stages of the disease. In this study, Pde6β expression after recombination was considered optimal/not limiting. 84
The potential of next-generation gene delivery tools was illustrated in animal models of other forms of severe early-onset rod–cone dystrophies (Supplementary Table S2). The PDE6α-deficient mouse has a faster rate of photoreceptor degeneration compared with the rd10 mouse, with the loss of 30% of rods already evident by PN14. Using AAV8(Y788F)-RHO-Pde6α, Wert et al. showed that treatment at PN5 and PN21 resulted in a dramatic preservation of retinal structure and cone function. 85,86 These results were very exciting, although the overall rescue of rod function was too low to result in a detectable difference by ERG. The murine model of CNGB1 retinopathy also benefited from subretinal delivery of AAV8(Y733F)-RHO-mCngb1a. 87 Like rd1 mice, Cngb1 −/− mice have no recordable rod response by ERG. However, rod degeneration progresses more slowly than in murine models of PDE6-deficiency, as 50–70% of rods are still present at 6 months of age. Treatment of Cngb1 −/− mice at PN14 resulted in dramatic restoration of rod function, accounting for 33% of wild-type levels corresponding to the surface of the retina directly exposed to the vector. Improvement of the functional component of the disease was associated with a preservation of 50–70% of rods in treated eyes at 12 months of age. More recently, Palfi et al. demonstrated that AAVrh10 transduces rods comparably to AAV8 in a degenerating retina. 88 Using this serotype and an optimized murine rhodopsin promoter, they showed great progress in the treatment of the rhodopsin knockout mouse, compared with similar doses of AAV5. 89 Nonetheless, despite the fact that therapeutic benefits were observed up to 11 months after treatment with AAVrh10, degeneration was not arrested in treated retinas. 88
While most of the gene replacement therapies for photoreceptor diseases discussed here target either cones or rods, it is useful to treat the two photoreceptor subtypes simultaneously. This is because many inherited photoreceptor degenerations are caused by mutations in genes expressed in both rods and cones. The short human rhodopsin kinase 1 (hRK1) promoter was the first well-defined promoter able to drive efficient transgene expression in both cell types, when used in conjunction with AAV. 90 While the efficiency of cone transduction remains very low as compared with rod transduction, validation of this promoter in small 91 and large animal models, 91 –93 along with the development of next-generation of vectors, led in the past few years to an exponential growth of retinal gene transfer studies targeting both rods and cones.
A case in point is the gene replacement therapy for the retina-specific guanylate cyclase (GUCY2D), which is expressed exclusively in rods and cones and constitutes one of the most common causes of LCA. 61 The GC1-deficient mouse undergoes severe cone dysfunction before cone degeneration. Rods retain 30–50% of their function and do not degenerate, because of the presence of a second guanylate cyclase (GC2). Subretinal injection of AAV5-CBA-bovine Gc1 at PN21 had no effect in the GC1-deficient mouse model. 94 In contrast, AAV5-CBA-mGucy2e and AAV5-hRK1-mGucy2e at PN14 restored 45% of cone ERG responses and preserved cone survival for at least 9 months postinjection. 95 Subsequently, injection of AAV8-hRK1 vectors carrying the murine Gucy2e or human GUCY2D cDNA at PN10 provided a 65% rescue of cone ERG responses, cone vision and cone survival for up to 6 months, as well as a 35% rescue of rod function. 96 A more recent study reported a restoration of 54% and 38% of cone ERG responses in mice treated at PN21 with AAV8-CMV-hGUCY2D and AAV8-hRK1-hGUCY2D, respectively. 91
Comparison of AAV5-hRK1-mGucy2e and AAV8(Y733F)-hRK1-mGucy2e/hGUCY2D confirmed the superiority of AAV8(Y733F) in restoring cone function in Gucy2e −/− mice 97 and Nrl −/− Gucy2e −/− mice. 98 This difference likely reflects the ability of AAV8(Y733F) to drive faster transgene expression than AAV5, and indicates that this temporal difference is important in terms of the ability to restore function. However, it is unclear whether there is a higher overall number of preserved cones and/or a better functional rescue of transduced cones. Regarding rod function, there were no differences between the AAV5 and the AAV8(Y733F) vectors.
