
Abstract
At present, the clinically most advanced strategy to treat Duchenne muscular dystrophy (DMD) is the exon-skipping strategy. Whereas antisense oligonucleotide-based clinical trials are underway for DMD, it is essential to determine the dystrophin restoration threshold needed to ensure improvement of muscle physiology at the molecular level. A preclinical trial has been conducted in golden retriever muscular dystrophy (GRMD) dogs treated in a forelimb by locoregional delivery of rAAV8-U7snRNA to promote exon skipping on the canine dystrophin messenger. Here, we exploited rAAV8-U7snRNA-transduced GRMD muscle samples, well characterized for their percentage of dystrophin-positive fibers, with the aim of defining the threshold of dystrophin rescue necessary for normalization of the status of neuronal nitric oxide synthase mu (nNOSμ), inducible nitric oxide synthase (iNOS), and ryanodine receptor-calcium release channel type 1 (RyR1), crucial actors for efficient contractile function. Results showed that restoration of dystrophin in 40% of muscle fibers is needed to decrease abnormal cytosolic nNOSμ expression and to reduce overexpression of iNOS, these two parameters leading to a reduction in the NO level in the muscle fibers. Furthermore, the same percentage of dystrophin-positive fibers of 40% was associated with the normalization of RyR1 nitrosylation status and with stabilization of the RyR1–calstabin1 complex that is required to facilitate coupled gating. We concluded that a minimal threshold of 40% of dystrophin-positive fibers is necessary for the reinstatement of central proteins needed for proper muscle contractile function, and thus identified a rate of dystrophin expression significantly improving, at the molecular level, the dystrophic muscle physiology.
Introduction
D
A preclinical trial has been conducted in dystrophic GRMD dogs treated in a forelimb by locoregional delivery of rAAV8-U7snRNA inducing specific exon skipping in order to restore an in-frame dystrophin mRNA. 12 This study aimed to optimize the injection protocol, identify the safety profile, and define a therapeutic dose, and allowed the definition of a threshold of 40% dystrophin-expressing fibers needed to detect a significant improvement of strength. 12
Although the recovery of muscle strength is an essential parameter for quantifying the benefit of therapies, the definition of a rate of dystrophin-positive fiber expression capable of improving muscle pathophysiology at the molecular level remains an important point to investigate. In the present work, we used rAAV8-U7snRNA-transduced GRMD muscle samples, well characterized for their percentage of dystrophin-positive fibers, to define a threshold of dystrophin rescue necessary to recover the status of the neuronal nitric oxide synthase μ isoform (nNOSμ), inducible nitric oxide synthase (iNOS), and ryanodine receptor-calcium release channel type 1 (RyR1). Indeed, it is well known that muscle function is dependent on the integrity of the dystrophin complex-associated proteins, including the presence of nNOSμ. Therefore, we paid particular attention to its expression and its location. nNOSμ binds to dystrophin, via its rod domain at spectrin-like repeats 16 and 17 (R16/17). 13 The lack of dystrophin leads, directly or indirectly, to the delocalization of nNOSμ from the sarcolemma and its accumulation in the cytosol, inducing vasoconstriction and necrosis in contracting muscles, 14 as well as to structural defects of the neuromuscular junction. 15 Another isoform of NOS, called iNOS, is absent or present at a low level in normal skeletal muscles 16 ; nevertheless, its expression is increased in inflamed tissues. 17,18 Interestingly, iNOS protein is up-expressed in muscles of mdx mice and patients with DMD, 19,20 and its expression is reduced by gene transfer of dystrophin or utrophin in mdx mice. 20 In dystrophic muscle, both nNOSμ mislocalization and iNOS up-regulation lead to abnormal NO production in the cytosol, altering skeletal muscle contractility via the excessive S-nitrosylation of proteins, including RyR1. RyR1 contains a large number of free thiols whose nitrosylation influences channel function, 21 and its hypernitrosylation is associated with depletion of immunophilin FKBP12 (calstabin1) from the RyR1 complex. 22 The role of calstabin1 is to stabilize the RyR1 complex and it is required for its optimal function especially to facilitate coupled gating. 23,24 Consequently, dissociation of calstabin1 from the RyR1 complex, described in the muscle of mdx mice, 25 results in leaky calcium channels, which alters the excitation–contraction coupling and thus impacts the strength/function of the muscle.
Here, taking advantage of a preclinical study achieved in the GRMD model that provided well-characterized muscular samples displaying an increasing percentage of dystrophin-positive fibers, we defined the threshold of dystrophin-expressing fibers necessary for normalization of the status of two NOS proteins and the RyR1–calstabin1 complex, central proteins for efficient contractile function.
Materials and Methods
Animals and study design
Dog muscle samples used in the present study were obtained from a preclinical study, designed in the Atlantic Gene Therapies laboratory, which was based on the delivery of rAAV8-U7snRNA for specific exon skipping in order to restore an in-frame dystrophin mRNA.
12
Dogs were part of the breeding Center of Domaine des Souches (Mezilles, France) or the Boisbonne Center for Gene Therapy (ONIRIS, Atlantic Gene Therapies, Nantes, France). The Institutional Animal Care and Use Committee of the Region des Pays de la Loire (University of Angers, Angers, France) approved the protocol (authorization 2011.31, obtained in 2011). The dogs were injected, via locoregional transvenous injection in one forelimb, with 5 × 1013 vector genomes (VG)/kg of a therapeutic rAAV8-U7snRNA-E6/E8 vector, as described in Le Guiner and colleagues.
