
Abstract
A ready source of autologous myogenic cells is of vital importance for drug screening and functional genetic studies in Duchenne muscular dystrophy (DMD), a rare disease caused by a variety of dystrophin gene mutations. As stem cells (SCs) can be easily and noninvasively obtained from urine specimens, we set out to determine whether they could be myogenically induced and useful in DMD research. To this end, we isolated stem cells from the urine of two healthy donors and from one patient with DMD, and performed surface marker characterization, myogenic differentiation (MyoD), and then transfection with antisense oligoribonucleotides to test for exon skipping and protein restoration. We demonstrated that native urine-derived stem cells express the full-length dystrophin transcript, and that the dystrophin mutation was retained in the cells of the patient with DMD, although the dystrophin protein was detected solely in control cells after myogenic transformation according to the phenotype. Notably, we also showed that treatment with antisense oligoribonucleotide against dystrophin exon 44 induced skipping in both native and MyoD-transformed urine-derived stem cells in DMD, with a therapeutic transcript-reframing effect, as well as visible protein restoration in the latter. Hence MyoD-transformed cells may be a good myogenic model for studying dystrophin gene expression, and native urine stem cells could be used to study the dystrophin transcript, and both diagnostic procedures and splicing modulation therapies in both patients and control subjects, without invasive and costly collection methods. New, bankable bioproducts from urine stem cells, useful for prescreening studies and therapeutic applications alike, are also foreseeable after further, more in-depth characterization.
Introduction
D
Current treatment via corticosteroid drugs is beneficial, and, ataluren, marketed under the trade name Translarna in Europe, has been provisionally approved as an orphan drug for boys with DMD with nonsense mutations. 1 However, the large number of other mutations identified in the dystrophin gene mean that novel personalized medicine strategies are a fundamental research target, in this and other rare diseases. 2 Furthermore, preclinical studies aiming at accurately evaluating the efficacy of emerging treatments, based, for example, on splicing modulation (antisense), gene correction (gene editing), or dystrophin analog upregulation (utrophin), would greatly optimize the clinical trials process settings. 3 Indeed, although regulatory authorities currently sanction only human trials of drugs whose safety and efficacy have previously been tested in animal models, often nonhuman primates, preliminary studies in disease-specific in vitro models would offer huge benefits in terms of both costs and accuracy. 4
In this context, induced pluripotent stem cells (iPSCs), which are able to differentiate into various cell types that express the same genotype and phenotypic features of their donors, represent an appealing avenue of research. iPSCs, obtained by reprogramming somatic cells, were first used to obtain differentiated murine skeletal and cardiac myocytes in 2006. 5 iPSCs were generated from mouse somatic cells by means of a combination of four transcription factors (Oct3/4, Sox2, Klf4, and c-Myc) transduced by retroviruses, a methodology replicated in human cells soon thereafter. 6
The importance of this approach lies in its suitability for human in vitro modeling, cell-based therapies, and tissue regeneration. Despite the technical feasibility and obvious appeal of this methodology, inducement remains a challenge, especially for iPSCs derived from adult tissues, which present their own unique complexity. New and more efficient strategies and technical advances are therefore required before such methods can be applied to the clinical setting. In particular, the identification of noninvasive cell sources that lend themselves to both simple, repeated expansion in culture and rapid and efficient reprogramming is essential. 7
To this end, Zhou and colleagues 8 reported that human urine could be a novel source for iPSC derivation, and, more recently, Guan and colleagues 9 showed the feasibility of rapid iPSC generation and differentiation into beating cardiomyocytes that recapitulate the DMD phenotype from the urine samples of a patient with DMD.
The use of urine as a source of stem cells to generate an in vitro model of human disease has also been validated by Jouni and collaborators 10 in a patient with long QT syndrome, a cardiac arrhythmic disease.
Compared with other mesenchymal stem cells (MSCs), urine stem cells (USCs) have many advantages. Indeed, they allow for inexpensive, easy, noninvasive access, without surgical procedures, and can be obtained from people of different ages, sexes, and health status. Furthermore, a single USC can be expanded into many additional cells, retaining telomerase activity and replication potential, but without generating teratomas or other types of tumor. 11 USCs also show the capacity for multipotent differentiation and express pericyte/mesenchymal markers, although they do not express hematopoietic stem cell markers (except for MHC-1), endothelial cell markers, or human leukocyte antigen (locus) DR. 11,12
Even though next-generation sequencing (NGS) techniques now allow rapid and comprehensive identification of DMD gene mutations, 13 muscle biopsy is often required of boys with DMD if no mutations are detected by NGS or other DNA-based techniques, if there is no genotype–phenotype correlation, and in cases of complex phenotype, characterized by nonmuscle symptoms (i.e., intellectual disability) or other atypical features. Moreover, muscle tissue or myogenic cells are needed for transcriptome profiling to disclose RNA pathways or other signatures that characterize the DMD phenotype. 14 Therefore, muscle tissue is quite frequently requested from patients with DMD as a source of myogenic cells, despite the observed low compliance with this painful, invasive procedure. An alternative, noninvasive source of cells would therefore be an extremely useful model in DMD.
We show here that USCs derived from the urine samples of a patient with DMD recapitulate the dystrophin genotype/phenotype. In addition, we observed dystrophin protein restoration via antisense exon skipping in transformed myogenic DMD USCs. These results all together suggest that USCs could represent a noninvasive human cell model for studying DMD, to be used in genetic DNA and RNA profiling, and screening of new therapeutic molecules and drug candidates.
Materials and Methods
Study design and isolation of human USCs
Urine samples were collected from three individuals: two healthy control subjects and one patient with a diagnosis of Duchenne muscular dystrophy, for research purposes (Table 1). All individuals signed informed consent before urine collection.
Patients enrolled for isolation of urinary cells
DMD, Duchenne muscular dystrophy.
Progenitor cells were isolated and cultured from urine specimens as described in Zhou and colleagues, 8 albeit with some modifications. Briefly, cells were collected from the first morning urine specimen (200–350 ml) by centrifugation at 400 × g for 10 min at room temperature, and washed with phosphate-buffered saline (Gibco PBS; Thermo Fisher Scientific, Waltham, MA) supplemented with an antibiotic/antimycotic solution (Sigma-Aldrich, St. Louis, MO). After discarding the supernatant, 1 ml of primary medium was added, and each sample was plated into a single well of a 12-well plate, coated beforehand with 0.1% gelatin (Millipore, Billerica, MA). Every 24 hr, 1 ml of primary culture medium was added to each cell preparation. The primary culture medium was removed 96 hr after plating, and 1 ml of proliferation medium was added to 1 ml of primary culture medium in each well.