The ability of AAV8(Y733F)-hRK1-mGucy2e to rescue both cones and rods was confirmed in the GC1/GC2 double-deficient mouse, which exhibits complete loss of both cone and rod ERG responses, as well as slow degeneration of rods. Both cone and rod ERG responses were restored to 42–44% of wild-type levels after treatment at PN18, and to 26–29% of wild-type levels after treatment at PN108. The rescue remained stable for at least 1 year posttreatment. However, only intervention before PN108 slowed photoreceptor degeneration. 99
Interestingly, benefits of AAV8-based vectors over AAV5 have also been observed in the RPGRiP1 (retinitis pigmentosa GTPase regulator interacting protein 1)-deficient 100,101 and AiPL1 (aryl hydrocarbon receptor interacting protein-like 1)-deficient 102 –104 mouse models of early-onset severe photoreceptor dystrophies, but not in the RPGRIP1-deficient dog, in which AAV5 and AAV8 gave similar therapeutic effects, 93 probably reflecting differences in the kinetics of photoreceptor loss and timing of therapeutic transgene expression between these species.
The first evidence of the efficacy of gene replacement therapy in a large animal model of severe photoreceptor dystrophies was obtained in the XLPRA1 (X-linked progressive retinal atrophy) and XLPRA2 canine models of RPGR-X-linked RP. 105,106 In these dogs, Beltran et al. evaluated the efficacy of AAV5-hRK1-hRPGR and AAV5-IRBP-hRPGR to mitigate retinal degeneration. 105 The XLPRA1 dogs were treated at 28 weeks of age, before any apparent signs of photoreceptor degeneration. In contrast, XLPRA2 dogs were treated at the onset of the disease, which is at 5 weeks of age. In both cases, treatment resulted in convincing preservation of retinal structure in the vector-exposed area for a 2-year period. 105 However, the benefits seen in XLPRA2 dogs treated with AAV5-hRK1-hRPGR or with low dose of AAV5-IRBP-hRPGR were smaller likely because these animals were treated at the onset of the disease. 106 Early evaluation of the effects of gene transfer on retinal function was difficult, because of the long-term preservation of functional photoreceptors in areas not exposed by the vector. 106 Nonetheless, retention of rod and cone function was clearly seen in XLPRA2 dogs up to 3 years of age with improved long-term vision. 105
Interestingly, XLPRA2 dogs treated at 12 weeks of age (loss of ∼40% of the total photoreceptors) showed an initial decline in outer nuclear layer (ONL) thickness, but ONL loss in the vector-exposed area was halted from 31 weeks to 2.7 years of age. XLPRA2 dogs treated at 26 weeks of age (loss of 50–60% of total photoreceptors) showed a similar profile, but with a decrease and stabilization of ONL thickness after 52 weeks of age. A higher proportion of nontreated photoreceptors could explain this delay in ONL stabilization after late intervention. Remarkably, ERG analysis showed preserved rod function in all 3 dogs treated at this late stage, and preserved cone function in 2 out 3 dogs, accounting for 8% of wild-type ERG levels, providing very exciting proof-of-concept to support a future clinical trial. 105
Additional improvements
Extensive efforts have also been made to expand the applicability of gene-specific therapy in the eye, including the treatment of photoreceptor dystrophies caused by mutations in large genes, the treatment of autosomal dominant retinal diseases, and the delivery of transgene to a large retinal surface by intravitreal injection. The reader is referred to recent reviews for further information about the strategies that are being devised to overcome these limitations. 107 –109
Broadening the Spectrum of Treatable Patients by the Development of Gene-Independent Therapies
Over the last two decades, proof-of-concept studies of corrective gene therapy have been established in many different animal models of inherited retinopathies, strongly supporting further translation from animal models to human. However, evidence of the potency and efficacy of gene transfer to the retina at late stages of the disease is less robust. Early physiological alteration and loss of photoreceptors cells remain important factors limiting the therapeutic window for corrective gene therapy. In addition, with over 240 genes associated with retinal degeneration in humans, targeting individually each group of patients will likely not be possible. Mutation-independent therapies that allow treating entire families of retinal degenerative diseases would represent a more cost-effective approach (Fig. 1). Current strategies that utilize gene therapy and might be the more broadly applicable are detailed below.