12
The injection was done via the cephalic vein after having placed a tourniquet above the elbow of the injected forelimb. Fourteen weeks postinjection, euthanasia was performed by intravenous injection of pentobarbital sodium (pentobarbital sodium [Dolethal]; Vetoquinol, Buckingham, UK). The samples were collected in the area located below the tourniquet, from paws injected or not with vector. Two untreated GRMD dogs that received vehicle only and one wild-type dog were used as controls. Dog tissue samples flash frozen in liquid nitrogen were used for Western blot and immunoprecipitation experiments (dogs D8, D13, and D14), and histology was carried out on sections collected from muscle samples frozen in isopentane cooled in liquid nitrogen (dogs D8 and D13). These muscles were obtained either from the injected forelimb or the noninjected forelimb, as summarized in Supplementary Table S1 (supplementary data are available online at
Antibodies
For dystrophin immunolabeling, we used a rabbit polyclonal antibody (Thermo Scientific, Waltham, MA) or a goat polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA). For nNOS, we used a rabbit polyclonal antibody (BD Biosciences, San Jose, CA) for immunolabeling and a mouse monoclonal antibody (BD Biosciences) for Western blotting. For iNOS detection, we used a rabbit polyclonal antibody (Abcam, Cambridge, UK). We used a mouse monoclonal antibody anti-GM130 (Golgi matrix protein 130; BD Biosciences) and a rabbit polyclonal antibody anti-CD11b (Abcam) as markers of Golgi complex and macrophages, respectively. To check nitrosylation of RyR1, both a mouse monoclonal antibody anti-RyR1 (Thermo Scientific) and a rabbit polyclonal anti-S-nitroso-cysteine (Cys-NO) antibody (Sigma-Aldrich, St. Louis, MO) were used. For RyR1 immunoprecipitation, the mouse monoclonal antibody (Thermo Scientific) was used, and for the detection by Western blotting we used a polyclonal antibody anti-RyR1 (a kind gift from I. Marty, Grenoble Institut des Neurosciences, Université Joseph Fourier, La Tronche, France) and a polyclonal antibody anti-calstabin1 (Abcam). For actin, we used a mouse monoclonal antibody (Sigma-Aldrich). For Na/K ATPase, a marker of membranes, we used a monoclonal antibody (GeneTex, Irvine, CA) and for glyceraldehyde 3-phosphate dehydrogenase (GADPH), a marker of cytosol, we used a polyclonal antibody (Santa Cruz Biotechnology). Secondary antibodies for immunolabeling were from Life Technologies (Carlsbad, CA) (Alexa Fluor 488, 594, and 633 conjugates). For Western blotting, secondary antibodies coupled to horseradish peroxidase were from Jackson ImmunoResearch Laboratories (West Grove, PA), and for RyR1 nitrosylation experiments we used IRDye 680-conjugated goat anti-mouse IgG and IRDye 800CW-conjugated goat anti-rabbit IgG from LI-COR (Lincoln, NE).
Histology and immunolabeling
For muscle analysis, tissue sections were cut at 8 μm on a cryostat, fixed in 4% paraformaldehyde for 10 min, and stained with hematoxylin–eosin. Images were acquired with a Leica DM R microscope.
For immunolabeling, cryosections of 8 μm were permeabilized with 0.25% Triton X-100 and blocked in phosphate-buffered saline (PBS)–10% fetal bovine serum (FBS) for 1 hr. Sections were incubated in PBS–2% FBS with primary antibodies overnight at 4°C and washed in PBS. Sections were then incubated with secondary antibodies for 1 hr, washed in PBS, incubated with 4′,6′-diamidino-2-phenylindole (DAPI) for nuclear staining, and mounted in Fluoromount-G (CliniSciences, Nanterre, France). Images were acquired with a macroscope Nikon AZ100 or Leica SPE confocal microscope.
Costaining dystrophin/NADPH diaphorase activity
To assess NADPH diaphorase activity, tissue sections were fixed in 4% paraformaldehyde for 8 min, permeabilized with 0.25% Triton X-100 for 10 min, and then incubated with a 0.2-mg/ml concentration of NBT (nitroblue tetrazolium with phosphate buffer with NaCl) and 2 mg/ml NADPH (β-nicotinamide adenine dinucleotide phosphate reduced) (Sigma-Aldrich) in PBS–0.25% Triton X-100 for 3 hr at 37°C in the dark. Sections were then washed in PBS and treated for dystrophin immunolabeling. Sections were mounted in Fluoromount-G (CliniSciences). Images were acquired with a Nikon Ti microscope.
Muscle tissue lysate
For nNOS and iNOS detection in Western blotting and for RyR1 protein immunoprecipitation, muscle tissues were homogenized with a Dounce homogenizer in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 100 mM NaCl, and 0.5% NP-40 with a mix of protease inhibitors. For RyR1 nitrosylation experiments, tissues were homogenized in lysis buffer containing 200 mM sucrose, 20 mM HEPES (pH 7.4), and 0.5 mM CaCl2 with a mix of protease inhibitors.
Membranes and cytosol fractionation
We performed membrane–cytosol fractionation with a Mem-PER Plus membrane protein extraction kit (Thermo Scientific). Total lysate, membrane fraction, and cytoplasmic fraction were isolated from 35 mg of muscle, in accordance with the manufacturer instructions for hard tissues. Forty micrograms of total and membrane proteins and 60 μg of cytoplasmic proteins were used for Western blotting. Quantification was performed with Quantity One software.
Immunoprecipitation
Samples were centrifuged for 10 min at 14,000 × g, and 600 μg of proteins was precleared with 20 μl of washed protein G–Sepharose (PGS 4 fast flow; GE Healthcare, Piscataway, NJ) and then incubated with mouse monoclonal anti-RyR1 antibody or with mouse anti-IgG overnight at 4°C. Then, 40 μl of protein G–Sepharose beads was added for 2 hr at 4°C. Beads were collected by centrifugation and washed with lysis buffer. Proteins were eluted in Laemmli buffer.
Western blotting
For nNOS and iNOS detection and for RyR1 protein immunoprecipitation, samples were centrifuged for 10 min at 14,000 × g and denatured at room temperature for 30 min with Laemmli buffer. For RyR1 nitrosylation experiments, samples were centrifuged for 5 min at 1500 × g and remained under nondenaturing and nonreducing conditions. Proteins were separated by electrophoresis (NuPAGE 4–12% Bis-Tris gel; Life Technologies) and then transferred to nitrocellulose membranes and labeled with primary antibodies and secondary antibodies coupled to horseradish peroxidase or IRDye secondary antibodies for the Odyssey infrared system (LI-COR). Quantification was performed with Quantity One software.
Functional assessment
The functional assessment was performed during the preclinical study conducted by Le Guiner and colleagues. 12 Briefly, movements of flexion and extension of the carpal joint were elicited by subcutaneous stimulation of the common branch of the medial and ulnar nerves for flexion measurements or of the radial nerve for extension measurements. Stimulations were performed at seven different frequencies from 5 to 200 Hz. Signals were analyzed at the end of the experiment and the maximum value obtained at each stimulation frequency was recorded to obtain the frequency–response curve for each function tested. Values were obtained in volts and converted to torque (in N·m). The maximal strength value obtained over all stimulation frequencies was then expressed as a percentage of predicted normal values. Three measurement sessions were planned: before injection, and 1.5 and 3 months after injection. The strength changes were expressed as a percentage of change per month.
Results
Description of muscular samples used in the study
To assess the percentage of dystrophin-positive fibers required for the improvement of muscle physiology at the molecular level, we used dog muscle samples obtained from a preclinical trial based on the delivery of rAAV8-U7snRNA for specific exon skipping in order to restore an in-frame dystrophin mRNA 12 ; more details on this study are reported in Materials and Methods. Because we were especially interested in selecting muscle samples with increasing amounts of dystrophin-positive fibers, we collected muscle samples previously well characterized for their percentage of dystrophin-positive fibers. Indeed, as reported in the study by Le Guiner and colleagues, the percentage of dystrophin-positive fibers was quantified after immunoperoxidase staining of frozen transverse sections of the muscles, using a specific anti-dystrophin antibody (NCL-DYS2), by manual count. For each section, a total of at least 250 fibers was counted. 12 A fiber was considered positive when more than one-third of its circumference exhibited staining more intense than that of negative controls (negative fibers in untreated GRMD dogs).