Medium reagents
The primary medium contained Dulbecco's modified Eagle's medium (DMEM)/high glucose (EuroClone, Pero, Italy) and Gibco Ham's F12 nutrient mix (1:1; Thermo Fisher Scientific), supplemented with 10% (v/v) fetal bovine serum (FBS), antibiotic/antimycotic solution (Sigma-Aldrich), and an REGM (renal epithelial cell growth medium) SingleQuot kit (Lonza, Basel, Switzerland). The main constituents of the proliferation medium, on the other hand, included an REGM BulletKit and RE cell basal medium (Lonza) and mesenchymal proliferation medium (DMEM/high glucose, 10% [v/v] FBS, 1% [v/v] Gibco GlutaMAX, 1% [v/v] nonessential amino acids [Gibco NEAA], 1% antibiotic/antimycotic solution, basic fibroblast growth factor [bFGF, 5 ng/ml; ProSpec, Rehovot, Israel], platelet-derived growth factor [PDGF-AB, 5 ng/ml; ProSpec], epidermal growth factor [EGF, 5 ng/ml; Lonza]) mixed at a 1:1 ratio.
Fluorescence-activated cell-sorting analysis
After isolation, USCs were expanded in DMEM/F12K medium. Aliquots of USCs at passage 5–7 were analyzed by flow cytometry to evaluate epithelial cells (CD13, CD324), fibroblasts (fibronectin), stem cells (c-Kit, CD90, CD105), and bone marrow markers (CD34, CD133). Antibodies, excluding c-Kit, were conjugated with fluorochromes CD324 (E-cadherin) eFluor 660 (eBioscience, San Diego, CA) 0.5 μg/100 μl; anti-human fibronectin, CD34, CD133, Alexa Fluor 488 (eBioscience) 10 μg/ml; anti-human CD90 and CD105 with phycoerythrin (PE) (eBioscience) 0.25 μg/100 μl; and anti-human CD13 allophycocyanin (APC) (eBioscience) 0.125 μg/100 μl. USCs were incubated with antibodies against surface antigens. The primary antibody, c-Kit anti-rabbit (Santa Cruz Biotechnology, Santa Cruz, CA), was diluted 1:100, and the secondary antibody, Alexa Fluor 488 anti-rabbit, was diluted 1:1000.
The presence of epithelial and stem cell markers was investigated. In all cases, USCs were incubated for 20 min at room temperature. Flow cytometry was performed with an FC 500 instrument (Beckman Coulter, Brea, CA), and cellular debris and aggregates were gated out on the basis of forward and side scatter. Isotype-matched negative control subjects helped to define the threshold for each specific signal and to establish the appropriate gate for positive cells. Data were analyzed with Kaluza software (Beckman Coulter). The viability of the cell preparations was evaluated by propidium iodide (PI) staining, and apoptosis was analyzed in each cell preparation by annexin V expression.
OCT3/4 and NANOG quantification
Total RNA was extracted from cultured USCs with an RNeasy microkit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. RNA yield and purity were determined by spectrophotometry absorption at 260 and 280 nm. To obtain cDNA, an equal amount of mRNA (1 μg) was reverse transcribed, using iScript reverse transcription supermix (Bio-Rad, Hercules, CA). mRNA expression was measured by quantitative real-time PCR, using a thermal cycler (iCycler; Bio-Rad). The reaction was performed in a 15-μl volume, using a SYBR green supermix kit (Bio-Rad). All measures were performed in triplicate. The reaction conditions were as follows: 95°C for 3 min (polymerase activation), followed by 40 cycles at 95°C for 5 sec and 60°C for 10 sec, and 81 cycles at 55°C for 15 sec. Quantified values were normalized against the input determined with the housekeeping gene β2-microglobulin (B2M), using the formula log 2−ΔC t .
The following oligonucleotides were used: B2M: Forward: 5′-AGGACTGGTCTTTCTATCTCTTGT-3′ Reverse: 5′-ACCTCCATGATGCTGCTTACA-3′ OCT3/4: Forward: 5′-AGGAGAAGCTGGAGCAAAA-3′ Reverse: 5′-GGCTGAATACCTTCCCAAA-3′ NANOG: Forward: 5′-GGTCCCGGTCAAGAAACAGA-3′ Reverse: 5′-GAGGTTCAGGATGTTGGAGA-3′
Telomerase activity
The catalytic activity of telomerase in USCs was assessed by quantitative PCR. USCs were homogenized in CHAPS (3
Statistical analysis
Continuous variables are expressed as means ± standard deviation, whereas categorical variables are expressed as numbers and percentages. An unpaired t test or Mann–Whitney U test was used as appropriate for comparison between two groups, and categorical variables were compared by χ2 test or Fisher exact test, as appropriate. All probability values reported are two-sided, and p < 0.05 was considered statistically significant. Statistical analysis was performed with STATA 11.1 software (Stata, College Station, TX).
DMD urine-derived stem cell genotype characterization
Genomic DNA was extracted from urine-derived stem cells with a Nucleon BACC3 kit (GE Healthcare Life Sciences, Piscataway, NJ). Mutation analysis was performed by multiplex ligation-dependent probe amplification (MLPA) as previously described. 15 MLPA analysis was carried out with SALSA probemix 034 and 035, according to the manufacturer's recommendations (MRC Holland; Amsterdam, The Netherlands), thereby achieving the copy number screening of all 79 dystrophin exons. PCR products were analyzed with an Applied Biosystems ABI 3130 automated sequencer, using GeneScan software (Thermo Fisher Scientific).
Myogenic transformation of USCs
Myogenesis of urine cells from both healthy donors and the patient with DMD was induced by infection with an adenovirus serotype 5 (Ad5)-derived, EA1-deleted adenoviral vector carrying the MyoD gene, as previously described. 16 MyoD-transformed USCs were cultured in high-glucose DMEM supplemented with 2% FBS and antibiotic/antimycotic solution (differentiation medium) during differentiation into myotubes.
Expression analysis of dystrophin transcript
RNA extraction
Total RNA was extracted from cells, using an RNeasy kit (Qiagen), and reverse-transcribed with an Applied Biosystems high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific), according to the respective manufacturers' instructions. Before cDNA synthesis, RNA was treated with DNase I (Roche, Indianapolis, IN) and checked for residual DNA contamination by real-time PCR analysis.