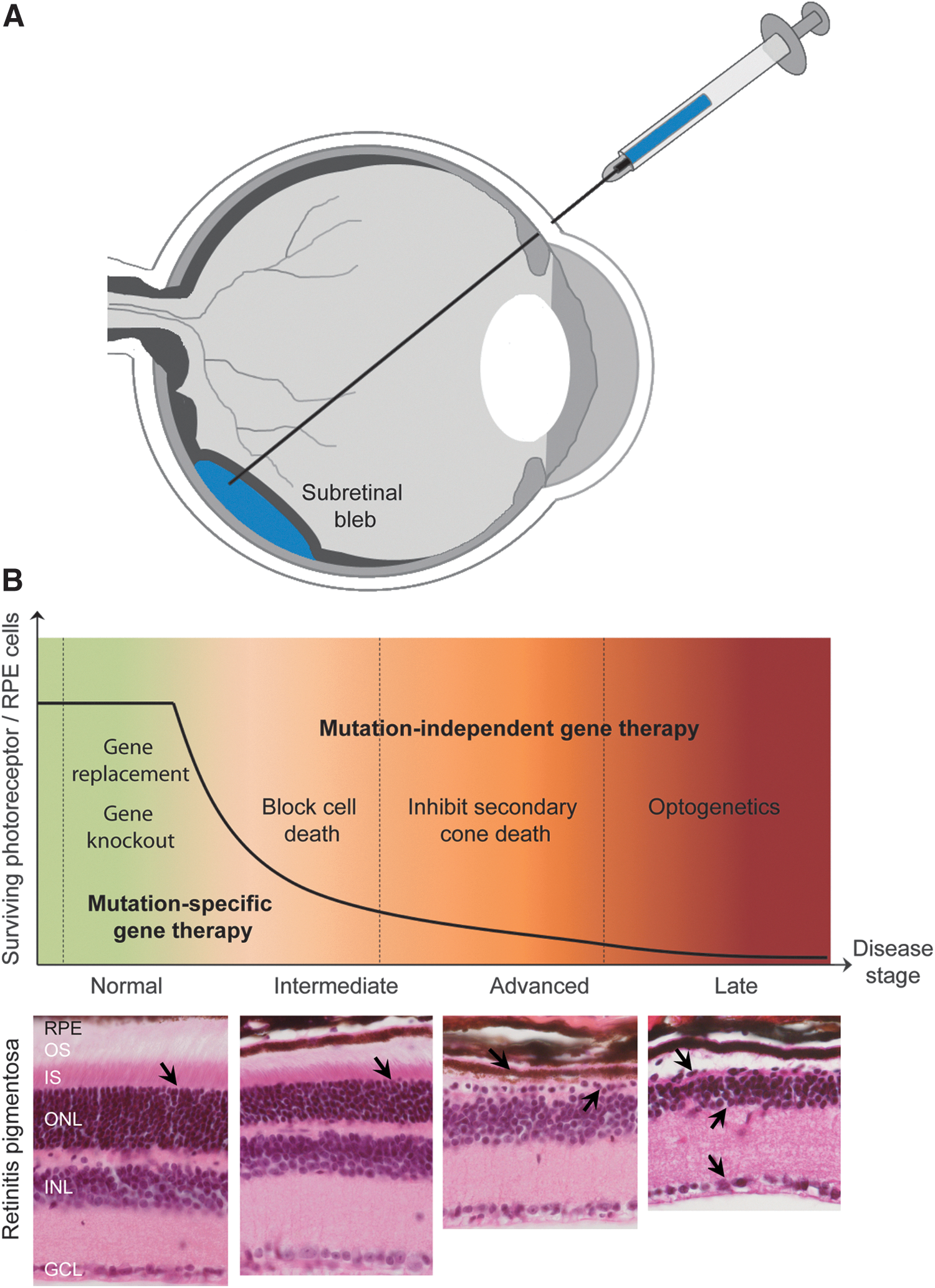
Gene therapy strategies for inherited retinal diseases.