On the basis of these data, we classified the collected muscle samples into four groups, depending on the percentage of dystrophin-positive fibers on each muscle section as follows: the percentage of dystrophin-positive fibers is less than 20% (<20%); between 20 and 30% (20–30%); between 40 and 50% (40–50%); or more than 65% (>65%) (Supplementary Table S1). One wild-type dog, two untreated GRMD dogs (dogs C1 and C2), and three treated GRMD dogs (dogs D8, D13, and D14) were included for molecular investigations. To validate the dystrophin expression in all the muscle samples used in the present study, immunofluorescence labeling analyses were performed (Fig. 1A).
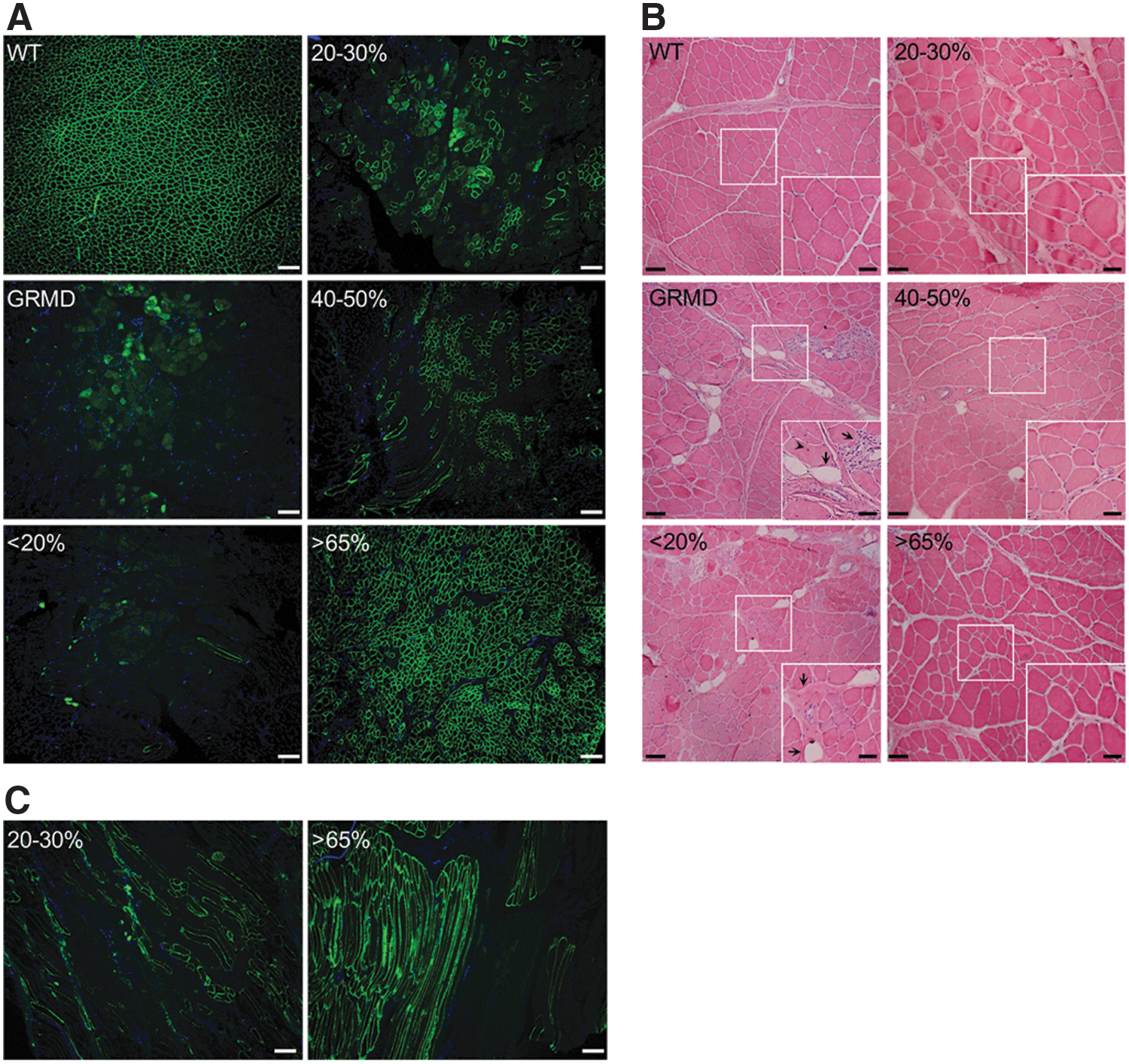
Restoration of dystrophin is associated with improvement of histopathophysiological features in canine muscle sections.
Positive dystrophin fibers were counted on sections of muscle excluding the diffuse labeling due to necrotic fibers. The estimating amount of dystrophin-positive fibers was in good accordance with the dystrophin quantification performed in the study by Le Guiner and colleagues. 12 Furthermore, to confirm that the dystrophin-positive fibers detected on sections were positive all along the fibers, reflecting thus the percentage of positive fibers relative to the entire muscle, we performed immunostaining for dystrophin on sections displaying longitudinal fibers. Analysis of dystrophin labeling on these sections confirmed that a fiber was dystrophin positive or dystrophin negative along its entire length (Fig. 1B). To investigate histopathophysiological features of muscles, hematoxylin and eosin staining was performed on sample cryosections (Fig. 1C). In muscles from the wild-type dog, fibers were uniform in both size and shape and nuclei were peripherally localized. GRMD muscles presented typical dystrophic features including heterogeneity in fiber size, few centralized nuclei, infiltrating mononuclear cells, endomysial fibrosis, and adiposis. In accordance with Le Guiner and colleagues, 12 histological improvement with more uniform fibers and less fibrosis and adipose tissue was observed in muscle samples presenting more than 40% dystrophin-positive fibers.
Determination of percentage of dystrophin-positive fibers needed for proper localization and expression of nNOSμ protein
To investigate nNOSμ protein expression in the various canine muscle samples, Western blotting was done on total protein extracts from the four classes of muscle samples (<20, 20–30, 40–50, and >65%) (Fig. 2A). Results showed a 160-kDa band corresponding to the muscular nNOSμ isoform, and the quantification of band intensity revealed equal levels in wild-type dogs and in untreated and treated GRMD dogs whatever the percentage of dystrophin-positive fibers. To answer the question concerning what percentage of dystrophin-positive fibers is needed to recover proper nNOSμ localization, immunolabeling of dystrophin and nNOSμ was investigated in the four classes of muscle samples (Fig. 2B). Immunolabeling carried out on sections of wild-type dog muscle confirmed nNOSμ colocalization with dystrophin at the sarcolemma. In untreated GRMD muscles, nNOSμ was detected in the cytosol as aggregates, with an accumulation under the sarcolemma and in the perinuclear zone.
