
Abstract
In addition to the ability to boost gene delivery efficiency in many therapeutically relevant cells, the capability of circumventing neutralizing antibody (NAb) inactivation is a key prerequisite that gene carriers must fulfill for their extensive applications as therapeutic agents in many gene therapy trials, especially for cancer treatments. This study revealed that a genetically engineered adeno-associated virus (AAV) variant, AAVr3.45, inherently possesses dual beneficial properties as a gene carrier: (i) efficiently delivering therapeutic genes to many clinically valuable cells (e.g., stem or cancer cells) and (ii) effectively bypassing immunoglobulin (IgG) neutralization. Detailed interpretation of the structural features of AAVr3.45, which was previously engineered from AAV2, demonstrated that the LATQVGQKTA peptide at the heparan sulfate proteoglycan binding domain, especially the presence of cationic lysine on the peptide, served as a key motif for dramatically enhancing its gene delivery capabilities, ultimately broadening its tropisms for many cancer cell lines. Furthermore, the substitution of valine on the AAV2 capsid at the amino acid 719 site to methionine functioned as a coordinator for promoting viral resistance against IgG inactivation. The NAb-resistant characteristics of AAVr3.45 were possibly associated with the LATQVGQKTA sequence itself, indicating that its synergistic cooperation with the point mutation (V719M) is required for maximizing its ability to evade NAb inactivation. The potential of AAVr3.45 as a cancer gene therapy agent was confirmed by provoking apoptosis in breast adenocarcinoma by efficiently delivering a pro-apoptotic gene, BIM (Bcl-2-like protein 11), under high titers of human IgG. Thus, the superior aspects of the NAb-resistant AAVr3.45 as a potential therapeutic agent for systemic injection approaches, especially for cancer gene therapy, were highlighted in this study.
Introduction
E
Adeno-associated virus (AAV) is a highly versatile gene carrier that has been extensively utilized in many gene therapy applications due to its lack of pathogenicity and its efficient delivery characteristics. 6,7 Thus, AAV-mediated gene delivery has demonstrated many successes in many human clinical trials for treating muscular dystrophy, 8 Leber's congenital amaurosis, 9 and hemophilia B. 10,11 Importantly, the first AAV-based gene therapy product for curing lipoprotein lipase deficiency (LPLD), alipogene tiparvovec (Glybera®; uniQure, Amsterdam, Netherlands), has been successfully approved for human trials by the European Medicines Agency, 12 demonstrating the safety aspects of AAV vectors. To raise the therapeutic efficacy of AAV-based gene therapy further, however, several hurdles associated with AAV vectors must be addressed: narrow tropisms to therapeutically relevant cell types (e.g., stem or cancer cells), 1,13 and the antigenic properties of AAV vectors against neutralizing antibodies. 14,15
Among the shortfalls of the AAV vectors, their rapid inactivation by neutralizing antibodies (NAbs) may be the most important barrier that considerably reduces the therapeutic efficacy of AAV-based gene therapy. Coating AAV capsids with surfactants 16 and associating them with extracellular vesicles 17,18 have been the prevailing ways to block immediate contacts between AAV vectors and NAb circuits. Furthermore, genetic modification of viral cap genes was attempted to extend the duration of AAV resistance within the circulatory system. 15 Although previous studies presented promising strategies for evading NAb inactivation, the post processes for modifying AAV exteriors can inevitably alter their carrier characteristics, and the beneficial outcomes (i.e., NAb evasion and enhanced gene delivery) may not be fully reproduced in on-spot usage, presumably due to complicated procedures or user-dependent variations. To this end, engineering or discovering AAV variants that inherently possess the beneficial features as gene therapy agents without post processes may be the optimum way to facilitate their successful translation into human clinical use.
This current study discovered an AAV variant, AAVr3.45, which inherently exhibits the dual beneficial gene delivery aspects: (i) inherently possessing NAb-resistant capacity and (ii) simultaneously enhancing transduction in therapeutically relevant stem or cancer cells. The AAVr3.45 was genetically engineered based on the AAV2 capsid specifically for infecting neural stem cells in a previous study. 19 The key structural motifs of AAVr3.45 eliciting the dual beneficial effects were deeply elucidated. Compared to the AAV2 capsid, the AAVr3.45 vector distinctively displays an LATQVGQKTA peptide at the heparan sulfate proteoglycan (HSPG) binding domain, with an additional point mutation of valine to methionine at the amino acid (aa) 719 site (i.e., V719M). Importantly, under high titers of a human immunoglobulin (IgG) cocktail, the AAVr3.45 variant inherently exhibited highly enhanced gene delivery properties in many cancer cell lines, confirming its dual performances, including immune resistance and promotion of gene delivery. Exploring the NAb resistance of AAVr3.45 in this study can be closely related to evaluating the potentials of AAVr3.45 as a cancer gene therapy agent because most cancer treatments have been primarily conducted by systemically injecting therapeutic agents through the human bloodstream, whose sera contain abundant NAbs. Thus, the dual features of the virus were further evaluated by inducing apoptosis in breast adenocarcinoma cells (i.e., MDA-MB-231) via delivering a pro-apoptotic gene (BIM; Bcl-2 like protein 11) under high titers of human IgG. This study therefore potentially presents a perspective of the AAVr3.45 as a safe and efficient therapeutic agent for many gene therapy applications, especially for cancer gene therapy.
Materials and Methods
Cell culture
Six different cell lines—HEK293T, AAV293, MDA-MB-231, A375, 526 mel, and HeLa—were used in this study. Four cell lines—HEK293T, AAV293, MDA-MB-231, and HeLa—which are derived from human embryonic kidney cells, transformed genetically for producing AAV vectors, isolated from human breast adenocarcinoma, and immortalized cervical cancer cells, respectively, were cultured in Dulbecco's modified Eagle's medium (DMEM; Corning Cellgro, Corning, NY) supplemented with 10% fetal bovine serum (FBS; Corning Cellgro) and 1% penicillin-streptomycin (pen-strep; Thermo Fisher Scientific, Waltham, MA) in a humidified atmosphere of 5% CO2. Additionally, both A375 and 526 mel cell lines, which were derived from human melanoma, were cultured in Roswell Park Memorial Institute (RPMI; Thermo Fisher Scientific) 1640 medium with 10% FBS and 1% pen-strep at 37°C and 5% CO2.
Production of AAVr3.45 vectors
To test both gene delivery efficiency and NAb evading capability, both AAV2 and AAVr3.45 vectors carrying cDNA encoding green fluorescent protein (GFP) driven by the cytomegalovirus (CMV; i.e., AAV2-GFP and AAVr3.45-GFP) were packaged using a calcium phosphate transient transfection method. 15 To estimate the fractions of empty vectors out of the total viral pools, comparative analysis of the ratios of capsid particles (cp) with viral genomes (vg) between AAVr3.45 and AAV2 was performed by measuring the absorbance at 260 nm/280 nm on the denatured viral capsids, as previously described. 20 Additionally, three different AAVr3.45-derivative vectors carrying GFP, r3.45-V719, r3.45-587InsK 8 E, and r3.45-587InsK 8 E/V719 were produced using the aforementioned transfection method. As shortly described in Table 1, these derivative vectors are AAVr3.45 vectors modified (i) by replacing methionine (M) with valine (V) at the aa 719 site (r3.45-V719), (ii) by substituting lysine (K) with glutamic acid (E) having a negatively charged side group on the peptide displayed at the HSPG binding motif (i.e., LATQVGQKTA to LATQVGQETA), and (iii) by having both mutations (i.e., V719 and K to E at the displayed peptide at the HSPG binding domain), respectively. Furthermore, to evaluate the capability of AAVr3.45 vectors as a potential therapeutic gene carrier that can be used for cancer gene therapy applications, additional AAV2 and AAVr3.45 vectors carrying a pro-apoptotic gene (BIM; Bcl-2 like protein 11) driven by the CMV promoter (i.e., AAV2-BIM, AAVr3.45-BIM) were packaged using the same transfection protocol. Subsequently, all seven resulting vectors, including AAV2-GFP, AAVr3.45-GFP, r3.45-V719, r3.45-587InsK 8 E, r3.45-587InsK 8 E /V719, AAV2-BIM, and AAVr3.45-BIM, were purified by heparin column chromatography and buffer-exchanged to phosphate-buffered saline (PBS) solution with 0.01% Tween 20 (Biosesang, Gyeonggi, Korea) using a 1 mL HiTrap heparin column (GE Healthcare, Waukesha, WI), according to the manufacturer's instructions. Because no substantial decreases in the genomic titers of all variants were observed (Table 1), all AAV derivatives were purified using the heparin column as for AAVr3.45. The titer of each eluted viral solution was concentrated using Ultra-15 Amicon tubes (MWCO: 10,000; Millipore, Billerica, MA). The resulting viral vectors, which were treated with DNase and benzonase (Sigma–Aldrich, St. Louis, MO) for 30 min at 37°C, were incubated with proteinase K (Thermo Fisher Scientific) for 90 min at 37°C to extract viral genomic DNA. Finally, viral genomic titers were determined by quantitative polymerase chain reaction (qPCR; Mini Opticon; Bio-Rad, Hercules, CA).
Structural deviations and genomic titers of each AAV vector
AAV, adeno-associated virus.