Preventing secondary cone death in retinitis pigmentosa
Diseases that benefit the most from a mutation-independent approach are those associated with photoreceptor loss, in particular RP. The reason why this disease context is attractive for a mutation-independent approach is that, in the vast majority of cases, the genetic defects are specific to rod photoreceptors; however, cones die as well. It is this secondary loss of cones that causes the severe visual disabilities and complete blindness in humans. Consequently, gene therapies that prolong cone function and survival for such patients would maintain the temporal window of useful vision in a large number of patients.
A first point of intervention that is being developed inhibits rod cell death, which should prevent or delay the secondary loss of cones. Because the onset of cone death always follows the major rod death phase, 62 even a small delay in the death of rods could translate into a high preservation of cones.
One of the most straightforward ways to inhibit cell death is to inhibit the execution of cell death itself. Photoreceptor cell death has been found under various stress conditions to be predominantly apoptotic 110 and executed by caspases. 111 The X-linked inhibitor of apoptosis (XIAP) is one of a series of proteins that can inhibit cell death by binding to the two executioner caspases 3 and 7 and to the initiator caspase 9. 112,113 Misexpression of XIAP by adenoviral- or AAV2-mediated gene transfer has been shown to delay ganglion cell death in various rodent models of optic nerve injury 114 –116 and protect neurons from retinal ischemia 117 and from the mutagen N-nitroso-N-methylurea. 118 Most recently, subretinal injection of AAV5-CBA-hXIAP has been shown to prevent photoreceptor death in the P23H rat model of dominant retinitis pigmentosa for at least 30 weeks 119 and confer photoreceptor protection 2 months after sodium hylauronate-induced retinal detachment. 120
Inhibition of photoreceptor degeneration using growth factors has also been tested. The ciliary neurotrophic factor (CNTF) is the most studied. CNTF has been shown to delay ganglion cell death through direct and AAV delivery 121 –125 and to delay photoreceptor death in various animal models of RP. 126 –129 A recent study in Rho −/− mice showed that intraocular delivery of AAV2-hCNTF can confer long-term rod and cone photoreceptors protection and significantly delay vision loss even when rod death is well advanced. 130 However, the mode of action of CNTF is not fully understood, and even though CNTF is protective in animals with photoreceptor degeneration, high doses are associated with an acute deconstruction of photoreceptor outer segments 131,132 and gene expression changes that are similar to those seen in light-induced photoreceptor plasticity. 133 Interestingly, the temporary deconstruction of photoreceptor outer segments appears to enhance gene transfer by AAV5 in a canine model of CNBG3-ACMH, making CNTF a potential therapeutic candidate to pretreat the eye before receiving the actual gene therapy. 29 Clinical evaluation of CNTF for the treatment of retinal degeneration has been already conducted using encapsulated cell implants in patients with advanced stages of RP (NCT00063765). In this phase 1 study, 10 patients received the implants in one eye for 6 months without major side effects. 134 Subsequently, three phase 2 clinical trials for early RP (NCT00447980, 68 patients), late RP (NCT00447993, 65 patients), and geographic atrophy (NCT00447954, 39 patients) were initiated. Data at 2 years postimplantation demonstrated that expression of CNTF was maintained over 24 months, with no serious adverse effects. 135,136 In patients with early-stage RP, no protection of rods has been demonstrated. Macular cone photoreceptors remained stable over 12–35 months, when sham-treated eyes experience a 9–24% decrease in cone number; however, this protective effect was not associated with detectable changes in visual acuity and ERG responses. 137 Determining final outcomes of the therapy on cone survival will require a longer follow-up because of the slow progressive nature of RP. If successful, AAV-mediated CNTF gene therapies may follow.