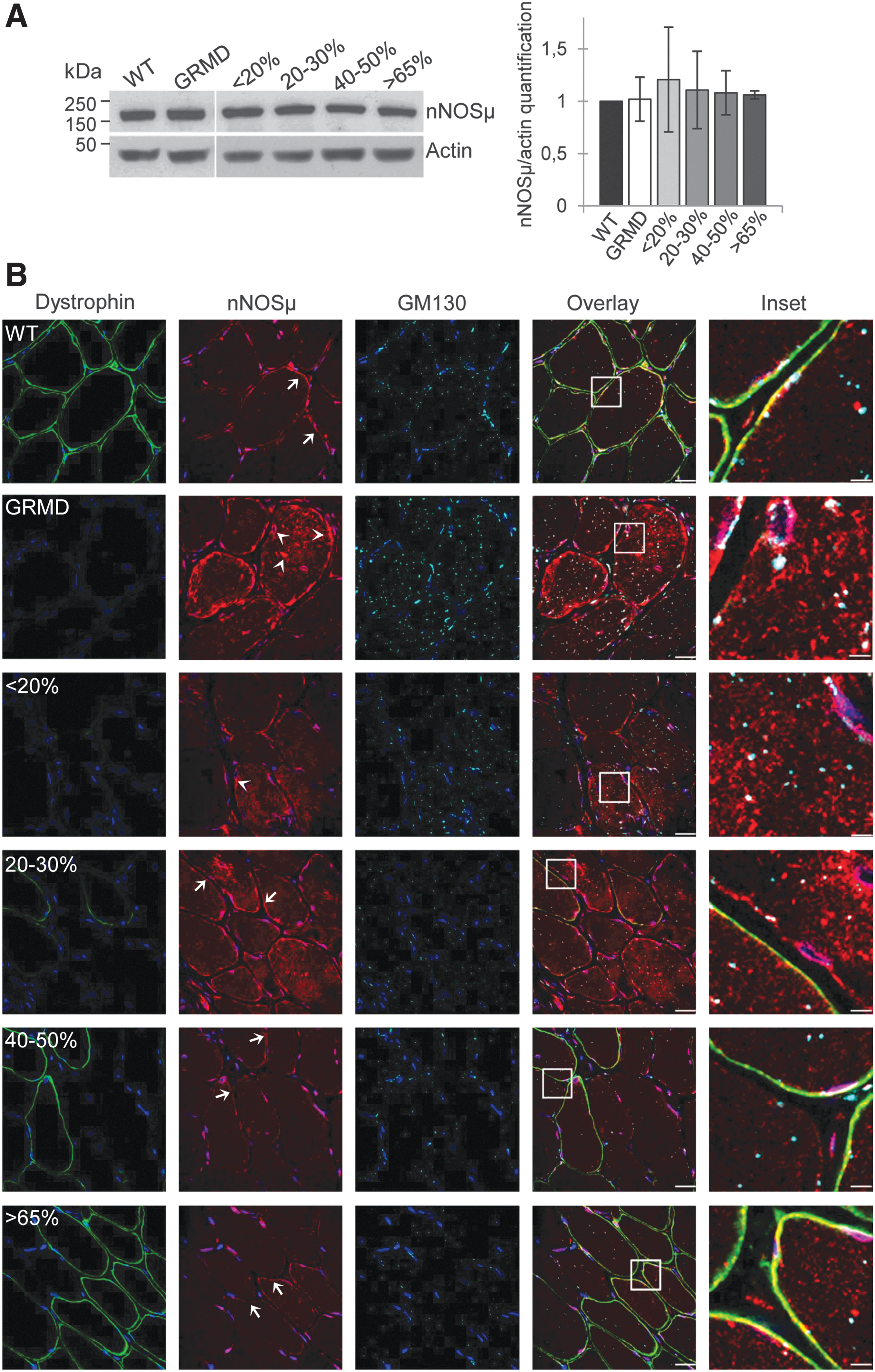
Restoration of dystrophin in 40% of fibers is needed to decrease cytosolic neuronal nitric oxide synthase mu (nNOSμ).
To support the imaging results, subcellular fractionation experiments were performed with muscle samples from control, GRMD, and treated muscle expressing >65% dystrophin-positive fibers. Data showed the presence of nNOSμ protein in the cytosol fraction obtained from GRMD muscle samples, whereas no protein could be detected in the cytosolic fractions from control or GRMD dog tissue samples expressing more than 65% dystrophin-positive fibers. Furthermore, nNOSμ protein was found in the enriched membrane fractions from control and treated GRMD >65% muscle samples whereas a weak band was observed for the nontreated GRMD muscle sample (Supplementary Fig. S1).
To precise identify the cytosolic nNOSμ location, we performed double-immunofluorescence labeling of this compartment and nNOSμ protein using GM130, a marker of Golgi apparatus, and nNOSμ antibodies, respectively (Fig. 2B). In wild-type muscles, Golgi puncta were localized principally around the nuclei, with a few localized deeper inside the fibers. In untreated GRMD muscles, the Golgi apparatus was disorganized and accumulated in the cytosol. Cytosolic nNOSμ was not colocalized with Golgi puncta but appeared near the Golgi apparatus as aggresome-like inclusions, as already described in primary cortical neurons. 26
In muscle samples displaying less than 30% dystrophin-positive fibers, nNOSμ was detected in the cytosol and in subsarcolemmal aggregates. Surprisingly, this abnormal location was observed in both dystrophin-negative and dystrophin-positive fibers. Nevertheless, a partial sarcolemmal location of nNOSμ was observed in a few fibers exhibiting strong positive dystrophin staining and a correct organization of the Golgi complex. In treated muscles presenting more than 40% dystrophin-positive fibers, a major reduction in nNOSμ staining in the cytosol and the subsarcolemmal region was detected. In addition, although proper location of nNOSμ to the sarcolemma requires the presence of dystrophin at the membrane, unexpectedly, the disappearance of cytosolic nNOSμ staining was observed in both dystrophin-positive and dystrophin-negative fibers. Similarly, in these muscle samples, Golgi apparatus organization was restored in all fibers.