Point mutation of r3.45 cap gene to produce AAVr3.45-derivative vectors
Two types of point mutations were conducted on the AAVr3.45 cap gene. First, the methionine (ATG) at the aa 719 site of the AAVr3.45 capsid was replaced with valine (GTG) using site-directed mutagenesis. The replacement of the ATG sequence with GTG was guided by two primers: 5′-CTGTGGACACTAATGGCGTGTATTCAGAGC-3′ and 5′-GCTCTGAATACACGCCATTAGTGTCCACAG-3′, under the Phusion high-fidelity DNA polymerase (Thermo Fisher Scientific). After the site-directed mutagenesis process, the resulting PCR products were treated with DpnI for 1 h at 37°C to digest the original templates. Next, the substitution of the positively charged lysine (K; AAA) displayed at the aa 587 site (i.e., LATQVGQKTA) to glutamic acid (E; GAA), which has a negatively charged side group under physiological conditions (i.e., pH 7.0–7.4), was performed with the following primers: 5′-CCCAAGTAGGACAAGAAACTGCTAGGCAAG-3′ and 5′-CTTGCCTAGCAGTTTCTTGTCCTACTTGGG-3′. The final PCR products were transformed into Escherichia coli (DH5α; Sigma–Aldrich), and clones having the intended sequences were selected to produce the final plasmids on a large scale. The plasmids that were finally chosen were utilized to package the resulting AAVr3.45-derivative vectors (i.e., r3.45-V719 and r3.45-587InsK 8 E). To examine the effect of the point mutation (K to E at the inserted peptide) on the viral physical properties, the electrostatic charges of AAVr3.45 and AAVr3.45-587InsK 8 E in PBS buffer (1 × 106 vg/mL) were measured using a zeta potential analyzer (ELS-2000ZS; Otsuka Electronics, Osaka, Japan). Additionally, to produce the additional AAVr3.45-derivative vector (i.e., r3.45-587InsK 8 E/V719), a point mutation replacing methionine with valine at the aa 719 site was performed on the plasmid that already had the mutation on the peptide at the aa 587 site (i.e., LATQVGQETA). All AAV production procedures were identical to the ones used for packaging the original AAVr3.45 vectors. Detailed information on the peptide sequence and viral genomic titers was summarized in Table 1.
Cellular transduction and AAV neutralization analysis
Cellular transduction efficiencies of all viral vectors carrying GFP, including AAV2, AAVr3.45, r3.45-V719, r3.45-587InsK 8 E, and r3.45-587InsK 8 E/V719, were determined by quantifying the number of GFP-expressing cells. For measuring cellular transduction in a normal culture condition, two different cell types, such as HEK293T and MDA-MB-231, with a designated cell density (HEK293T: 1 × 104 cells/100 μL; MDA-MB-231: 5 × 103 cells/100 μL) were cultured in 96-well tissue culture plates (TCP), transduced at 12 h post culture by the resulting viral vectors at various genomic multiplicities of infection (MOI; 1 × 104, 5 × 104, 1 × 105, and 2 × 105) and harvested at 2 days post transduction for analysis with flow cytometry at Yonsei University College of Medicine Medical Research Center (LSR II; Beckman Coulter, San Diego, CA). For analyzing the extent of AAV inactivation under various titers of human IgG purified from normal human serum (Sigma–Aldrich), each virus type was preincubated at 37°C for 1 h with human IgG, whose titers ranged from 0 to 800 μg/mL (0, 50, 100, 150, 200, 400, and 800 μg/mL; final titers in culture medium), and subsequently transduced cells (HEK293T or MDA-MB-231) at various MOIs (i.e., 1 × 104, 5 × 104, 1 × 105, and 2 × 105). Normal human serum indicates that human-derived serum samples contain numerous endogenous proteins, including a variety of immunoglobulins existing in healthy humans. Different IgG sets from a different vendor (Thermo Fisher Scientific) and a A20 monoclonal antibody set (GeneTex, Irvine, CA) possessing AAV-specific antibodies were purchased to demonstrate further the AAVr3.45's resistance against NAb inactivation. At 2 days post transduction, the percentages of GFP-expressing cells were counted by flow cytometry analysis, and images of GFP-positive cells in each condition were acquired using a fluorescence microscope (Nikon Eclipse Ti-S; Nikon, Tokyo, Japan) to determine the performance of each AAV vector at evading neutralizing antibodies. Additionally, to recognize readily the reduction rates of transduction efficiencies under human IgG or to identify the fractions of infectious viral titers having NAb-resistant properties, the percentages of GFP-expressing cells obtained at each titer of human IgG were normalized to those measured in the absence of human IgG (i.e., % of GFP+ cells normalized to no IgG control).
Apoptosis and viability analysis
To examine the apoptotic effects of BIM expression in MDA-MB-231 cells, the fractions of cells entering the apoptotic cycle were quantified by an annex in V/7-amino-actinomycin D (7-AAD) assay (BD Biosciences, San Jose, CA). The appearance of phosphatidylserine (PS), which is typically exposed to the external cellular environment in the early phase of apoptosis, was detected by quantifying the emission of the phycoerythrin (PE)-tagged annexin V, which has a high affinity for PS, thereby representing the hallmarks of cellular programmed death (i.e., early apoptosis). 21 The specificity of 7-AAD for DNA guanine-cytosine base-pair fragments, which can be exposed once the integrity of the cellular membrane is sufficiently disrupted (i.e., late apoptosis/cell death), was employed to detect the signals of cells in the necrotic phase. 22 The percentages of live (annexin V–/7-AAD–), early apoptotic cells (annexin V+/7-AAD–), and late apoptotic cells (annexin V+/7-AAD+) were demonstrated to assess the cells' fates against BIM expression mediated by AAVr3.45-BIM under high titers of human IgG (1,000 μg/mL). In parallel with the apoptosis analysis, the viability of MDA-MB-231 cells that were infected by AAVr3.45-GFP or AAVr3.45-BIM was quantified using a WST-1 assay kit (Sigma–Aldrich) according to the manufacturer's guidelines. Briefly, MDA-MB-231 cells were seeded on a 96-well plate (5,000 cells/well) and infected with AAVr3.45-GFP or AAVr3.45-BIM at a genomic MOI of 5 × 104. The cell culture medium was replaced with fresh medium at 12 h post transduction, and a 0.1 volume of the WST-1 reagent out of the total volume of medium was dropped onto each well. Subsequently, the intensity of the colorimetric changes was quantified after 4 h by measuring the absorbance at 440 nm using a spectrophotometer (Nanodrop 2000; Thermo Fisher Scientific, West Palm Beach, FL). Each absorbance value was compared to those obtained from cells grown without viral infection.
Western blotting
The quantification of BIM expression from MDA-MB-231 cells transduced by AAVr3.45 vectors carrying the BIM gene was confirmed by performing Western blot analysis. At 2 days post transduction with AAVr3.45-BIM or AAV2-BIM (MOI 5 × 104), MDA-MB-231 cells were harvested and lysed to extract BIM for running sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels. Briefly, once electrophoresis had been completed to induce the BIM to migrate through the PAGE gels, each band was blotted, transferred to a nitrocellulose membrane (Thermo Fisher Scientific), and exposed to rabbit anti-human BIM polyclonal antibody (1:1,000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) and mouse anti-human beta-actin polyclonal antibody (1:1,000; Sigma–Aldrich) at 4°C overnight. Subsequently, the membranes were further exposed to secondary antibodies, namely, Alexa Fluor 488 anti-rabbit and Alexa Fluor 594 anti-mouse (Thermo Fisher Scientific), at room temperature for 1 h, and the immunoblots were visualized by chemiluminescence using an ECL Detection system at Yonsei University College of Medicine Medical Research Center (mini LAS 3000; FujiFilm, Tokyo, Japan).
Constructing three-dimensional structure models of AAVr3.45 derivatives
The capsid structure of AAVr3.45, which was originally created based on the AAV2 capsid structure
19
and modified in Fig. 1A, was used to generate the three-dimensional (3D) structure models of the AAVr3.45-derivatives by aligning their peptide sequence with the AAV2 peptide structure coordinates (RCSB PDB No. 1LP3). Each viral capsid model was built in the SWISS MODEL online 3D modeling server (
Brief characterization of adeno-associated virus (AAV) variant AAVr3.45 as a gene carrier for cancer gene therapy applications.
Statistical analysis
All the conditions were performed in triplicate and expressed as the mean and the standard deviation (SD). A one-way analysis of variance (ANOVA) with a post hoc Dunnett's test using the PASW Statistics for Windows v18.0 (SPSS, Inc., Chicago, IL) was used to test for statistical significance.