Besides the classical neuroprotective factors, both AAV-mediated delivery of erythropoietin (EPO), a cytokine that is upregulated during hypoxia, and AAV-mediated delivery of proinsulin have been shown to have neuroprotective properties in several models of RP. 138 –140 Interestingly, however, only AAV-mediated systemic administration of EPO but not intraocular administration appears to be protective. 141 To translate this into a human gene therapy, EPO derivatives have been used that were protective after subretinal delivery by AAV in various models of retinal degeneration. 139 However, how applicable EPO derivatives and proinsulin are for a human therapy remains to be determined.
A second point of intervention targets cones directly to protect them from degeneration once the majority of rods have died. This requires an understanding of the mechanism of cone death, for which several models have been proposed. Some of these models seem to converge around energy availability and the associated redox potential (for review see ref. 142 ).
Originally, it was believed that rods produce a trophic factor that is required for cone survival simply because cone death always follows rod death, regardless of the circumstances that lead to rod death. Identification of such factor would inevitably provide a unifying therapy for all forms of RP that are caused by mutations in rod-specific genes. In 2004, the rod-derived cone viability factor (RdCVF), 143 encoded by the nucleoredoxin-like 1 (NXNL1) gene, was identified and since has moved from the proof-of-concept as an injectable trophic factor that can delay cone death 143,144 into an AAV-mediated gene therapeutic approach. 145 RdCVF is a thioredoxin-like protein with two isoforms 146 and was initially believed to reduce oxidative stress, a disease condition that accompanies the degeneration of rods and cones because of the massive loss of rods. 142 Meanwhile, the protein has been shown to indirectly interact with the glucose transporter-1, thereby promoting glucose uptake in cones during the period of cone degeneration. 147 This finding is in line with one of the proposed mechanisms of secondary cone death, which postulates that cones are nutrient deprived; in particular, they are short of glucose. 62,148 Systemic intravenous delivery to rd10 mice of AAV92YF-CAG-Nxnl1 at PN1 resulted in improved cone-mediated ERG responses and cone survival at 1 month of age. In the same mouse model, intravitreal injection at PN14 of a novel AAV2 variant with increased photoreceptor cells tropism, 7m8-CAG-Nxnl1, also resulted in good preservation of cone function and survival 1 month posttreatment. 145 Clinical trials will undoubtedly determine if RdCVF becomes a therapeutic agent with broad applicability in humans.
The realization that oxidative stress is a contributing factor to secondary cone death 149 led to extensive research in antioxidant therapies, many of which have shown promising results in mouse. 150,151 In humans, various combinations of vitamins and omega-3 fatty acids have been tested, showing a slight effect in delaying the disease progression. 152 –154 However, the problem with orally supplemented antioxidants is that they may never reach critical concentrations in the tissue of interest. This circumstance, as well as the finding that antioxidant enzymes may need to be present in the right combination 155 directly in sick cones to reduce oxidative stress, 156 led to the first AAV-mediated approaches in which various enzymes and transcription factors that regulate the expression of detoxifying enzymes were tested. 155,157 The most promising of these candidates, nuclear factor erythroid-derived 2 like 2 (NRF2), showed a remarkable delay in cone death in two mouse models of RP, opening the door for a new mutation-independent approach for secondary cone death.