DMD gene micro-fluidic card (FluiDMD) analysis
The dystrophin transcript analysis was performed as described by Bovolenta and colleagues. 17 In brief, 250 ng of RNA from each sample was reverse-transcribed, using an Applied Biosystems high-capacity cDNA reverse transcription kit, in a volume of 20 μl. To this quantity 80 μl of sterile water and 100 μl of Applied Biosystems 2 × universal master mix were added. A 100-μl volume of mix per port was loaded into the fluidic cards, which were run on an Applied Biosystems real-time 7900HT appliance (Thermo Fisher Scientific). The cycle threshold (C t) values obtained for all exon junctions and dystrophin isoform systems were normalized, using human β-actin as the housekeeping gene (ΔΔC t = C t exon junction system – C t β-actin).
Expression analysis of dystrophin protein
Western blotting
Protein extracts were isolated from cell cultures, using 150 μl of lysis buffer (20 mM Tris-HCl [pH 6.8], 0.1% sodium dodecyl sulfate [SDS], 150 mM NaCl, 1% Nonidet P-40 [NP-40], 0.5% deoxycholic acid, 1 mM NaF, 0.1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl sulfate [PMSF], protease inhibitors), at 3 days postdifferentiation. Protein concentration was determined by the Bradford method. Aliquots corresponding to 20 μg (myoblasts derived from a control muscle, WT) and 60 μg (MyoD-transformed USCs) of protein were loaded on a 6% polyacrylamide gel and separated by electrophoresis. Samples were transferred to a nitrocellulose membrane that was blocked with nonfat dried milk for 60 min at room temperature and incubated overnight at 4°C with Dys2 antibodies (diluted 1:50; Leica Biosystems, Nussloch, Germany). Amersham horseradish peroxidase-conjugated anti-mouse secondary antibody (diluted 1:1000; GE Healthcare Life Sciences) was used as the secondary antibody. Western blots were developed with an Amersham ECL Plus Western blotting detection system (GE Healthcare Life Sciences).
Immunostaining analysis
Cell cultures were grown on coverslips, and fixed in −20°C methanol. Samples were incubated for 30 min in PBS mixed with 4% bovine serum albumin (Sigma-Aldrich). All samples were labeled with both polyclonal anti-dystrophin antibody (H300; Santa Cruz Biotechnology) diluted 1:100 and monoclonal anti-myosin antibody diluted 1:40 (Dako, Carpinteria, CA), washed with PBS, and revealed with tetramethylrhodamine isothiocyanate (TRITC)-conjugated anti-rabbit and fluorescein isothiocyanate (FITC)-conjugated anti-mouse secondary antibody (diluted 1:100; Dako), respectively. The slides were mounted with a Molecular Probes antifade mounting medium (Thermo Fisher Scientific), and analyzed with a Nikon Eclipse 80i fluorescence microscope (Nikon Instruments, Melville, NY).
Antisense treatment
Myotubes obtained from MyoD-transformed DMD USCs were transfected with 2′-O-methyl phosphorothioate (2′OMePS) antisense oligoribonucleotide for dystrophin exon 44 skipping (IDT Technologies, Coralville, IA) in the presence of polyethylenimine (PEI) (ExGen 500; Biomol, Hamburg, Germany) as transfection reagent. 18
Exon-skipping quantification
Exon skipping was quantified with an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA). The region surrounding the DMD mutation, spanning exons 43–46, was amplified, and PCRs were performed via 30 cycles at 94°C (30 sec), 60°C (45 sec), and 72°C (80 sec), with Invitrogen Platinum Taq DNA polymerase (Thermo Fisher Scientific). PCR products were analyzed with an Agilent high-sensitivity DNA chip that performs capillary electrophoresis and uses a fluorescent dye to bind DNA and determine both DNA concentration and size. The skipping percentages were calculated as the ratio between skipped transcript and total transcript (skipped transcript/total transcript [skipped transcript + unskipped transcript] × 100).
Skipping was also quantified with FluiDMD cards, and the skipping percentage was calculated by the ΔΔC t method: a comparison of the ΔC t of ex43–44 and the ΔC t of total transcript (ΔΔC t = ΔC t ex43–44 – ΔC t of total transcript [mean of ΔC t from ex1–2 junction to ex28–29]). ΔΔC t values were used to calculate 2−ΔΔC t and the skipping percentage as 2−ΔΔC t × 100.
Results
Morphology and CD marker evaluation in USCs
USCs were isolated from both healthy donors (two) and a patient with DMD known to have a deletion of dystrophin exon 45. The USCs displayed a typical MSC phenotype, in accordance with previous publications. 8,9 Two types of USC morphology were apparent: one type (named type I) had a regular and rounded shape, and the second type (named type II) consisted of spindle-shaped cells (Fig. 1a). No differences between the cells of control subjects and the subject with DMD were found in terms of morphology, time of expansion, or in vitro survival: 90% of cells were viable at P7, and fewer than 5% of cells expressed the apoptosis-associated marker annexin V (Fig. 2a).