Determination of percentage of dystrophin-positive fibers needed for proper expression of iNOS protein
Untreated GRMD muscle sections stained with hematoxylin and eosin revealed the presence of strong cellular infiltrates (Fig. 1C) confirmed by the immunolabeling of CD11b-positive macrophages (Supplementary Fig. S2). This prompted us to focus on the expression of iNOS, which has been described to be up-regulated in inflamed tissues and thus strongly expressed in dystrophic muscles. 20 To evaluate its expression in the GRMD dog model, Western blot analysis was performed on total protein extracts from the various muscle samples (Fig. 3A). Results showed that untreated GRMD muscles displayed an 8-fold increase in iNOS protein expression compared with wild-type muscles. Furthermore, we observed that under 30% of dystrophin-positive fibers, treated muscles displayed also an 8-fold increase in iNOS expression. The restoration of dystrophin in more than 40% of fibers resulted in a significant halving of iNOS expression in the muscle extracts.

Restoration of dystrophin in 40% of fibers is needed for the reinstatement of inducible nitric oxide synthase (iNOS) expression.
These findings were confirmed by an immunolabeling experiment (Fig. 3B) showing, as expected, strong iNOS expression in infiltrating cells, but also up-expression in the cytosol and at the sarcolemma of all muscle fibers of the GRMD muscles. This is in stark contrast to wild-type muscles, where iNOS was detectable only in interstitial cells. These data confirmed that iNOS expression was associated with infiltration by inflammatory cells and was up-expressed in untreated GRMD muscle fibers. Furthermore, the data showed that in muscle samples containing less than 20% dystrophin-positive fibers, iNOS was highly expressed in fibers and in interstitial cells as observed in untreated GRMD dogs (Fig. 3B). In muscle sections displaying between 20 and 30% dystrophin-positive fibers, iNOS expression detected by Western blotting was not significantly different compared with the GRMD muscle samples (Fig. 3B); however, an immunofluorescence experiment allowed us to discriminate the iNOS protein location: decreasing in the fibers while intense labeling remained obvious between the interstitial cells (Fig. 3B). When more than 40% of fibers were positive for dystrophin, the rescue of dystrophin was correlated with a reduction of iNOS expression in the whole muscle.
Determination of percentage of dystrophin-positive fibers for proper NOS enzymatic activity
To correlate NOS protein expression and location with enzymatic activity, we detected NADPH diaphorase activity as a marker for nitric oxide synthase 27 by colorimetric staining concomitantly with the detection of dystrophin by immunofluorescence (Fig. 4). As expected, the labeling of NO production in the fibers was strictly sarcolemmal in wild-type muscles, because of the nNOSμ presence at the membrane. On the other hand, in GRMD muscle fibers, NO accumulation was found in the cytosol, in the subsarcolemmal region and in interstitial areas, results that were well correlated with what was observed for nNOSμ and iNOS expression and location. In muscle samples displaying less than 30% dystrophin-positive fibers, NO was found accumulated in the cytosol and in the subsarcolemmal region in both dystrophin-negative and dystrophin-positive fibers. In treated muscles expressing more than 40% dystrophin-positive fibers, NOS enzymatic activity was detected at the sarcolemma in dystrophin-positive fibers associated with a major reduction in cytosolic NO production. This decrease was even observed in some dystrophin-negative fibers. When more than 65% of fibers were positive for dystrophin, NADPH diaphorase staining was similar to that observed in wild-type muscles. In accordance with the preceding data, these results revealed that a restoration of dystrophin in 40% of muscle fibers was needed to lead to a reduction of the NO level within the fibers.
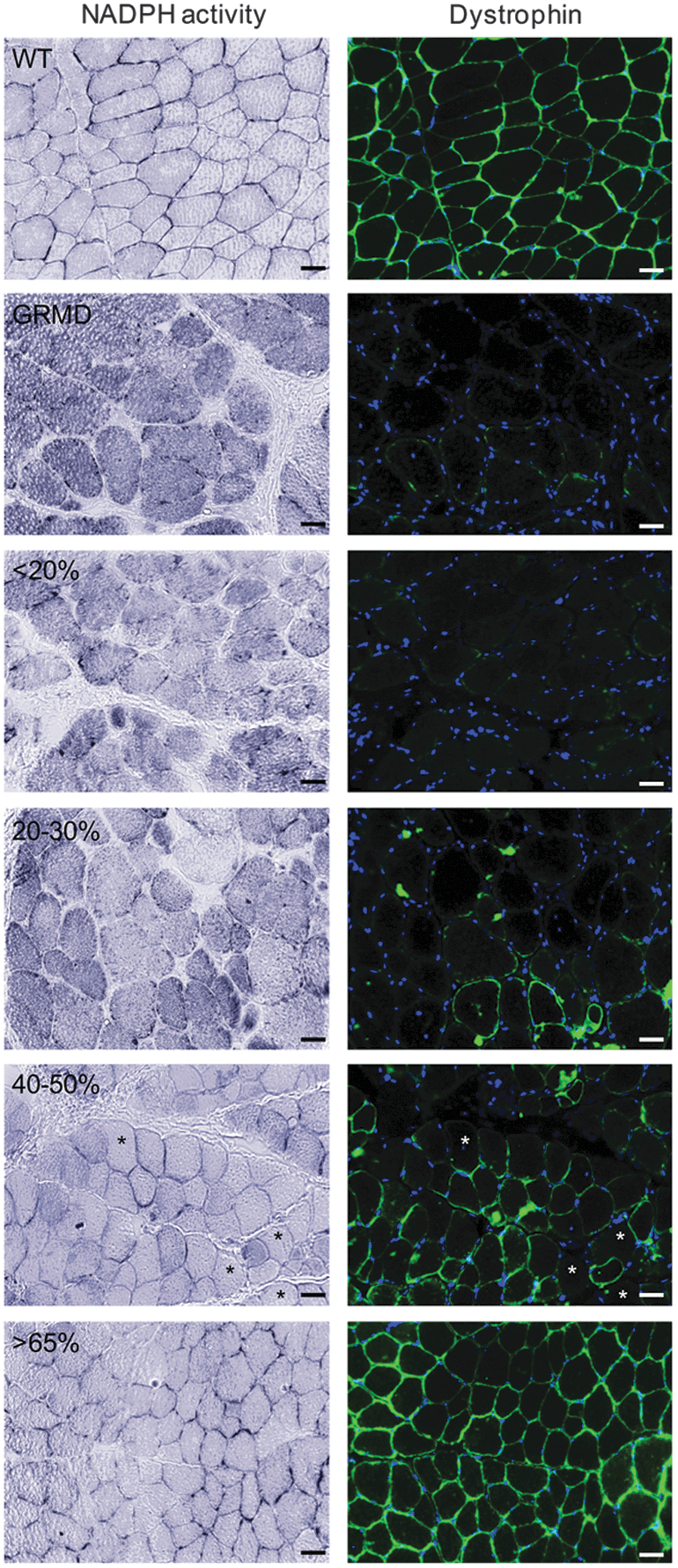
Restoration of dystrophin in 40% of fibers leads to a major reduction in cytosolic nitric oxide (NO) level. NADPH diaphorase activity staining was visualized with the colorless soluble tetrazolium salt (blue staining) and dystrophin labeling on transverse cryosections of muscle samples from WT, untreated dog C2 (GRMD), and treated dog D13 expressing <20, 20–30%, 40–50, or >65% dystrophin-positive fibers. Asterisks indicate dystrophin-negative fibers displaying a major reduction of cytosolic NO production. Scale bars: 25 μm.