By investigating the molecular mechanisms of secondary cone death, we recently revealed that cones are nutrient deprived in RP, 62 a finding that is in line with the increase in oxidative stress seen in cones during degeneration, 149 since lack of glucose reduces the redox potential of a cell. 142 Because rods account for over 95% of all photoreceptors, we proposed that once a critical threshold of rod death is breached, cone death initiates a cell autonomous event caused by the collapse of the relatively few cone–RPE interactions reducing nutrient flow to cones. 142 This model explains why cones may survive for extended periods in the cone-rich central retina of patients and large animal models of RP, despite the total loss of rods, and why cone-specific diseases do not lead to rod degeneration. Genetic hyperactivation of a key kinase that promotes cell metabolism, the kinase mechanistic target of rapamycin complex 1 (mTORC1), has led to a remarkable delay of cone death for at least 8 months of age in two mouse models of RP. 148 The data represent the most profound and long-lasting effect of cone survival seen thus far, strongly suggesting that boosting cell metabolism in cones is a viable strategy to prolong cone survival in RP. 158 A targeted gene therapy approach that augments mTORC1 target genes in cones is currently under development. Interestingly, extending cell autonomous cone survival may go beyond RP and hold promise for many other degenerative diseases, such as age-related macular degeneration (AMD). 159 In these diseases, once a critical number of photoreceptors have died in a specific region, the remaining healthy photoreceptors in that region may be affected by reduced nutrient flow, as are cones in RP. In addition, the degeneration of rescued photoreceptors observed in RPE65-treated patients may involve a similar mechanism of cone death to the one in RP. 28
Restoration of visual sensitivity by optogenetics
Restoration of vision through optogenetics provides an interesting strategy to restore visual perception in blind retinas (see reviews 160,161 ). This therapeutic approach relies on delivering a gene encoding a light-sensitive channel protein (microbial opsins, endogenous opsins, or synthetic light-sensitive ion channels) to either reactivate dormant cones or activate other retinal neurons at late stages of degeneration.
Opsin-like microbial proteins have the advantage to reversibly isomerize a vitamin A derivate as the chromophore. 162 In response to light, microbial opsins can thus independently induce changes in the membrane potential of a cell allowing the electrical signal to propagate to the brain. Channelrhodospin (CHR2) was the first microbial opsin to be used in the retina. CHR2 pumps cations upon excitation by light and produces excitatory currents. AAV-mediated expression of CHR2 at the level of ON bipolar cells 163 –167 and ganglion cells 168 in mouse models of RP successfully restored light-evoked potentials in treated retinas. While transduction of ganglion and bipolar cells is still difficult in large animal models, novel vectors and promoters with better affinity for the inner retina have been recently reported. 166,169,170 Moreover, good preservation of these cells has been demonstrated in patients with advanced RP and LCA. 171 Another approach is to reactivate nonfunctional “dormant” cone photoreceptors using halorhodospins, which produce inhibitory currents. 172 This strategy may restore visual processing in all layers of the retina. However, it remains to be determined who is an appropriate patient for this procedure as many RP patients keep functional cones for decades and the remodeling of the retinal network upon loss of photoreceptors could make this approach challenging in humans. 173 In addition, long-term effects of membrane potential depolarization/repolarization on nutrient-deprived cones remain unexplored.