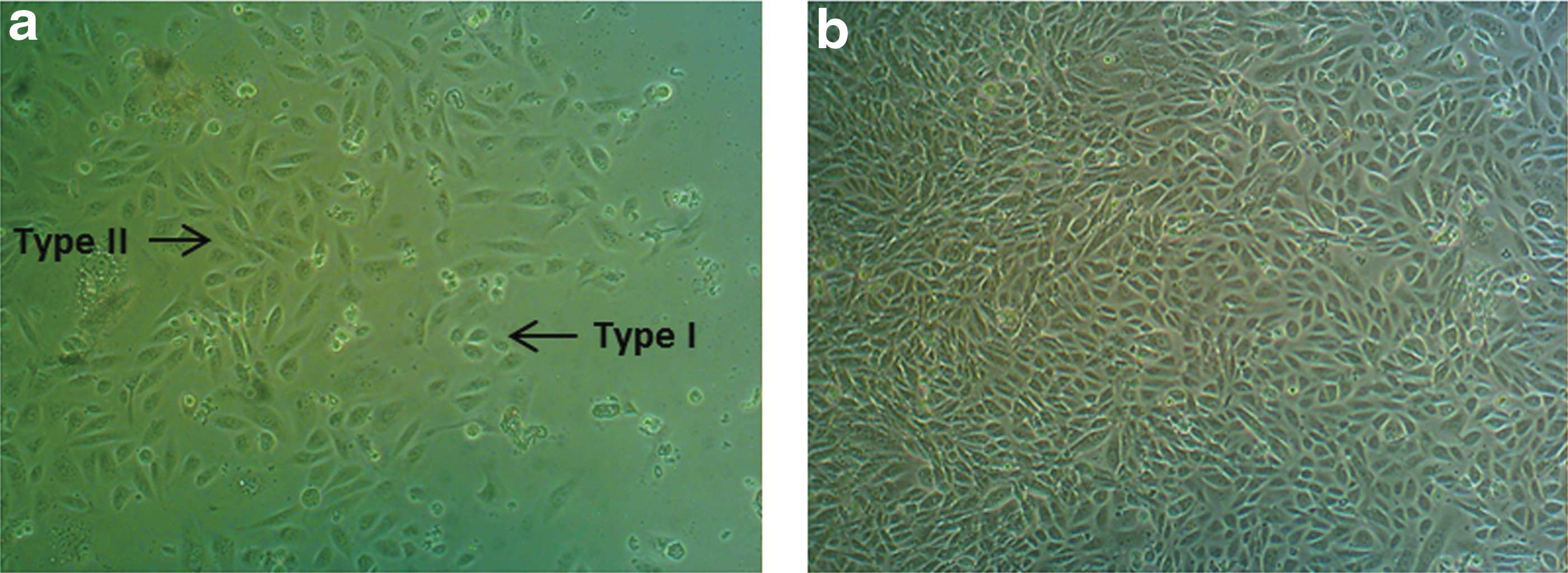
Morphology of urine stem cells (USCs) obtained from fresh Duchenne muscular dystrophy (DMD) urine samples at various time points after collection. Shown are type I and type II cells (arrows) on day 12
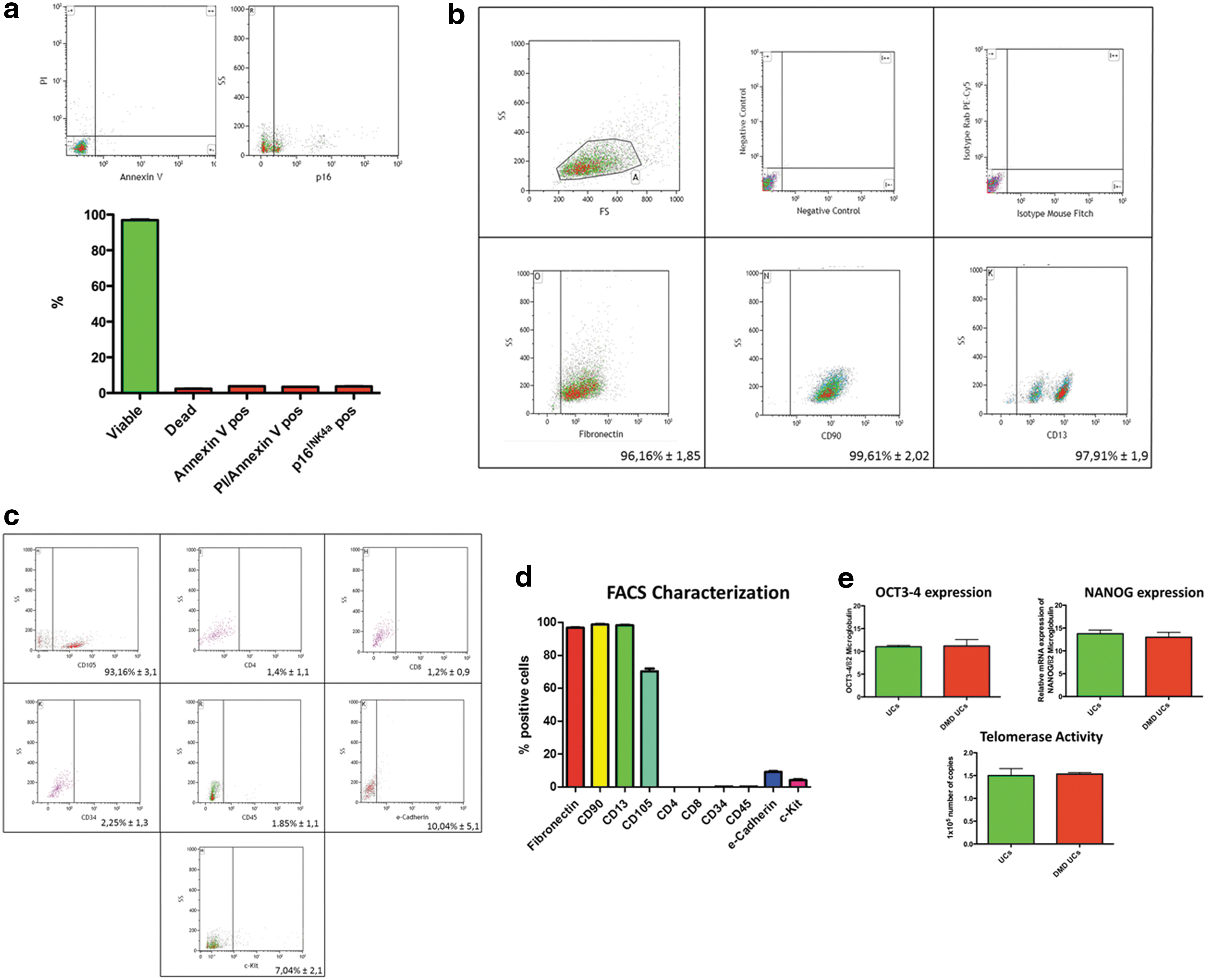
A variable percentage of cells (85–95%) were positive for markers of mesenchymal origin (CD90 and CD105), and for the fibroblast-associated protein fibronectin. USCs expressed low levels of the stem cell factor receptor (CD117) and bone marrow-derived epitopes such as CD34. Importantly, a high percentage of cells showed the presence of CD13, a renal epithelial marker, also known as aminopeptidase N. Unspecific positive labeling was excluded by isotype and negative control staining (Fig. 2b–d). At P5 and P7, a small fraction of USCs, varying from 1 to 5%, expressed lymphocyte and mast cell surface markers, namely CD4, CD8, and CD45 (Fig. 2b–d).
Subsequently, in order to evaluate the proliferation capacity in vitro, stemness-related gene expression (NANOG and Oct3/4) and telomerase activity were evaluated and found to be present in all preparations analyzed. Notably, no differences were found between healthy and DMD donors (Fig. 2e).
Thus, we established the conditions for the isolation and expansion of SCs from human urine samples in healthy control subjects and the patient with DMD.
DNA analysis in DMD urine-derived cells
MLPA analysis of DMD USCs confirmed the presence of an isolated exon 45 deletion, as previously identified in peripheral blood cells of the patient during routine diagnostic procedures (Fig. 3).

Multiplex ligation-dependent probe amplification (MLPA) profile of the whole DMD gene shows the isolated exon 45 deletion (red dot).