Determination of percentage of dystrophin-positive fibers needed to normalize RyR1–calstabin1 complex status
Cytosolic NO produced by cytoplasmic nNOSμ and iNOS could alter skeletal muscle contractility via the S-nitrosylation of RyR1 and could subsequently reduce the affinity of calstabin1 for RyR1. 21,22 In this regard, RyR1 expression and its nitrosylation state were investigated by multiplex Western blotting of muscle sample lysates from untreated and treated GRMD muscles and compared with wild-type muscles (Fig. 5A). Although the total RyR1 levels were identical in untreated GRMD and wild-type muscle extracts, S-nitrosylation of the RyR1 cysteine residues was detected only in untreated GRMD muscles. The data revealed that the expression levels of RyR1 were lower in treated GRMD muscles than in wild-type and untreated GRMD muscles; this difference is probably due to the sensitivity of the RyR1 protein to the sample storage conditions. The nitrosylation status of RyR1 was investigated in the four classes of treated GRMD muscle samples and showed that RyR1 was hypernitrosylated, in muscle samples presenting a level below 30% dystrophin-positive fibers, as observed in untreated GRMD samples. Interestingly, S-nitrosylation of RyR1 decreased when muscles expressed more than 40% dystrophin-positive fibers and was almost undetectable in muscles displaying more than 65% dystrophin-positive fibers. These data were confirmed by confocal microscopy analysis of S-nitrosocysteine (Cys-NO) expression achieved on muscle sections of untreated and treated GRMD muscles compared with wild-type muscles (Fig. 5B). Indeed, a higher Cys-NO expression level was observed in untreated GRMD muscles in comparison with wild-type muscles. In accordance with Western blotting, the level of the nitrosylated RyR1 was inversely proportional to the level of dystrophin rescue in muscles from treated GRMD dogs. These data demonstrated that a minimal percentage of 40–65% dystrophin-positive fibers was needed to return to the physiological RyR1 state.
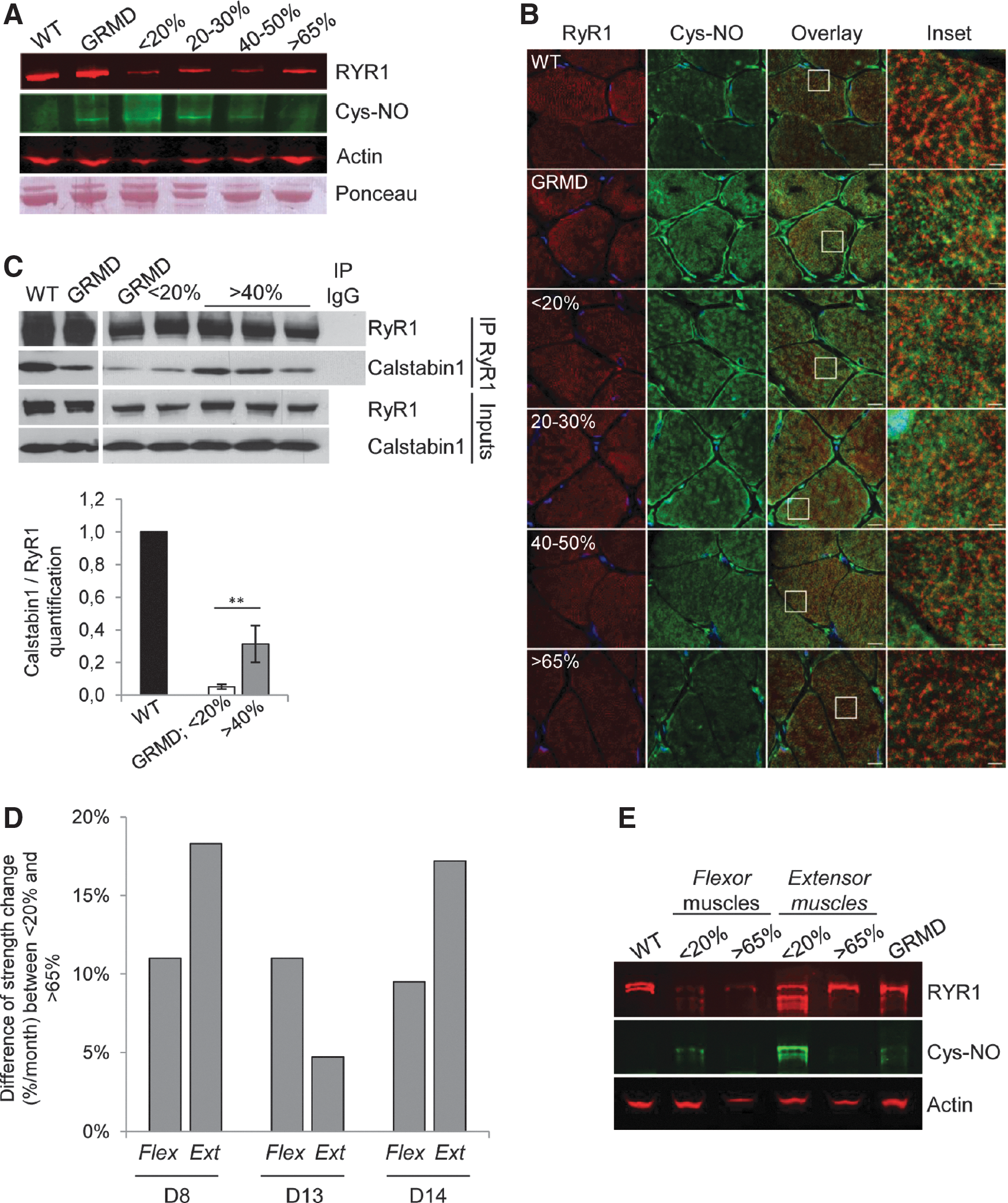
Restoration of dystrophin in 40% of fibers is needed to stabilize ryanodine receptor-calcium release channel type 1 (RyR1)–calstabin1 complex.