Results and Discussion
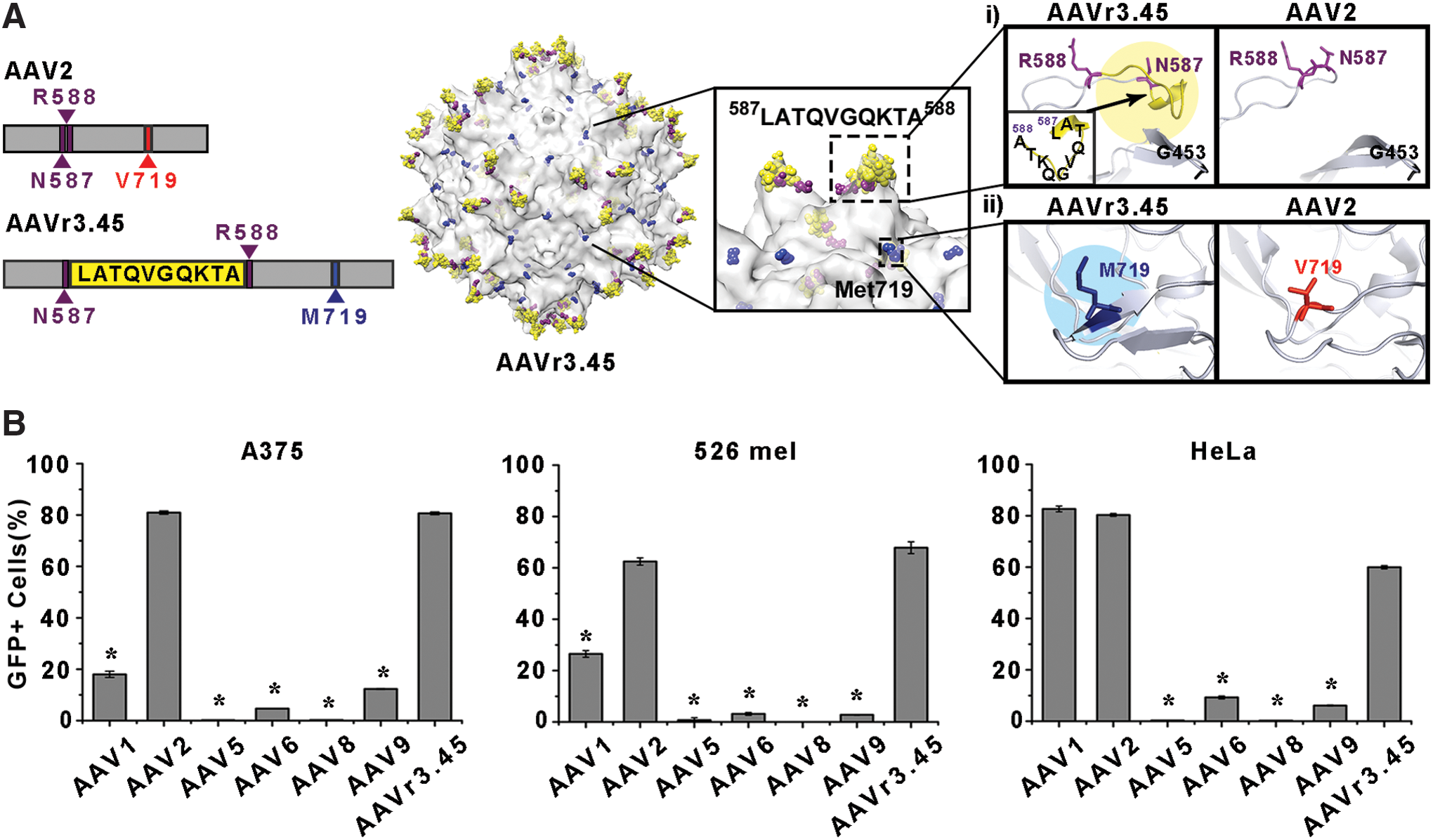
The potential of AAVr3.45, which was originally selected from random viral libraries for specifically infecting neural stem cells in a previous study, 19 was evaluated as a therapeutic agent for many gene therapy applications, especially for cancer gene therapy approaches, by assessing two essential therapeutic capabilities: (i) delivering genes of interests efficiently to several key cancer cell lines and (ii) circumventing neutralization by a purified human IgG cocktail. AAVr3.45 was genetically engineered from AAV2, which has been widely used in many clinical trials, and demonstrated remarkable deviations in its gene delivery capabilities compared to the parental AAV2, namely, its superior gene delivery efficiency in many therapeutically relevant cells, such as stem cells, which are mostly non-permissive to the AAV2 vector. 19 Detailed understanding of the structural characteristics of AAVr3.45 compared to the parental vector may therefore provide crucial clues for interpreting its distinguishing properties, potentially expanding the scope of its application for many clinical trials. Thus, the structural roles of both key peptides distinctively classified compared to those of AAV2 were investigated: (i) the LATQVGQKTA peptide that was externally displayed at the HSPG binding domain (i.e., between asparagine (N) at the aa 587 site and arginine (R) at the aa 588 site) and (ii) the methionine (M; Met719) that was replaced with valine (V) located at the aa 719 site (Val719) of the AAV2 capsid 19 (Fig. 1A). The effects of these characteristics on both gene delivery efficiency and NAb evading capabilities were assessed. The unique features of AAVr3.45 were examined by evaluating its performance as a versatile gene carrier specifically for cancer gene therapy applications, where the immune resistance of viral vectors is critical for successfully delivering therapeutic genes via systemic routes to cancer cells scattered within the body.
Initially, as a basic requirement for a gene carrier that can be used for cancer gene therapy applications, the ability of AAVr3.45 to transduce several cancer cell lines, which have been frequently used as model cells in many cancer studies,
26
–29
was evaluated by quantifying the percentages of GFP-expressing cells transduced by AAVr3.45 along with those of other AAV serotypes. The AAVr3.45 vectors demonstrated great potential as a versatile candidate for genetically stimulating cancer cells, exhibiting enhanced transduction efficiency in two types of melanoma cells (i.e., A375 and 526 mel), and cervical cancer cells (i.e., HeLa), and comparable to the efficiency of AAV2 (Fig. 1B), which has been extensively used in preclinical cancer gene therapy trials.
7,30
–32
The transduction efficiency by AAVr3.45 was significantly enhanced compared to the other representative AAV serotypes (i.e., AAV 5, 6, 8, and 9; Fig. 1B), which have also been widely used in AAV-mediated gene therapy applications. A previous study, in which AAVr3.45 was originally developed, also showed enormously improved gene delivery efficiency (>80%) in a murine metastatic melanoma cell line (i.e., B16F10) compared to other wild-type AAV serotypes.
19
Importantly, the superior capability of AAVr3.45 to enhance transduction efficiencies in T lymphocyte-resistant MDA-MB-231 (Supplementary Fig. S1; Supplementary Data are available online at
The structural deviations of AAVr3.45 compared to wild-type AAV2 resulted in its dramatic alterations in gene delivery characteristics. All the key modified peptides that are observed on AAVr3.45 (i.e., LATQVGQKTA at the HSPG binding domain and V719M) are primarily surface-exposed motifs, as shown in a previous study, thereby likely functioning as links that can specifically interact with external substances (e.g., receptors or antibodies). The previous study in which the AAVr3.45 was initially developed revealed that its dramatically enhanced gene delivery capability in neural stem-cell (NSC) infection was primarily attributable to the singular effect of the external LATQVGQKTA peptide at the HSPG binding domain rather than synergistic coordination between the peptide and Met719. 19 Thus, an interpretation of the potential function of the point mutation (i.e., V719M) is still lacking. Furthermore, the reasons for the beneficial effects of the inserted 10 amino acids on the robust gene delivery observed in many cell types were not identified because the peptide sequence had no partial sequence matches with known enzymes or proteins. 19
It was anticipated that the substitution of Val719 of AAV2 capsid to Met719 in AAVr3.45 capsid would render it resistant against inactivation by NAbs, thereby reducing the probability of AAVr3.45's exclusion from the human circulatory system. In a previous study, multiple AAV variants capable of escaping neutralization from preexisting immunity were selected from random viral libraries, and one of the mutated peptides that were observed from the resulting AAV variants was an alanine (A) that was substituted for a threonine (T) at the aa 716 site of AAV2 capsid, 15 which nearly coincides with the location of the aa 719 site. Thus, it was anticipated that the specific 3D structure constructed by the surrounding peptides adjacent to the aa 716 site, including the aa 719 site, or characteristic properties of side groups on the designated amino acids (i.e., methionine and valine) might serve as one of the primary epitopes that can be readily recognized by NAbs. To examine this aspect, the Met719 of the AAVr3.45 was reversely mutated to the original sequence of the aa 719 site observed on the AAV2 capsid, Val719, using a site-directed mutagenesis protocol (Fig. 2A). As shown in Fig. 2B, the amino acids surrounding the 716–719 site were exposed on the external surface, and the point mutation from methionine to valine, or vice versa, did not result in substantial alterations in the electrostatic properties of the designated surfaces (Supplementary Fig. S2). Instead, the difference in each side chain, sulfur, and propyl group on the methionine and valine, respectively, was distinctively recognized in Fig. 2B. The AAVr3.45-derivative containing the original V719, referred to as r3.45-V719, was newly synthesized. Thus, it possesses the LATQVGQKTA sequence inserted at the HSPG binding domain (i.e., aa 587 site), which is its only difference from the AAV2 capsid (Table 1). Viral vectors carrying GFP, including AAV2, the original AAVr3.45, and r3.45-V719, were pretreated with a series of various human IgG titers (i.e., 0–800 μg/mL) for 1 h and incubated with medium containing HEK293T cells that are highly permissive to both AAV2 and AAVr3.45. Subsequently, the reduction profiles of cellular transduction by the resulting vectors were examined to verify the role of the point mutation at the aa 719 site for evading antibody neutralization.