Expression profile of dystrophin transcript in native USCs and differentiated myogenic USCs
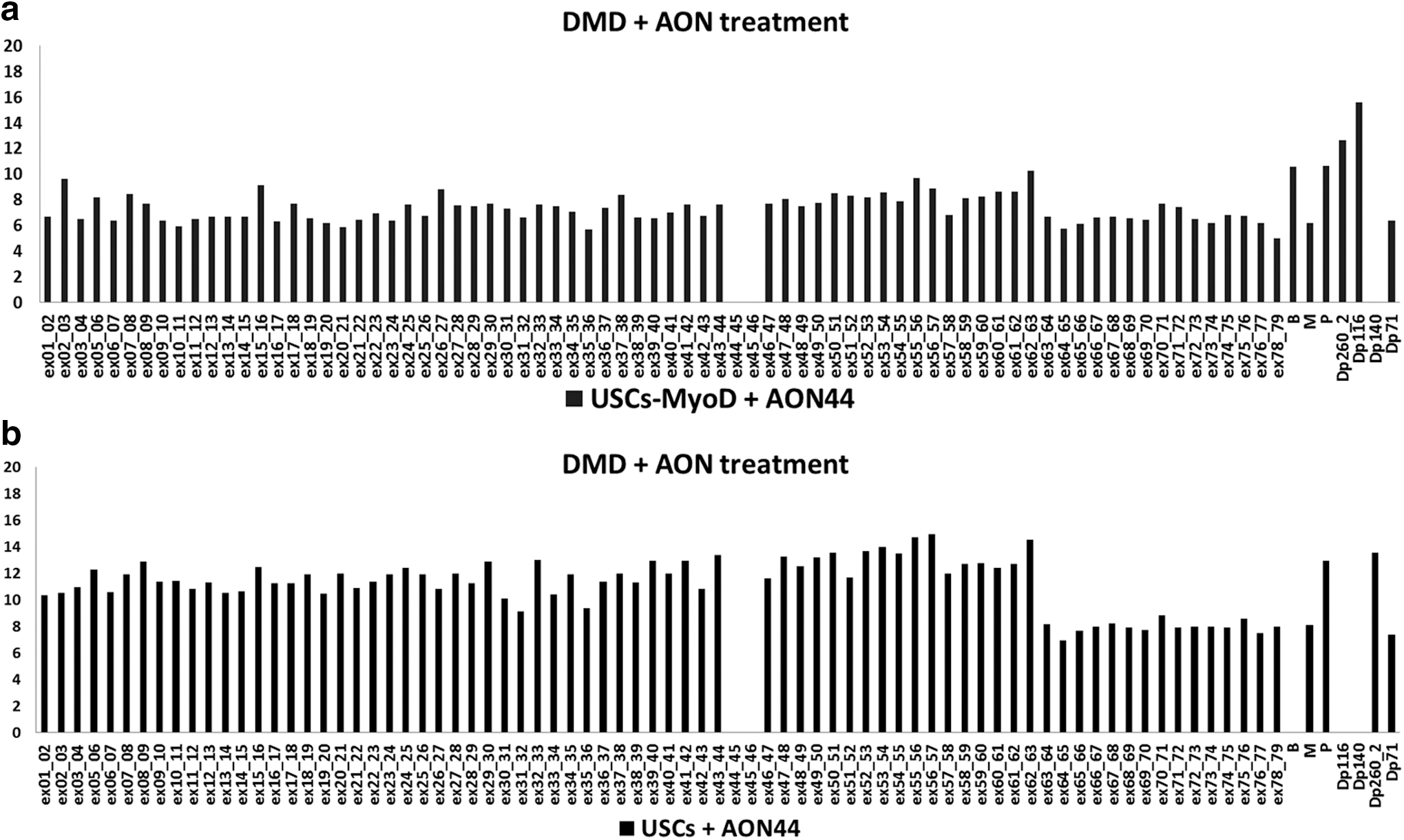
To establish whether USCs could be a good in vitro model for muscle disease studies, we first needed to establish the presence of the dystrophin transcript as a muscle cell-specific transcript. Hence, we studied both native USCs and MyoD-transformed USCs from control subjects and the patient with DMD: USCs from both healthy donors and the patient with DMD were transformed into myogenic cells by infection with an Ad5-derived vector carrying the MyoD gene, and the resulting USCs-MyoD were allowed to differentiate into myotubes. The dystrophin transcript was then evaluated in both USCs and differentiated USCs-MyoD, using the FluiDMD card, a TaqMan gene expression assay providing ABI real-time exon–exon junction systems that cover the whole dystrophin transcript (including all the dystrophin isoforms).
In the healthy USC sample, this revealed the presence of the full-length dystrophin transcript (Fig. 4a, light gray bars), in which all the junction–junction systems were detected, albeit at low levels, including the full-length Dp427m (M), Dp427p (P), and the short Dp260, Dp116, Dp71 dystrophin isoforms. The Dp427b (B) isoform was detected only after myogenic transformation. After myogenic transformation, the expression of all analyzed dystrophin transcripts was increased for both single junction–junction systems and the specific dystrophin isoforms (Fig. 4a, dark gray bars).

DMD gene microfluidic card (FluiDMD) profiles of USCs and USCs-MyoD (i.e., USCs transformed into myogenic cells by infection with an Ad5-derived vector carrying the MyoD gene) from healthy donor 1
In the patient with DMD, the FluiDMD card precisely identified the deletion of exon 45 (Fig. 4b), demonstrating that the USCs maintain the mutation in the dystrophin transcript. As in the healthy sample, the differentiation into myogenic cells enhanced the expression levels of all analyzed dystrophin transcripts.
These findings not only confirm that USCs could be useful tools for identifying the mutation at the genomic level, as previously reported, 9 but also suggest their application in transcriptomic profiling.
Exon skipping in USCs and USCs-MyoD treated with antisense oligonucleotide
The main goal of our work was to determine the feasibility of using USCs for testing and screening therapeutic compounds, without the need for iPSC reprogramming. To this end, both USCs and USCs-MyoD from a boy with DMD, carrying the deletion of exon 45, were treated with the 2′OMePS antisense oligoribonucleotide (AON) for exon 44 skipping. Skipping was then quantified by means of two different methods: (1) the ΔΔC t method, using FluiDMD card analysis; and (2) the Agilent 2100 bioanalyzer, using their high-sensitivity DNA chip.