Despite these advances, one problem microbial optogenetics faces is the light sensitivity and dynamic range of these various channels. Recently, a new generation of synthetic light-acting channels has been described that use azobenzene-based light switches fused to ion channels or receptors. 174 –176 One such approach remodeled the ionotropic glutamate receptor 177 restoring visual function at the level of ganglion cells 178 and bipolar cells. 179 Initially, optogenetics was introduced in 2002 by simultaneous coexpression of the Drosophila rhodopsin, arrestin-2, and the alpha subunit of the G-protein to activate culture hippocampal neurons by light. 180 The approach never gained much favorability over the microbial opsins because it requires a triple transduction, something that remains difficult to achieve in the retina. However, the idea of using endogenous G-protein-coupled receptors (GPCR) such as melanopsin, 181 which is involved in circadian rhythm and the pupillary reflex 182 and is naturally expressed in a subset of ganglion cells, has been revisited on the premise that other cells may express some of the components needed to activate the cascade with a GPCR. Recent experiments have shown that, when rhodopsin is delivered to bipolar cells, light sensitivity is restored by 2–3 orders of magnitude lower than what can be achieved by the microbial channels, making this a feasible approach to restore vision in late stages of RP. 183
Antiangiogenic gene therapy for acquired retinopathies
The most successful mutation-independent approach thus far has been applied to two diseases, neither of which is caused by a specific mutation but rather by a combination of genetic and environmental risk factors. Wet AMD 184 and diabetic retinopathy (DR) 185 are both characterized by formation of new blood vessels of the choroidal and retinal vasculature, respectively. The subsequent leakage of fluid from these newly formed blood vessels into the retinal proper is what generally causes massive neuronal loss. The realization that vascular endothelial growth factor (VEGF) is one of the key culprits in promoting the disease pathology has led to the development of anti-VEGF therapies with first-generation therapies utilizing anti-VEGF antibodies (Lucentis [approved in 2006 for wet AMD] and Avastin [Genetech], Avastin being the parent molecule of Lucentis) that are administered intravitreally. However, the therapy requires continuous administration on an interval of 4–8 weeks, is costly, is associated with potential side effects from the repeated injections, and is a burden for patients because of the many clinical visits. Hence, gene therapeutic approaches have been established to either target VEGF itself, by viral-mediated overexpression of the soluble ligand binding part of the vascular endothelial growth factor receptor 1 gene (FLT1), 186 –192 or by inhibiting its action through use of angiogenesis inhibitors such as pigment epithelium-derived factor (PEDF) 193 –195 or a combination of two angiostatic factors, angiostatin and endostatin. 196 All three gene therapy approaches have since moved to the clinic. Because of its vast experimental evidence in animal models 186 –192 and preclinical trials, 197 expression of sFLT1 by AAV2-mediated gene transfer is the most advanced approach with two ongoing clinical trials. Avalanche-Lions Institute is evaluating an alternatively spliced form of FLT1, using the subretinal route (NCT01494805). Published results at 1 year posttreatment indicate that 4 out of 6 patients did not require any rescue injections with anti-VEGF antibodies. 198 Genzyme uses intravitreal delivery (NCT01024998). In parallel, Oxford Biomedica is evaluating the efficacy of simultaneous expression of angiostatin and endostatin by subretinal injection of EIAV-LV (RetinoStat, NCT01301443).
Conclusion and Future Prospects
Despite existing limitations and questions that still need to be addressed and resolved, gene therapy for ocular diseases continues to show great promise for the future. Current clinical studies of gene replacement therapy, although not all are convincing in terms of longevity of therapeutic effects, have demonstrated a good safety record. They provide evidence that it is possible to obtain clinically relevant visual improvements after gene replacement. With the recent improvements in AAV vectors and the increase in therapeutic benefits observed in preclinical studies, new clinical trials should translate into even more encouraging results in patients. During the next decade, a better understanding of the mechanisms that commonly limit the efficacy of gene replacement therapy over the long-term will be a key aspect that needs to be solved to move forward with retinal gene replacement therapies. Simultaneously, lessons learned from the biology of retinopathies should enable the development of viable therapeutic strategies to prolong vision independently of the disease-causing mutations, further encouraging transfer to clinical trials. With the advent of genome editing technology for gene and cell therapy, future studies should also be aimed at safe and long-term AAV-mediated delivery of genome-modifying components to the target cell types in the eye, as well as other organs.
Footnotes
Acknowledgments
The following funding support is gratefully acknowledged: the “Information Recherche Retinite Pigmentaire” research grant, the Fulbright/Fondation Mohanan Fellowship, the Fondation de France/Fondation Berthe Fouassier Fellowship, and the “Association Francaise contre les Myopathies” Fellowship. Work in the H.K. lab is supported by grants from the National Institutes of Health (EY022372), University of Massachusetts Center for Clinical and Translational Sciences (UMCCTS), Massachusetts Lions Eye Research Fund, and Foundation Fighting Blindness (FFB). Work in the C.P. lab is supported by the NIH (EY023570) and the Massachusetts Lions Eye Research Fund.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.