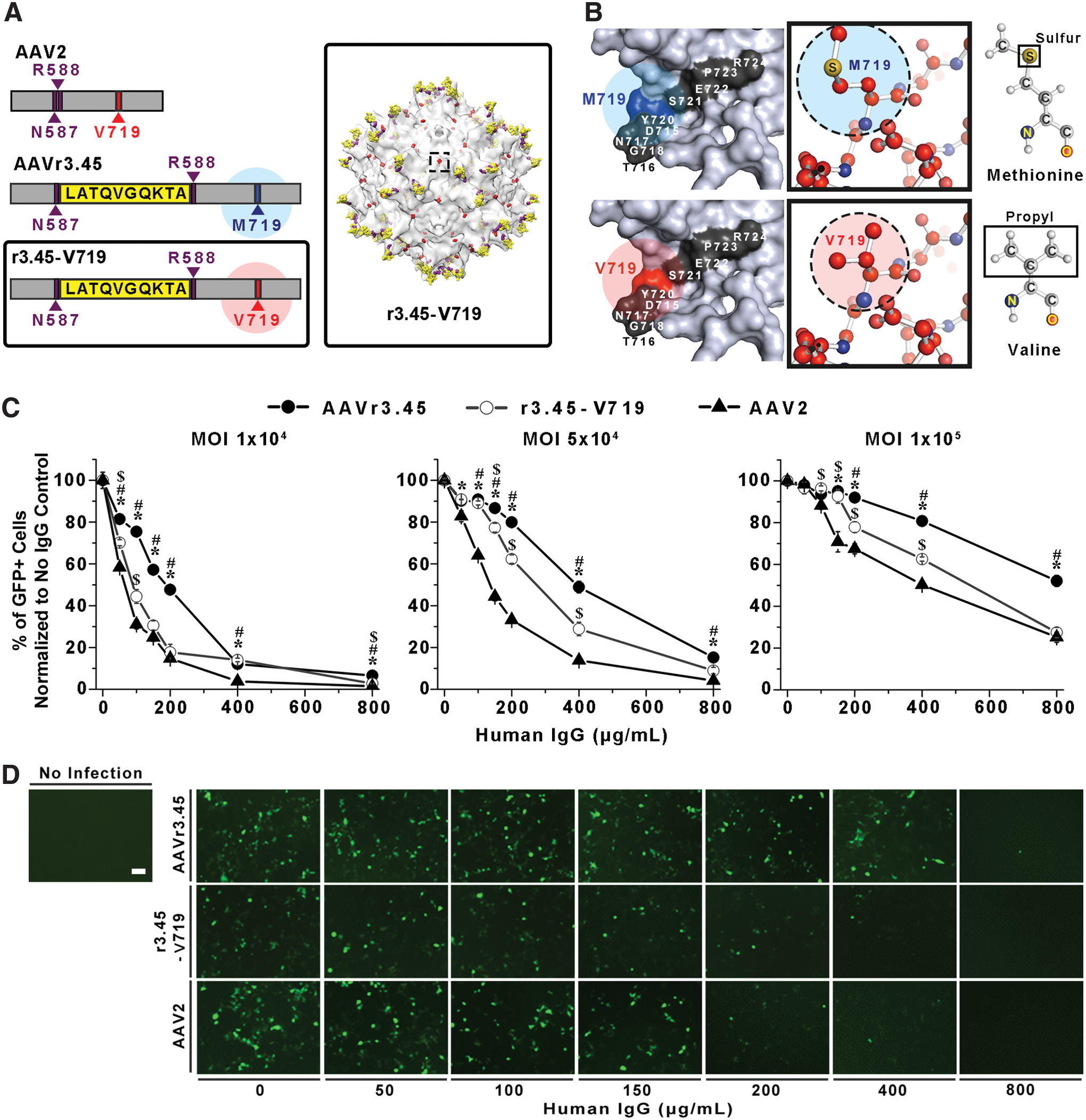
Characterization of r3.45-V719 vectors.
The original AAVr3.45 containing Met719 demonstrated superiority in evading antibody neutralization compared to both AAV2 and r3.45-V719. To evaluate the NAb-evading capability of each AAV vector, the percentages of GFP-expressing cells transduced by viral vectors treated with various titers of IgG were normalized to those obtained in the absence of IgG. These values were defined as the percentages of GFP-positive cells normalized to no IgG control for each AAV vector at the designated IgG titers, possibly dictating the portions of infectious AAV vectors with IgG resistance out of the total viral quantities. Thus, the comparisons in the percentages of GFP-positive cells normalized to no IgG control for each viral vector can provide relevant criteria to determine the differences in the degree of IgG resistance of AAV vectors. At 2 days post transduction, the percentages of GFP-positive cells normalized to no IgG control were quantified by varying viral MOIs (1 × 104, 5 × 104, and 1 × 105) as a function of the human IgG titers (Fig. 2C). When HEK293T cells were infected by each AAV vector (i.e., AAVr3.45, r3.45-V719, and AAV2) at a genomic MOI of 2 × 105 in the absence of human IgG, the percentages of GFP-expressing cells almost exceeded 90% (Supplementary Fig. S3). Unlike the results reported in the previous study, in which no beneficial contribution of the point mutation (V719M) to gene delivery in neural stem cells was confirmed, significantly enhanced transduction by AAVr3.45 with Met719 compared to that by r3.45-V719 with Val719 was observed when HEK293T cells were infected at low genomic MOIs (i.e., 1 × 104 and 5 × 104; Supplementary Fig. S3). These results indicate that the beneficial effects of the point mutation (V719M) on cellular transduction may be cell or MOI dependent. Importantly, at a genomic MOI of 1 × 105, the IgG-resistant capabilities of AAVr3.45 vectors compared to AAV2 or r3.45-V719 vectors appeared initially at 150 μg/mL of IgG or 200 μg/mL of IgG, respectively (p < 0.01; right panel of Fig. 2C). Upon the highest IgG treatment (i.e., 800 μg/mL), AAVr3.45 at a MOI of 1 × 105 demonstrated approximately twofold improvement of IgG resistance (i.e., % of GFP+ cells normalized to no IgG control) compared to those of the other vectors (<30%; MOI 1 × 105).
The deviations in IgG-evading capabilities of each virus were even more distinctive when lower viral quantities (i.e., MOI 1 × 104 and 5 × 104) were used for transducing cells (Fig. 2C). At genomic MOIs of 1 × 104 and 5 × 104, significantly enhanced evasive capabilities of AAVr3.45 relative to both AAV2 and r3.45-V719 were observed almost over the entire ranges of human IgG (p < 0.01). While the IgG-dependent slopes in the percentages of GFP-positive cells normalized to no IgG control for AAVr3.45 vectors were slowly and gradually reduced over the IgG titers, the reduction rates by AAV2 and r3.45-V719 were dramatically steep, even at the low IgG titers (<100 μg/mL). At genomic MOIs of 1 × 104 and 5 × 104, AAVr3.45 exhibited approximately 1.1- to 5-fold enhancement of IgG-resistant capabilities (i.e., % of GFP+ cells normalized to no IgG control) compared to its parental AAV2 and r3.45-V719 over the entire range of IgG titers. Importantly, at the genomic MOI of 5 × 104, AAVr3.45 vectors that were pretreated with 400 μg/mL of human IgG still demonstrated >48.9 ± 2.8% infectious properties, while only 13.8 ± 0.9% of AAV2 and 28.8 ± 3.2% of r3.45-V719 remained infectious under the same IgG titer (400 μg/mL). As shown in Fig. 2D, while moderate levels of GFP signals driven by the AAVr3.45 delivery were readily observed >400 μg/mL of human IgG, substantially reduced numbers of GFP-expressing cells were visualized at the high IgG titers (>200 μg/mL) when cells were transduced by AAV2 or r3.45-V719. All these observations indicate that the amino acid mutation (i.e., V719M) observed on the AAVr3.45 capsid disrupts the epitopic function of the surrounding peptide sequences, ultimately rendering AAVr3.45 vectors effectively resistant against NAb inactivation. The mutation of valine (V) to methionine (M) at aa719 of AAV2 (AAV2-V719M) resulted in enhanced NAb-evading properties compared to the non-modified AAV2, possibly confirming the role of the point mutation at aa719 for NAb evasion (Supplementary Fig. S4). It has been well known that even a single point mutation can sufficiently induce significant alterations in the overall structures or functions of polypeptides. 33,34 Thus, the substitution of Val719 of AAV2 to Met719 of AAVr3.45 may apparently cause epitopic functions at the designated peptide locus. Importantly, the NAb-evading capabilities of AAVr3.45 compared to those of AAV2 were further confirmed in different sets of human IgG samples (Supplementary Fig. S5). Further NAb-evading studies were performed with the human IgG used in Fig. 2.