The FluiDMD platform quantification of dystrophin exons showed the absence of off-target effects induced by the antisense treatment (Fig. 5a and b). To calculate skipping percentages by ΔΔC t, we compared the expression level of the ex43–44 junction with respect to the total dystrophin transcript, which was calculated as the mean ΔC t from junctions ex1–2 to ex28–29. We took the exon region 1–29 as representative of the total transcript driven by the full-length promoters, because the distal promoters lie within intron 29 (Dp260), intron 44 (Dp140), intron 55 (Dp116), and intron 62 (Dp71) and we needed to rule out the effect of any additional expression on the total dystrophin transcript. This was especially important in the case of Dp71, which was abundant in the USC culture. The ΔΔC t method revealed 7% physiological skipping of exon 44 in DMD-USCs, and 30% new skipping induced by AON treatment.
FluiDMD analysis of dystrophin transcript profile after antisense treatment in USCs-MyoD
Figure 6 summarizes the results obtained with the Agilent high-sensitivity DNA chip, including the run on the bioanalyzer microgel (Fig. 6A), which shows the skipped and unskipped fragments, and the chromatogram representation, which indicates the size and the concentration of the amplified fragments, for both DMD USCs-MyoD (Fig. 6B) and DMD USCs-MyoD treated with AON (Fig. 6C). The concentrations estimated by the chip were used to calculate the skipping percentages, as described in Materials and Methods.
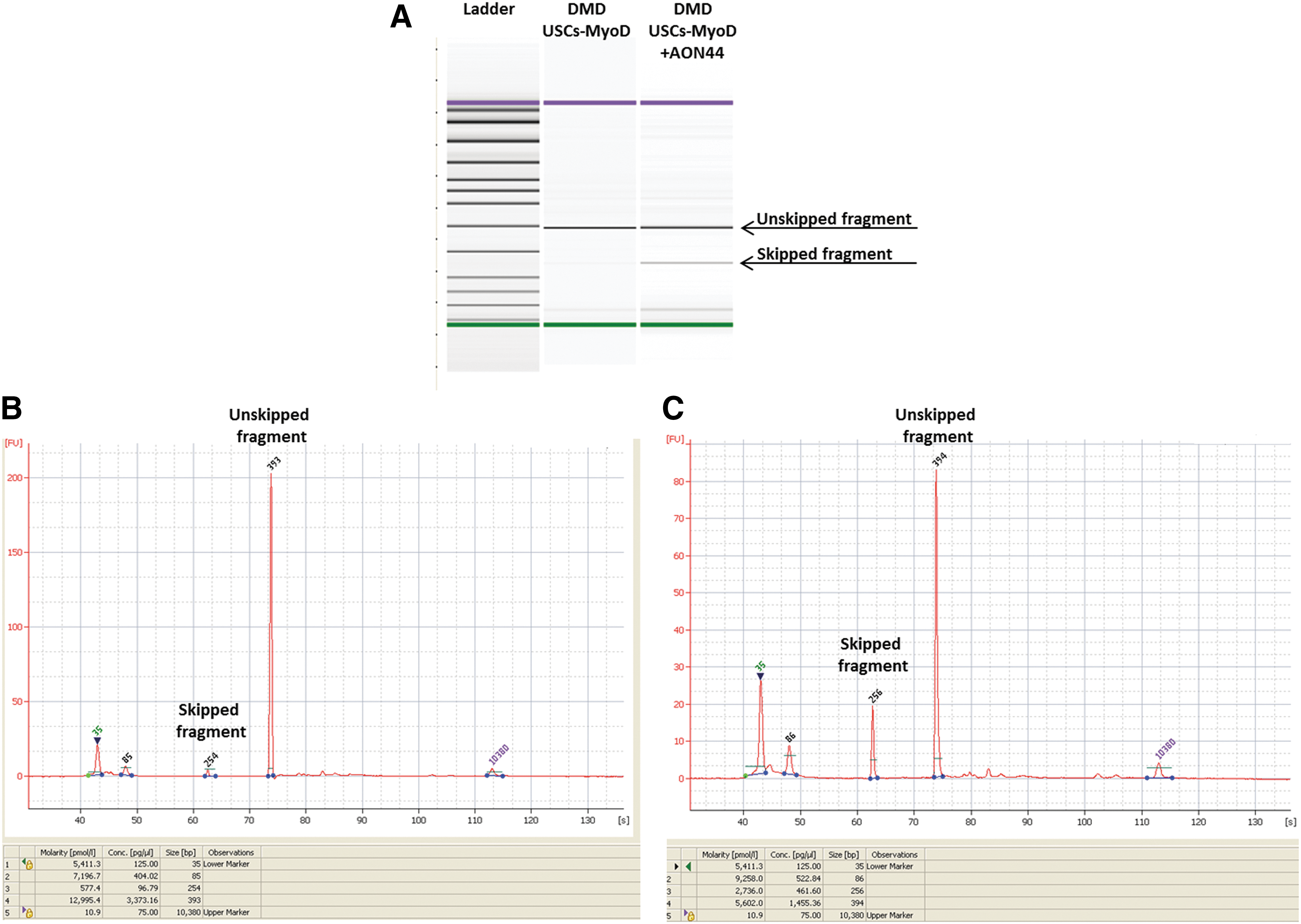
Agilent 2100 bioanalyzer microgel
The data obtained with the Agilent high-sensitivity DNA chip revealed percentages of exon 44 skipping similar to those obtained with the FluiDMD card, specifically 4% physiological and 33% AON-induced, and the use of two validated methods and their relative calculations lends robustness to our data.
The skipping quantification in native DMD USCs, using the high-sensitivity DNA chip, showed 9 and 35% skipping percentages, as physiological and AON-induced skipping, respectively, thus similar to what was observed in the DMD USCs-MyoD.
FluiDMD data did not allow us to accurately quantify the skipping, because of the low abundance of dystrophin transcript in these native cells.
Muscular protein analysis
All cells, both USCs and USCs-MyoD, derived from both healthy donors and the patient with DMD, were analyzed for dystrophin protein expression. Immunofluorescence analysis, performed with anti-dystrophin antibody, revealed the absence of protein in all the USCs analyzed (Fig. 7A), and protein rescue only in the MyoD-transformed control USCs (Fig. 7B). The molecular weight of the protein was verified by Western blot, and the full-length 427-kDa dystrophin protein was detected only in USCs-MyoD derived from healthy controls (Fig. 8). The Western blot of USCs-MyoD from the patient with DMD was negative, as expected (data not shown).