To study the impact of RyR1 hypernitrosylation on the calstabin1–RyR1 interaction, RyR1 was immunoprecipitated (IP RyR1) from muscle samples of wild-type, untreated, and treated GRMD dogs (Fig. 5C). Both total and immunoprecipitated proteins were immunoblotted with antibodies against RyR1 and calstabin1. Inputs confirmed that the total amounts of RyR1 and calstabin1 were identical in all muscle extracts. A 20-fold decrease in calstabin1 bound to RyR1, compared with wild-type muscle extracts, was observed in nontreated GRMD muscles and when the number of dystrophin-positive fibers was inferior to 20%. However, more than 40% dystrophin-positive fibers was associated with a stabilized calstabin1–RyR1 interaction with a significant 6-fold enhancement of calstabin1 binding to RyR1. Overall, these data revealed that restoration of dystrophin in at least 40% of muscle fibers allowed increased RyR1 channel stability and suggested an improvement of the excitation–contraction coupling.
To establish a link between these molecular data and muscular function, RyR1 nitrosylation status was investigated precisely in untreated or treated flexor carpi ulnaris muscles and extensor carpi radialis muscles previously used for measurement of force generation in the three treated GRMD dogs (dogs D8, D13, and D14) included in the preclinical previous study. 12 We collected data of force measurements and the corresponding muscle samples for S-nitrosylation of the RyR1 status investigation for two (dogs D8 and D14) of these three dogs. Flexion and extension strengths were evaluated at the wrist, using a custom-made torque measurement device (Fig. 5D). Three measurements were carried out to determine strength change: before injection, 1.5 months postinjection, and 3 months postinjection. In muscles from noninjected control forelimb, the percentage of dystrophin-positive fibers may be 20%, due to diffusion of the therapeutic vector in the contralateral limbs, whereas the treated muscles displayed at least 65% dystrophin-positive fibers. As described by Le Guiner and colleagues, 12 treated muscles exhibiting more than 65% dystrophin-positive fibers displayed an increase in strength between 4.7 and 18.3% for extensor and between 9.5 and 11% for flexor muscles compared with noninjected contralateral muscles. RyR1 nitrosylation status was then precisely investigated in both flexor and extensor (from dogs D8 and D14) expressing (1) less than 20% dystrophin-positive fibers or (2) more than 65% dystrophin-positive fibers and compared with muscles from wild-type and untreated GRMD dogs (Fig. 5E). The profile of RyR1 expression observed on multiplex Western blotting was slightly different between the two treated GRMD dogs (D8 and D14); this difference was probably due to the sensitivity of the RyR1 protein to the conditions of sample storage. Nevertheless, muscles that expressed less than 20% dystrophin-positive fibers exhibited increased S-nitrosylation of RyR1 that was almost undetectable when muscles presented more than 65% dystrophin-positive fibers. These data strongly suggested that RyR1 status normalization observed in treated GRMD muscles was correlated with an improvement in muscle strength.
Discussion
The present study indicates, for the first time in the dystrophic GRMD dog model, that restoration of dystrophin in 40% of muscle fibers is needed for reinstatement of molecular actors reported to be essential for ensuring efficient contractile function and for improving the global physiopathologic state of the treated muscle.
Previous investigations to determine dystrophin levels required to improve muscle function have already been performed in the mdx mouse model by using phosphorodiamidate morpholino oligomer (PMO) to modulate dystrophin pre-mRNA by exon skipping. 28,29 These studies concluded that 15–20% of dystrophin expression was sufficient to protect dystrophic muscle against exercise-induced damage but was not enough to enhance muscle strength. However, a series of repeated injections for 20 weeks allowed an increase in specific isometric force correlated with an average of 50% dystrophin-positive fibers in the tibialis anterior muscles. It is well known that mdx muscles are less affected by the absence of dystrophin compared with GRMD muscles, which display a more severe phenotype mimicking the progressive degeneration observed in human DMD muscles and resulting, for the GRMD dogs, in an important reduction in life span. 30 Differences between the species could account for the variable thresholds of dystrophin levels needed to obtain physiological benefits. Indeed, in the GRMD dog model, two studies have described benefits of dystrophin rescue with the exon-skipping strategy in skeletal muscles from GRMD dogs. 11,12 The first showed that intramuscular injection of 3-week-old GRMD puppies allowed dystrophin rescue to about 80%; however, this protocol was not designed to define the dystrophin threshold level needed to improve muscular contractile function. Second, dogs at 3–4 months of age were injected by locoregional transvenous perfusion with various therapeutic doses and showed a significant decrease in myofiber regeneration and endomysial and total fibrosis when the percentage of dystrophin-positive fibers was at least 33%. Nevertheless, the strength increase was observed when muscles displayed a restoration of dystrophin of at least 40% of fibers. Finally, regarding the correlation between the levels of dystrophin-positive fibers and muscle force, 40–50% dystrophin-positive fibers are required to observe an improvement in muscle strength whatever the studies and animal models.
Today, there is growing interest in restoring dystrophin in patients with DMD; although the major evaluation of therapeutic benefit relies on measurement of the improvement in muscle strength, a need remains to determine the level of dystrophin restoration required to improve muscle pathophysiology at the molecular level. Here, in an attempt to answer this question, we focused the assessments on the status of two NOS proteins and the RyR1–calstabin1 complex because of their crucial roles for ensuring the efficiency of contractile function.
Dystrophin rescue leads to proper NOS protein expression and location
In DMD, the lack of dystrophin leads to nNOS mislocalization from the sarcolemma to the cytosol, causing functional ischemia and an exacerbating fatigue response to exercise. 14,31 nNOSμ is then accumulated in the cytosol, inducing nitrosative stress and impaired muscle contraction. 32 In mdx mice, restoration of dystrophin expression by gene therapy allowed the recovery of nNOSμ at the sarcolemma, which was associated with physiological benefits. 33 –35 In our previous study involving muscle samples from patients with Becker muscular dystrophy, carrying a spontaneous 45–55 deletion in the DMD gene affecting the nNOSμ-binding domain, we correlated the severity of the dystrophic phenotype with the degree of cytosolic nNOSμ localization. 36 In this study, proper relocation of the sarcolemmal nNOSμ was systematically observed as concomitant with the rescue of dystrophin expression. In addition, the abnormal cytosolic nNOSμ observed in untreated GRMD was totally absent in muscles with 40% dystrophin-positive fibers, and in whole muscle comprising both dystrophin-positive and dystrophin-negative fibers. This observation was consistent with what was observed when the activity of NADPH was measured concurrently with dystrophin expression (Fig. 4), where in some fibers the NADPH activity was low although dystrophin was not expressed. Moreover, analysis of the histopathophysiological features of this class of muscle expressing 40% dystrophin-positive fibers revealed a remarkable improvement in phenotype, suggesting that it is linked more to the decrease in cytosolic nNOS than to proper sarcolemmal localization.