The ability of AAVr3.45 evade antibody neutralization was dependent on the quantity of viral vectors used for transducing cells. At ranges exposed to high IgG titers (i.e., >400 μg/mL), >79.8 ± 1.0% of AAVr3.45 at an excessively high MOI of 2 × 105 were resistant against the highest IgG titer (i.e., 800 μg/mL; Supplementary Fig. 6). The percentages of GFP-positive cells normalized to no IgG control for AAV2 and r3.45-V719 were decreased to 39.3 ± 0.7% and 71.1 ± 1.4%, respectively, at 800 μg/mL of IgG, all of which were decent levels, even at the harsh condition, but significantly lower than those for AAVr3.45 (Supplementary Fig. S6). The administration of viral vectors in large quantities might solely increase the absolute quantity, incidentally preventing their entrapment by neutralizing antibodies, 10,35 thereby raising their therapeutic efficacy. Additionally, a previous study demonstrated that the inclusion of mock vectors along with vehicles carrying therapeutic genes simply expanded the quantities of viral vectors that successfully approached the target spot. 36 Therefore, the infusion of large viral quantities, which can adversely cause detrimental effects, such as high frequency of random genomic insertion or acute immune responses, 37,38 may be currently an indispensable means for elevating the therapeutic efficacy of gene therapy treatment. No decoy effect of AAVr3.45 for the elevated NAb-evading capability was confirmed by the fact that no apparent deviations in the ratio of capsid particle to viral genome for AAVr3.45 were observed compared to that of AAV2 (Supplementary Table S1). Quantifying the ratio of capsid particles to viral genome by UV spectrophotometry analysis to estimate the fractions of empty AAV vectors was performed, as previously described. 36 Exploring NAb-resistant viral vectors can be highly beneficial to circumvent the safety issues associated with the infusion of large viral quantities because the dosages of the NAb-resistant AAVr3.45 can be remarkably reduced compared to the non-resistant AAV vectors. For example, below the 800 μg/mL IgG titer, approximately 50% reduced AAVr3.45 quantity (MOI 1 × 105) was required for yielding the same transduction efficiencies (i.e., ∼40%) obtained by AAV2 infection at a MOI of 2 × 105 (Supplementary Fig. S7).
Importantly, the fact that, in most ranges of IgG titers, the percentages of GFP-positive cells normalized to no IgG control for the r3.45-V719 were significantly greater than those for AAV2 possibly implies that the LATQVGQKTA at the HSPG binding domain may synergistically assist the point mutation (V719M) in evading antibody neutralization (Fig. 2C). It is obvious that the deviations in NAb-evading capabilities between AAVr3.45 and r3.45-V719 are caused by the point mutation (V719M) because the only structural difference between AAVr3.45 and r3.45-V719 is the replacement of Met719 with Val719. The increases in the percentages of GFP-positive cells normalized to no IgG control for r3.45-V719 over the IgG titer ranges compared to those for AAV2 might be attributed to the presence of the LATQVGQKTA on the r3.45-V719 because the LATQVGQKTA at the aa 587 site is the sequence distinctively not matched with the AAV2 capsid. Based on these results, in addition to the point mutation at the aa 719 site, it could be implied that the disruption of the HSPG binding domain by inserting the LATQVGQKTA peptide might work as a block against NAb attacks. Displaying the peptide on the protruding spikes of the viral capsid might substantially alter the steric features of AAVr3.45 compared to the non-resistant AAV2 structure, presumably causing the high probabilities of NAb misrecognition of the viral capsid. Similarly, several previous studies demonstrated that inserting external substances, such as laminin fragment 39 or QQNTAP, 40 into the three-fold spike locus (i.e., HSPG binding domain) led to persistent transduction in its systemic administration against the NAb inactivation. Furthermore, the substitution of AAV8's extruded locus into AAV2's HSPG binding motif resulted in the activation of CD8+ T lymphocytes, possibly demonstrating the antigenic aspects of the threefold spike HSPG binding domain existing on the AAV2 capsid. 41 Thus, disguising the HSPG binding spike locus by displaying the LATQVGQKTA motif might contribute to the ability of AAVr3.45 to circumvent NAb inactivation.
To confirm the roles of the LATQVGQKTA in determining the characteristic features of AAVr3.45, the replacement of the positively charged lysine (K) with glutamic acid (E), which possesses a negatively charged side group under physiological conditions, was conducted, and the transduction profiles by the AAVr3.45-derivative vector having the LATQVGQETA sequence under the various IgG titers were investigated by comparing them to those by AAVr3.45 (Fig. 3A). As shown in Fig. 3B, the overall amino acids comprising the LATQVGQKTA sequence that was inserted adjacent to the HSPG binding motif were exposed externally, thereby possibly acting as an additional motif that can closely interact with outer environments. Previous studies demonstrated that linking either the full or partial sequence of the LATQVGQKTA onto cationic molecules resulted in distinctively improved gene delivery efficiency in many cell types, presumably due to the robust association of the LATQVGQKTA-tagged vectors with the cellular membrane. 42,43 It was assumed that among all the amino acids in the LATQVGQKTA sequence, the lysine (K), which is shown in orange in Fig. 3B, would be the most active amino acid that effectively coordinates the interactions of the peptide sequence with external substances due to its electrostatic charge. The electrostatic potential analysis performed in Fig. 3C depicts that the replacement of the lysine (K) with glutamic acid (E; shown in green in Fig. 3B) clearly alters the electrostatic properties of the surrounding peptides, from positive (blue color) to negative surfaces (red color), which was further confirmed by the zeta potential analysis (Supplementary Fig. S8). Thus, it was hypothesized that charged peptide domains on a viral capsid would function as a critical driving force to arrange its attractive or repulsive interactions with numerous charged molecules on the cellular membrane, ultimately dominating its transduction properties. The newly packaged virus, referred as r3.45-587InsK 8 E, contained the LATQVGQETA sequence at the HSPG binding domain with an additional point mutation at the aa 719 site (i.e., V719M; Table 1). Thus, the presence of the glutamic acid (E) instead of the lysine (K) on the peptide inserted at the HSPG binding domain is a unique structural characteristic of r3.45-587InsK 8 E compared to AAVr3.45 (Fig. 3A). Alterations in amino acids on the inserted peptide, from K to G (r3.45-587InsK 8 G) or Q to E (r3.45-587InsQ 4 E), elicited nearly identical NAb-evading properties compared to those for r3.45-587InsK 8 E, whose GFP percentages relative to no IgG control were significantly lower than those for AAVr3.45 containing the original inserted peptide (LATQVGQKTA; Supplementary Fig. S9). These results indicate that the inserted peptide sequence itself might provoke inhibitory effects on the NAb recognition of one of the epitope sites (i.e., N587), possibly by steric hindrance or deviations in the ionic environment near the virus. The capabilities of the r3.45-587InsK 8 E for evading NAb inactivation and for promoting gene delivery efficiency were assessed by quantifying the percentages of GFP-expressing HEK293T cells under the various titers of human IgG.
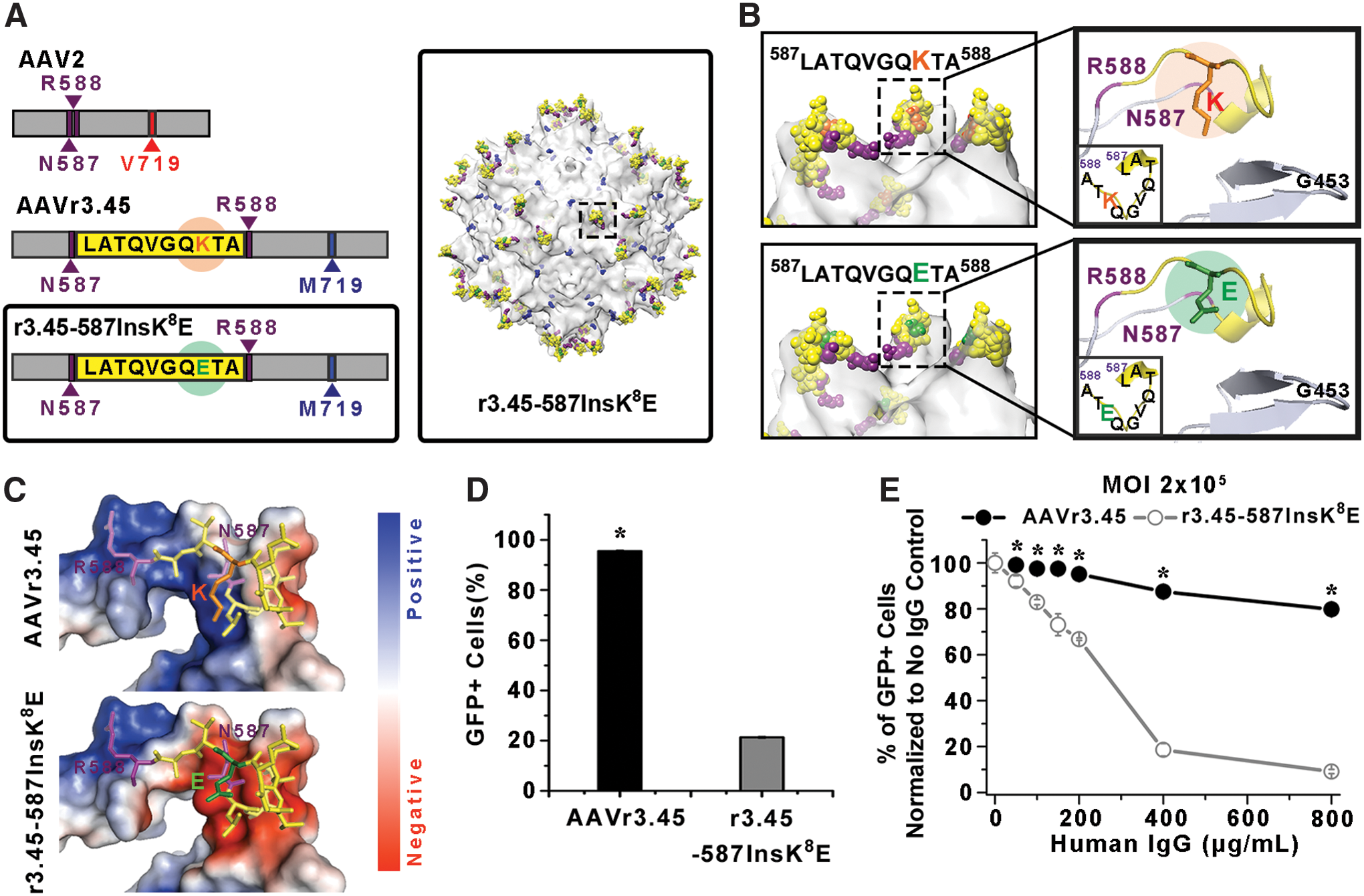
Characterization of r3.45-587InsK
8
E vectors.