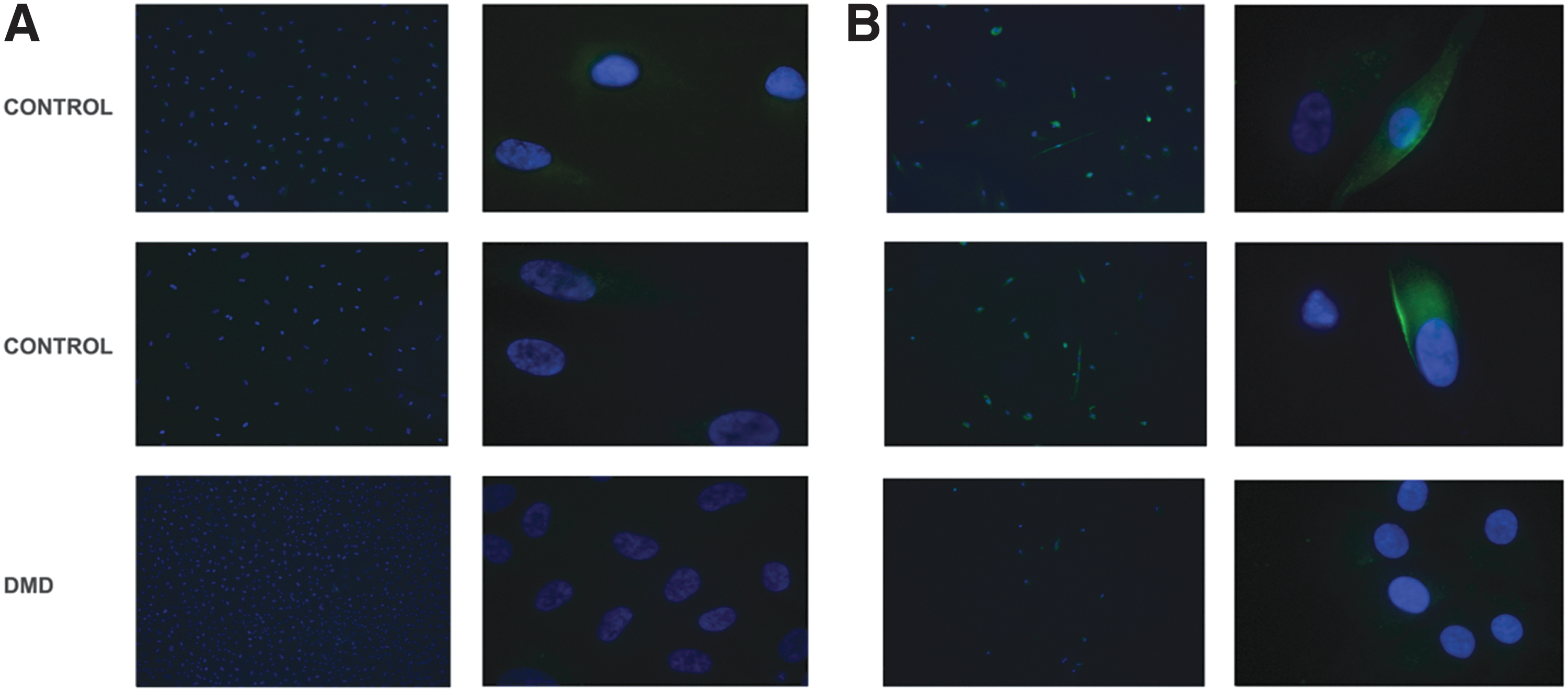
Immunofluorescence analysis of dystrophin (DYS) protein in USCs
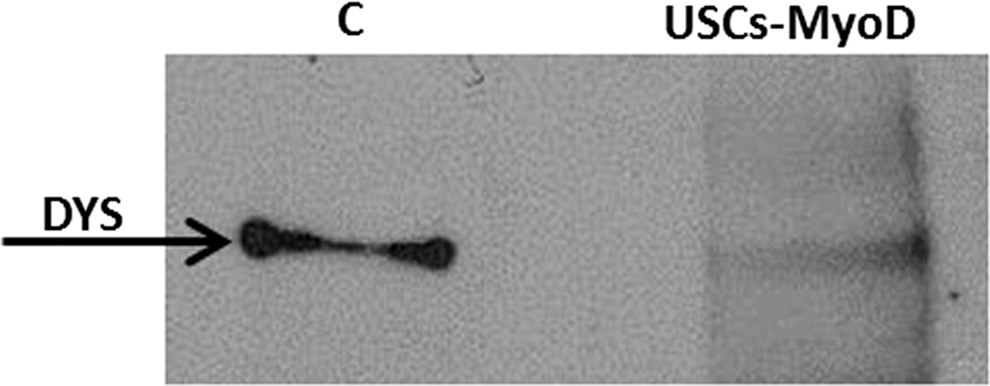
Western blot analysis of USCs-MyoD derived from a healthy donor (C, control) shows the presence of the dystrophin protein with the full-size molecular weight as a control.
Antisense treatment of DMD USCs-MyoD induced translation of the dystrophin protein, which was seen to be correctly localized at the subsarcolemma by immunofluorescence (Fig. 9); the Western blot did not detect any dystrophin because of the low skipping percentage and consequently low amount of rescued protein.

Immunofluorescence analysis of dystrophin protein in USCs-MyoD from the patient with DMD after antisense treatment. Original magnification
Figure 10 shows the double labeling of USCs-MyoD with anti-myosin and anti-dystrophin antibodies, which appeared to be colocalized at the subsarcolemmal membrane.