Dystrophin expression and loss of cytosolic nNOSμ in treated muscles were correlated with a restoration of normal Golgi apparatus organization. Previous studies have suggested that the cytosolic nNOS protein could correspond to the nNOSβ isoform, an nNOS splice variant. This isoform was first described as being localized at the Golgi complex in mdx skeletal muscle, and suggested to be involved in compensation mechanisms such as hypertrophy. 37 In GRMD dogs, we showed that cytosolic nNOS seems to form aggresome-like inclusions localized adjacent to Golgi cisternae as previously observed in primary cortical neurons, 26 but was not colocalized with the Golgi complex. Moreover, we were not able to detect the presence of the nNOSβ isoform described to be associated with the Golgi, either by RT-PCR or with a specific antibody, suggesting that this isoform does not correspond to the cytosolic nNOS observed in GRMD muscles.
The other member of the NOS protein family studied here, iNOS, was found to be overexpressed in dystrophic muscles displaying strong inflammation. In mdx mice, dystrophin expression or utrophin overexpression by gene transfer reduces iNOS expression. 20 Our findings also demonstrated that iNOS expression was inversely correlated with the dystrophin expression level, and allowed us to define a percentage of 40% dystrophin-positive fibers as totally suppressing its expression in interstitial cells and in myofibers. Nevertheless, the contribution of iNOS to the pathophysiology of DMD remains unclear. Two teams have independently generated iNOS/dystrophin double-null mice and could not observe changes in macrophage infiltration. 38,39 However, whereas Villalta and colleagues showed that ablation of iNOS in mdx mice reduced muscle fiber injury of the soleus muscle, Li and colleagues found that the removal of iNOS in mdx mice did not improve muscle histopathology and specific force of the extensor digitorum longus muscle. A possible explanation for such contradictory results could be linked to the different fiber type compositions (fast or slow twitch) of the muscles studied or to different muscle solicitation.
Dystrophin rescue leads to normalization of RyR1–calstabin1 complex status, correlated with improvement in strength
Previous studies have attempted to define the NOS, iNOS or nNOSμ, responsible for nitrosylation of RyR1. Bellinger and colleagues suggested that S-nitrosylation of RyR1 must be attributed to iNOS activity in mdx mouse on the basis that iNOS protein was immunoprecipitated with RyR1 and that nNOSμ expression was decreased in mdx muscle. 25 However, Salanova and colleagues showed a colocalization of RyR1 and nNOSμ in myofibers from normal human skeletal muscle, 40 and Li and colleagues showed that the absence of nNOSμ in dystrophin/nNOSμ double-deficient mice minimized RyR1 S-nitrosylation, improving muscle force. 32 Although this question remains open for the time being, our study showed that total nNOSμ was not reduced in GRMD muscle and that both NOS proteins disappeared concomitantly from the muscle cytosol after dystrophin rescue, leading to RyR1 status normalization.
Our findings show, for the first time, that dystrophin rescue in more than 40% of muscle fibers corrects RyR1 nitrosylation status, which is correlated with the return of calstabin1–RyR1 complex stability. However, several studies have shown that a default in RyR1 channel activity, due to the dissociation of calstabin1, led to defective calcium signaling and impaired skeletal muscle contractility. 23,24 Restoration of calstabin1 binding to RyR1 thus appears to be an interesting therapeutic strategy. In this context, drugs such as S107, which prevents the depletion of calstabin1 from the RyR1 complex, have been developed. In mdx mice, RyR1 status normalization using S107 was correlated with an improvement in grip strength. 25 In the dog model, force measurement protocols are not suitable to measure the force generated by a specific muscle. One protocol has been developed to record the strength of a single intact muscle in dystrophin-deficient dogs, 41 but it was not appropriate to our study because this procedure required an important surgical device and animals must be euthanized at the end of the experiment. In the present study, the protocol allowed attribution of the force generated mostly to one specific flexor and one specific extensor muscle. In concert with our hypothesis, we observed that RyR1 status normalization in these treated GRMD muscles resulted in an improvement in strength.
A threshold of 40% dystrophin-positive fibers is necessary to improve molecular parameters crucial for efficient contractile function
The present study raises a provocative question concerning whether dystrophin is the major provider of the global beneficial effect observed in muscle as a whole. Indeed, in these conditions, where 60% of the fibers do not express dystrophin, the muscle should remain susceptible to mechanical trauma with subsequent muscle fiber loss in an inflammatory environment. However, in muscles having 40% dystrophin-positive fibers or more, abnormal cytosolic nNOSμ was totally absent and NADPH activity was low, and this in whole muscle. Regarding iNOS expression, the iNOS level in dystrophin-positive fibers versus dystrophin-negative fibers was quantified in muscle sections colabeled for dystrophin and iNOS (Supplementary Fig. S3). This quantification confirmed that, even if iNOS staining significantly decreased in muscle samples presenting more than 40% positive fibers, the iNOS level in dystrophin-positive fibers versus dystrophin-negative fibers was not significantly different. We thus assume that the improvement in muscle dystrophic status should not be attributed restrictively to the local mechanical and structural role of dystrophin as being the single determinant of muscle fiber health. This question has already been discussed in a comment to the study by Le Guiner and colleagues. 42 A more general mechanism might involve a decrease in inflammatory status and, in particular, a reduction in the amount of macrophages leading to a decrease in the level of iNOS activation in all muscle fibers, as was observed in the present study. Moreover, a more diffuse mechanism must be considered, in which the regulation of calcium homeostasis would play a critical role in muscle fiber integrity. Indeed, it is known that high intracellular calcium concentration activates iNOS protein expression via cell lysis and macrophage activation 18,43 and nNOS via calcium–calmodulin kinase activation. 44 Finally, at the molecular level, NOS protein expression and location, RyR1 channel stabilization, and the improvement in muscle strength are all parameters correlated to the intracellular calcium level, which therefore would be a significant contributor to the general beneficial effect observed in the treated muscle even though more than half of the fibers do not express dystrophin.
To conclude, the present study defines a threshold of 40% dystrophin-positive fibers as necessary for improving the status of crucial proteins for efficient contractile function. The results also highlight the important point that dystrophin rescue is probably not the unique determining element for reinstating the physiological status of treated muscle, thus providing new information in the search for the development of successful therapeutic approaches to DMD.
Footnotes
Acknowledgments
The authors thank Prof. J.C. Kaplan, Dr. S. Vassilopoulos, and Dr. Y. Chérel for valuable contributions and Dr. B. Cadot for help in microscopy image acquisition. The authors thank M. Jorre, Dr. C. Falcone, and M. Dutilleul for technical assistance and Dr. I. Marty for kindly providing RyR1 antibody. This work was supported by the Association Française contre les Myopathies (AFM), the Association Institut de Myologie (AIM), the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé et de la Recherche Médicale (INSERM), and the Université Pierre et Marie-Curie Paris 6 (UPMC).
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.