The r3.45-587InsK 8 E led to dramatic reductions in cellular transduction efficiencies, even without its exposure to the human IgG cocktail. In the absence of IgG, 21.3 ± 0.5% of GFP-expressing cells were observed when HEK293T cells were infected by r3.45-587InsK 8 E, even at the excessively high MOI of 2 × 105, while almost saturated transduction efficiency (i.e., 95.6 ± 0.5%) could be obtained by AAVr3.45 infection at the same MOI (Fig. 3D). Consistently, substantially low transduction efficiencies (<4%) by the r3.45-587InsK 8 E vectors in many cancer cell lines (i.e., A375, 526 mel, HeLa, and T-cell resistant MDA-MB-231), which were highly permissive to AAVr3.45, as shown in Fig. 1B and Supplementary Fig. S1, further confirmed the decay in the transduction capabilities of the r3.45-587InsK 8 E compared to those of AAVr3.45 (Supplementary Fig. S10). Additionally, the percentages of GFP-expressing human neural stem cells (hNSCs) transduced by r3.45-587InsK 8 E was approximately 3% (Supplementary Fig. S10), which was reported as a high value when hNSCs were transduced by AAVr3.45 in a previous study. 19 The sharp decrease in gene delivery efficiency by r3.45-587InsK 8 E provides a clue for interpreting the roles of the LATQVGQKTA peptide: positively charged side groups on the lysine in the LATQVGQKTA (Fig. 3C), which is located in the vicinity of the HPSG binding domain (Fig. 3B), may trigger close interactions with negatively charged many glycosaminoglycans (GAG; e.g., HSPG or chondroitin sulfate proteoglycan; CSPG) or receptors (e.g., integrin or fibroblast growth factor receptors) anchored on the cell surface, possibly accelerating viral entry across the cellular membrane for transduction. In a previous study, treating highly permissive cells (e.g., HEK293T or hNSCs) with heparinase II or chondroitinase ABC, which, respectively, cleave HSPG and CSPG from the cell surface, substantially curtailed the transduction efficiencies obtained by AAVr3.45 infection. 44 The result shown in the previous study possibly indicate that coordinating the interactions of gene delivery vehicles with the GAGs on the target cell surfaces can be a critical factor for adjusting gene delivery properties. Thus, further reinforcing the interactions of the HSPG binding motif with negatively charged GAGs through the assistance of the positively charged lysine (K) on the LATQVGQKTA peptide might work as a key driving force to broaden the tropisms of AAVr3.45 considerably to many therapeutically relevant cell types, which abundantly display GAGs on their surfaces.
Switching the lysine (K) to glutamic acid (E) in the LATQVGQKTA while preserving the Met719 did not protect the r3.45-587InsK 8 E vectors effectively against neutralization by IgG. Due to the significantly reduced transduction capabilities of r3.45-587InsK 8 E vectors, excessively high genomic quantities of both AAV vectors (i.e., AAVr3.45 and r3.45-587InsK 8 E; MOI of 2 × 105) were exposed to various titers of human IgG and subsequently used for infecting HEK293T cells. The percentages of GFP-positive cells normalized to no IgG control for the r3.45-587InsK 8 E treated with human IgG were significantly decreased compared to those for AAVr3.45 over the entire ranges of IgG (Fig. 3E). The declining rates in the percentages of GFP-positive cells normalized to no IgG control for r3.45-587InsK 8 E were much steeper than those of AAVr3.45 (Fig. 3E). These observations imply that the NAb-resistant features of AAVr3.45 might be conferred by the coordinative contributions of the LATQVGQKTA peptide and the point mutation (i.e., V719M) rather than by the singular effect of the mutated sequence (V719M). Consistently, a previous study verified that the mutations of asparagine (N) at the aa587 site into isoleucine (I) and threonine (T) at the aa716 into alanine (A) served as key moieties for NAb evasion of AAV2-based vectors. 15 These two mutated sites nearly overlapped with the featured domains of AAVr3.45 (LATQVGQKTA & V719M). The antigenic function of amino acids around the 716–719 sites for the antibody-evasion phenotype was further identified in several previous studies. 45,46 Peptide insertions or mutations can significantly alter the 3D peptide structures or their functions compared to their original structure. Thus, the mutated sites occurring at aa587 and aa719 might synergistically disrupt their recognition by neutralizing antibodies.
To examine the anticipated benefits of the LATQVGQKTA peptide itself on NAb-evading capabilities, both the lysine (K) on the LATQVGQKTA and the Met719 on the AAVr3.45 capsid were modified to glutamic acid (E; LATQVGQETA) and Val719, respectively, to create r3.45-587InsK 8 E/V719 (Fig. 4A). The vector r3.45-587InsK 8 E/V719 contains LATQVGQETA at the HSPG spike locus and also contains Val719 (Table 1). Then, the percentages of GFP-positive cells normalized to no IgG control for the r3.45-587InsK 8 E/V719 vector under the various IgG titers were compared in parallel to those by r3.45-V719, which contains the full LATQVGQKTA sequence with the point mutation (M719V). Thus, these two vectors differ in only one amino acid on the peptide inserted at the HSPG binding domain (i.e., glutamic acid (E): r3.45-587InsK 8 E/V719 and lysine (K): r3.45-V719). As a result, in the absence of IgG, infection with HEK293T cells with the r3.45-587InsK 8 E/V719 at a genomic MOI of 2 × 105 resulted in significantly lower transduction efficiencies than that with r3.45-V719 vectors at a genomic MOI of 2 × 105 but no significant differences in transduction efficiencies compared to infection with r3.45-587InsK 8 E vectors (Fig. 4B). These results further confirm the significant contribution of the LATQVGQKTA peptide, especially the lysine (K), for enhancing gene transfer capabilities of AAV vectors and the insignificant influence of the point mutation (V719M) on gene delivery properties. The comparisons between the percentages of GFP-positive cells normalized to no IgG control for r3.45-587InsK 8 E/V719 and those for r3.45-V719 over the whole ranges of human IgG titers demonstrated higher resistance in r3.45-V719 than r3.45-587InsK 8 E/V719 (Fig. 4C), indicating that the less antigenic features of AAV vectors containing the LATQVGQKTA at the HSPG binding motif might be caused by the specific peptide sequence. The vector r3.45-V719 demonstrated approximately 18-fold enhanced IgG resistance compared to r3.45-587InsK 8 E/V719 <800 μg/mL of IgG (Fig. 4C). Furthermore, the reduction rates of the percentages of GFP-positive cells normalized to no IgG control under the various IgG titers were substantially greater in cellular transduction by r3.45-587InsK 8 E/V719 than in r3.45-V719 (Fig. 4C). Thus, retaining the dual benefits of both the LATQVGQKTA peptide itself and the point mutation V719M rather than the single effect of V719M may be highly critical to maximize the IgG-resistant capabilities of viral vectors. The percentages of GFP-positive cells normalized to no IgG control for r3.45-587InsK 8 E were mostly greater than those for r3.45-587InsK 8 E/V719 over the range of IgG titers, further confirming the significance of the mutation of Val719 for promoting the IgG-resistance of the resulting AAV vectors.