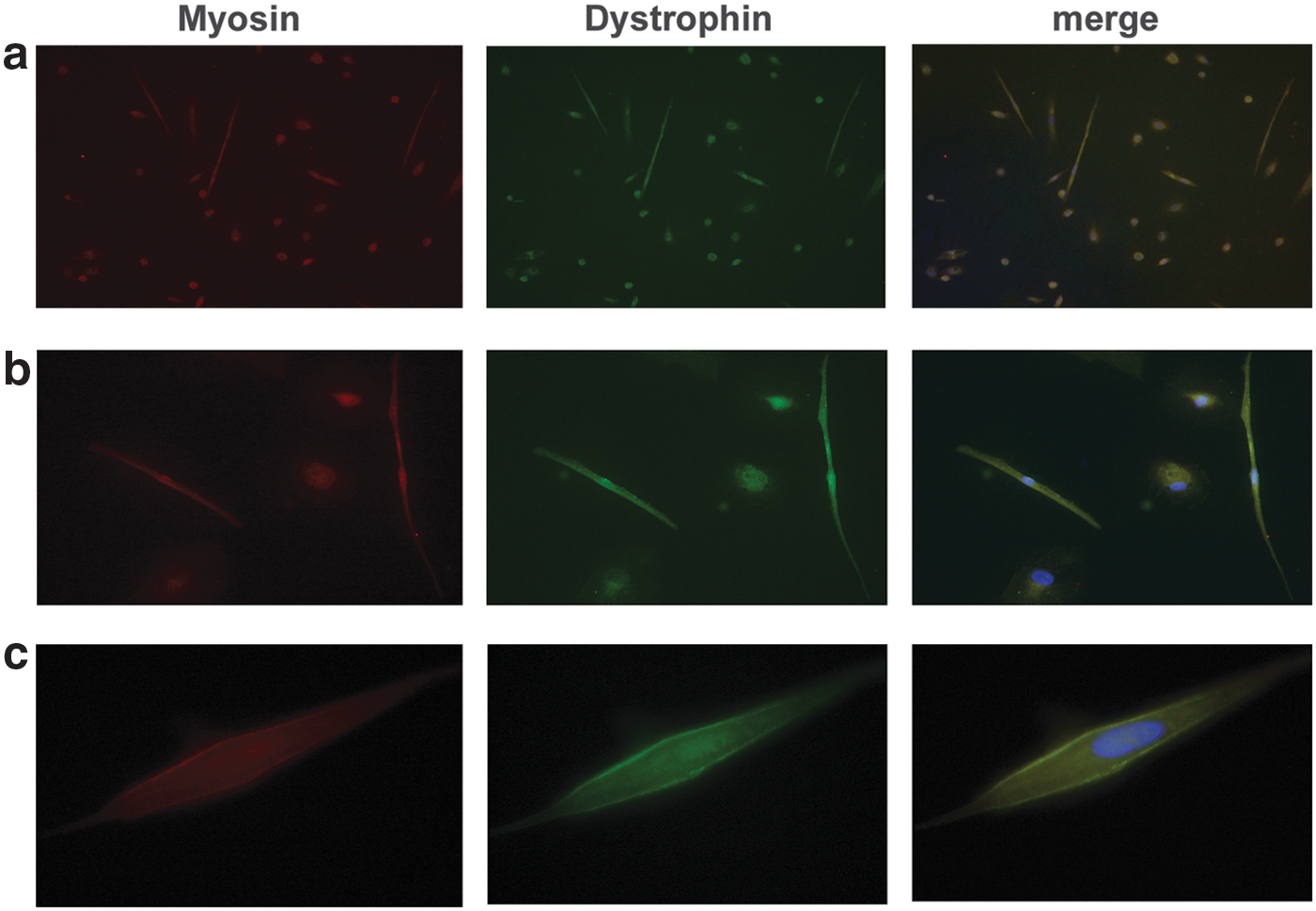
Immunostaining of USCs-MyoD from healthy donors, using double labeling with anti-myosin (red) and anti-dystrophin (green) antibodies. Original magnification:
Discussion
We found that USCs from both healthy donors and a patient with DMD were characterized by high expression of MSC surface markers, such as CD90 and CD105, together with markers typical of renal epithelial cells (i.e., CD13) and fibroblasts (fibronectin). Moreover, this class of cells retained a remarkable growth reserve in vitro, as shown by the low percentage of cells positive for the senescence-associated protein p16 INK4a and the apoptotic marker annexin V. The USCs also expressed a significant level of telomerase activity, indicating that they held characteristics compatible with stem/progenitor cells. These characterizations allowed us to qualify the urine-derived cells as mesenchymal stem cells, as previously described. 8
The USCs from our patient with DMD displayed the dystrophin mutation at both the DNA and RNA levels, and the dystrophin transcript was evidently detectable in both native and MyoD-transformed cells. The dystrophin transcript appeared to be correct in exon composition, with the expected exon–exon junctions, and therefore perfectly matched the RNA seen in skeletal muscle-derived cells. This finding is important, because it suggests that we may be able to use USCs for a variety of RNA analyses of patients with DMD. These cells may also prove useful in molecular genetic diagnosis, both for genotyping and for profiling the dystrophin transcript to detect exceptions to the frame rule, splicing choices, and, generally speaking, all the cohorts of atypical mutations in dystrophinopathies. 19
Furthermore, only USCs-MyoD derived from the control subjects were positive for correct localization of the full-length dystrophin protein at the subsarcolemma, as shown by myosin colocalization and Western blotting. The DMD USCs-MyoD carrying the out-of-frame dystrophin deletion (del.exon45) were dystrophin negative, as expected.
We were therefore able to recapitulate the dystrophin protein profiling in USCs, a finding that may make them suitable for drug discovery procedures. This may be of particular interest to those studying the consequences of utrophin upregulation (see the Summit Therapeutics website at
Indeed, we were also successful in modulating dystrophin gene splicing in DMD USCs-MyoD, using an antisense oligonucleotide. Treatment with 2′OMePS-backboned AON induced the skipping of exon 44 in native DMD cells, and the antisense exon-skipping strategy was seen to restore the dystrophin protein in DMD USCs-MyoD. These results suggest that USCs may provide not only a valid model for genetic diagnosis and studying disease mechanisms but also for testing drug efficacy, as a noninvasive source of myogenic cells, with consequent high patient compliance. Native USCs may foreseeably be used for diagnostic (DNA and RNA studies) and transcriptional profiling in patients with DMD, enabling the detection of a variety of mutations (splicing mutations, splicing choices, deep intronic mutations, complex phenotypes) that can escape routine genetic testing. In addition, we also propose that USCs might be adopted in other muscular diseases (limb girdle phenotypes, congenital myopathies, etc.) if the specific genotype and cell phenotype would be recapitulated.
Furthermore, the possibility of inducing iPSCs from these urine-derived cells may open new perspectives in a variety of research applications; in the post-omics era, in which an enormous number of variations and putative new causative genes remain devoid of functional validation, USCs may represent a valid tool for gene function exploration and genomic variation validation. Moreover, USCs might also be employed in tissue engineering, 20 particularly with a view to autologous transplantation.
In conclusion, we demonstrate here that USCs from a patient with DMD are able to recapitulate the cell phenotype of the disease, thereby legitimizing further investigations to fully explore the use of this methodology as a clinical tool for personalized medicine.
Footnotes
Acknowledgments
The Duchenne Parent Project Italy is gratefully acknowledged, and thanks are also due to Filippo Buccella and Fernanda De Angelis for their constant and affectionate support. The authors thank Dr. Paola Rimessi for providing the MLPA study of the patient with DMD.
Author Disclosure
Alessandra Ferlini is PI of ongoing BioMarin clinical trials. For all other authors, no competing financial interests exist.