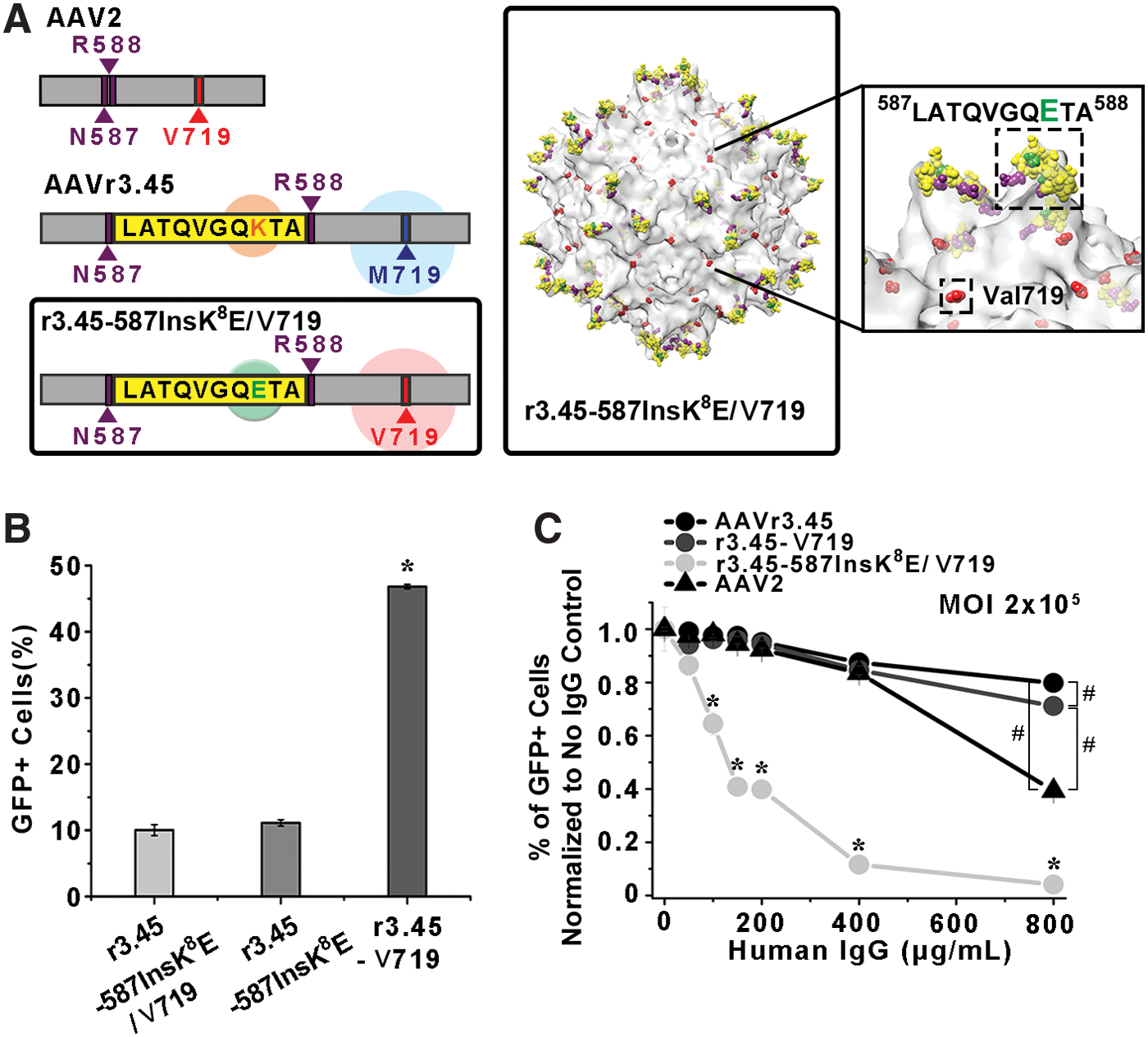
Characterization of r3.45-587InsK
8
E/V719 vectors.
In summary, AAVr3.45 demonstrated superior abilities in both gene delivery and IgG evasion, all of which were significantly improved compared to those for the other AAV vectors, including AAV2, r3.45-V719, r3.45-587InsK 8 E, and r3.45-587InsK 8 E/V719. Displaying the LATQVGQKTA peptide itself on the exterior served as a key clue for enhancing gene delivery as well as supplementally promoting NAb evasion. Importantly, one peptide mutation at the aa 719 site (V719M) was prerequisite for shielding the resulting AAV vectors against IgG inactivation rather than for promoting gene delivery capabilities, but its capability for blocking viral vectors against antibody neutralization could be further doubled by the assistance of the LATQVGQKTA peptide.
Based on the capabilities of AAVr3.45 in both enhancing transduction efficiencies in cancer cells and evading NAb inactivation, its potential as a cancer gene therapy vector capable of efficiently delivering therapeutic genes (Bcl-2-like protein 11; BIM) to breast adenocarcinoma cells (MDA-MB-231) was evaluated by examining the fractions of apoptotic breast cancer cells out of total cells under high titers of human IgG, as schematically illustrated in Fig. 5A. Importantly, AAVr3.45 led to significantly enhanced transduction in MDA-MB-231 cells compared to several representative wild-type AAV serotypes, demonstrating the basic requirement as a therapeutic agent for stimulating cancer cells. As the MOI of AAVr3.45 vectors increased, the percentages of GFP-positive cells were gradually improved, ultimately reaching approximately 68.2 ± 0.3% with a MOI of 1 × 105, whereas transduction levels by several representative wild-type AAV serotypes (AAV2, 5, and 8) remained <10% of GFP expression (Fig. 5B). Additionally, the other AAVr3.45-derivative vectors, including r3.45-V719, r3.45-587InsK 8 E, and r3.45-587InsK 8 E/V719, were used for infecting MDA-MB-231 cells to confirm the transduction profiles compared to the ones observed in HEK293T infection. As consistently observed in HEK293T transduction, vectors with the glutamic acid (E) in the peptide displayed at the HSPG binding motif infected MDA-MB-231 cells with dramatically low levels (i.e., <10%), and transduction with r3.45-V719 vectors at a MOI of 1 × 105 was not statistically different with that by AAVr3.45 (Fig. 5C). Finally, the percentages of GFP-positive cells normalized to no IgG control for AAVr3.45 in MDA-MB-231 cells under various titers of human IgG were investigated by varying viral MOIs. As observed in HEK293T cells, with a MOI of 2 × 105, no abrupt reductions in infectious titers were observed over the whole span of IgG titers (Fig. 5D). However, at a MOI of 5 × 104, the percentages of GFP-positive cells normalized to no IgG control reduced gradually as a function of IgG titers, ultimately reaching 40% drops in fractions of IgG-resistant AAV vectors <400 μg/mL of IgG (Fig. 5D). The percentages of GFP-positive cells normalized to no IgG control for AAVr3.45 vectors were significantly greater than those for r3.45-V719 vectors over the entire ranges of human IgG, further confirming the capabilities of AAVr3.45 for evading IgG neutralization (Fig. 5D). Significant differences yet relatively smaller deviations in NAb-evading properties between AAVr3.45 and r3.45-V719 in MDA-MB-231 cells were observed compared to those observed in HEK293T cells, which might be caused by different cellular transduction or transcription pathways, depending on the cell type. 46 The percentages of GFP-positive cells normalized to no IgG control for AAV2, r3.45-587InsK 8 E, and r3.45-587InsK 8 E/V719 could not be assessed due to their dramatically low transduction efficiencies.
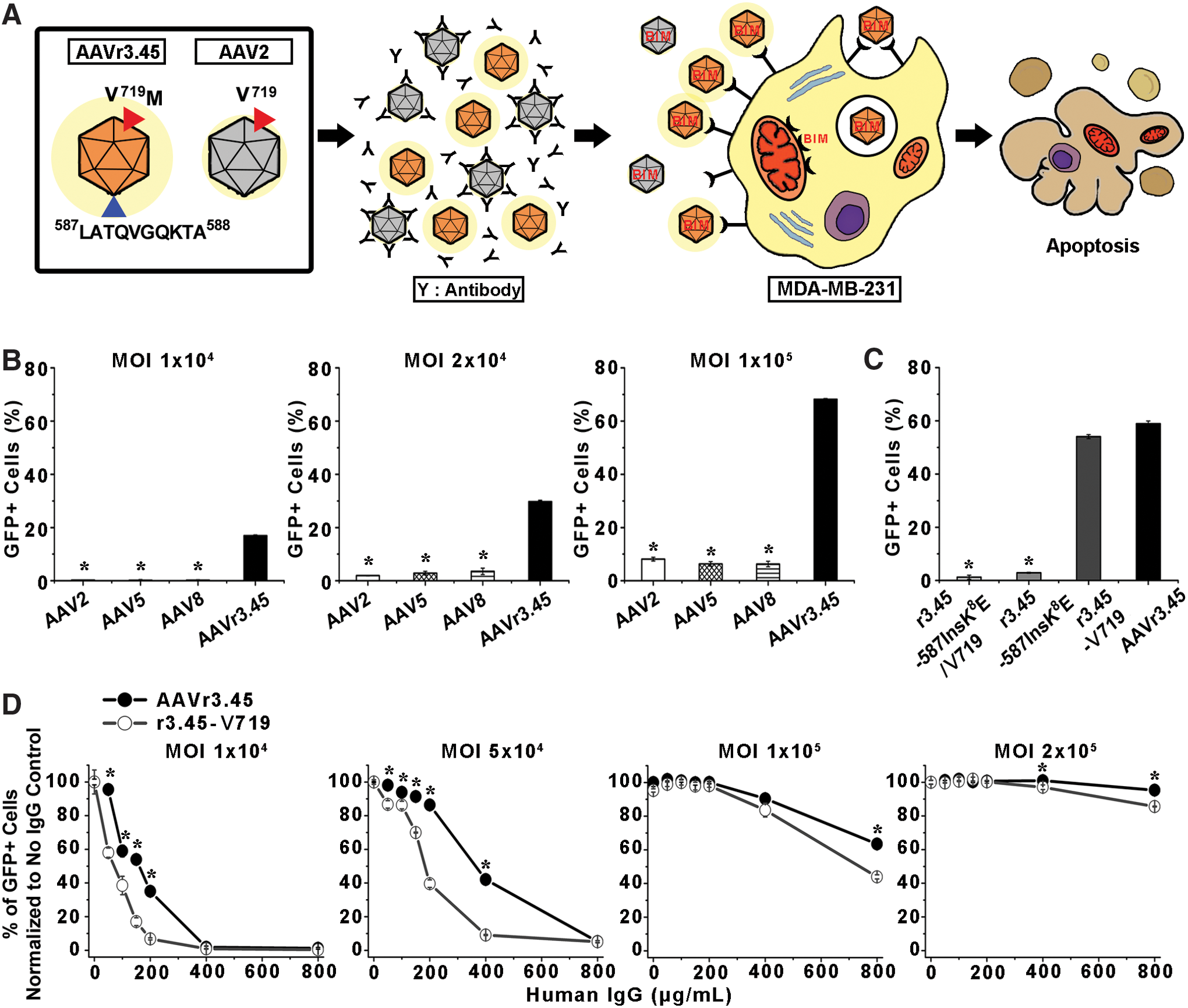
Transduction of MDA-MB-231 breast cancer cells by AAVr3.45 vectors.
The delivery of the pro-apoptotic gene BIM to MDA-MB-231 cells via AAVr3.45 (MOI 5 × 104) triggered the apoptosis of transduced cancer cells, even under a high titer of human IgG (1,000 μg/mL). Importantly, the IgG titer was elevated to 1,000 μg/mL in order to simulate closely the human blood sera further, where tremendously diverse antibodies that viral vectors must surpass to reach their targets (e.g., tumors) exist, thereby presumably mimicking actual body conditions. Exogenous BIM expression results in the upregulation of three apoptotic isoform inducers, including BIM-S, BIM-L, and BIM-EL, and the BIM-S has been known to be the most cytotoxic among the other isoforms, BIM-L and BIM-EL, which are normally expressed in most cell lines and normal tissues. 47,48 Thus, the BIM-s expression can accelerate cells' entry into the apoptotic cycle by inducing the mitochondrial outer membrane permeabilization (MOMP), 47 possibly causing chromatin condensation and DNA fragmentation. 49 As shown in Fig. 6A, both BIM-L and BIM-EL were expressed in all conditions (i.e., naïve cells, cells infected by AAVr3.45-GFP or AAVr3.45-BIM), but the BIM-s was detected solely in the MDA-MB-231 cells treated with AAVr3.45-BIM. Thus, successful induction of apoptotic cells by AAVr3.45-BIM, even under high titers of human IgG, can be a key criterion for evaluating the AAVr3.45 as a versatile therapeutic agent for cancer treatment (Fig. 5A). To test the functionality of BIM capable of determining cell fates initially, WST-1 assay was performed without exposing viral vectors to human IgG prior to viral infection (Fig. 6B). While the majority of cells transduced by AAVr3.45-GFP were viable, the metabolic activities of cells infected by AAVr3.45-BIM at a genomic MOI of 5 × 104 were significantly reduced compared to cells treated with AAVr3.45-GFP or cells without viral infection (Fig. 6B). Furthermore, once cells were infected by AAV2-BIM or AAVr3.45-BIM, the cellular fates were assessed by demonstrating the fractions of live and apoptotic cells, which were analyzed by quantifying the number of cells stained with 7-ADD/annexin V. Regardless of the presence of IgG, significantly smaller number of live cells (0 μg/mL: 45.3 ± 4.8%; 1,000 μg/mL: 64.0 ± 5.4%), which were simultaneously negative to annexin V and 7-AAD, was observed in AAVr3.45-BIM-treated cells than in AAV2-BIM (0 μg/mL: 71.9 ± 3.2%; 1,000 μg/mL: 81.1 ± 3.8%) or AAVr3.45-GFP-treated cells (0 μg/mL: 78.6 ± 3.8%; 1,000 μg/mL: 93.4 ± 0.4%; Fig. 6C). In the absence of IgG, the fractions of programmed cell death (i.e., early apoptosis: 49.2 ± 7.4%) were significantly promoted by AAVr3.45-BIM infection, compared to control conditions, including no infection (13.2 ± 1.7%), AAVr3.45-GFP (14.7 ± 4.7%), AAV2-GFP (1.0 ± 0.1%), and AAV2-BIM infection (19.0 ± 4.3%). Importantly, while <13.8 ± 4.2% or 1.3 ± 1.1% cells turned out to be programmed to death at 2 days post transduction by AAV2-BIM or AAVr3.45-GFP vectors, respectively, under the high titers of IgG (i.e., 1,000 μg/mL), very large percentages of cells (i.e., 31.3 ± 5.1%) were partitioned as early apoptotic cells under the high titers of IgG at 2 days post transduction by AAVr3.45-BIM vectors at a MOI of 5 × 104 (Fig. 6D). No statistical differences in the factions of the late apoptotic cells (7-ADD+/annexin V+) among all experimental sets were observed, but highly large fractions of cells (overall 35.6 ± 5.5%), which were stimulated by AAVr3.45-BIM under the high titer of IgG, were partitioned as apoptotic cells, confirming the superior aspects of the IgG-resistant AAVr3.45 vector as a potential therapeutic agent for cancer gene therapy trials.
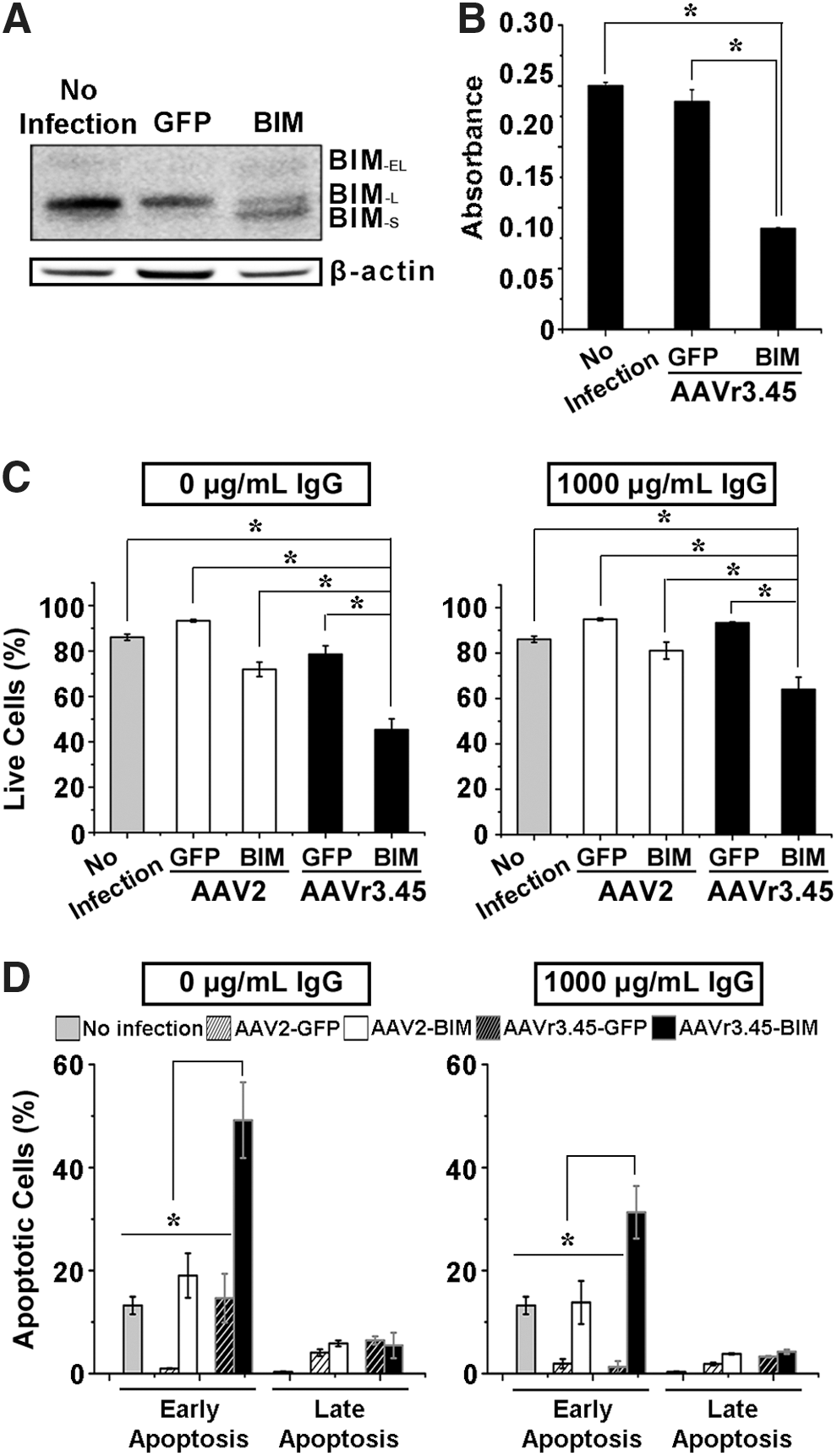
Apoptosis of MDA-MB-231 cells by AAVr3.45-BIM infection.
Conclusions
The structural features of a highly potential IgG-resistant AAV vector, AAVr3.45, compared to wild-type AAV2 were deeply interpreted, and its capability as a therapeutic agent that can be used for cancer gene therapy was evaluated in this study. It was confirmed that the two distinct structural deviations discovered on AAVr3.45 capsid, LATQVGQKTA displayed at the threefold spike of HSPG binding motif (i.e., aa 587 site) and the methionine replaced with valine at the aa 719 site, served their own functions, that is, they were key moieties for enhancing cellular transduction and for evading IgG neutralization, respectively. Thus, AAVr3.45 retains dual beneficial features as a therapeutic gene carrier for many gene therapy applications: it acts as (i) an enhancer for elevating gene delivery efficiency to therapeutically relevant cell types (e.g., stem cells or cancer cells) and (ii) a suppressor for reducing the probability of NAb inactivation, ultimately rendering AAVr3.45 highly versatile as a powerful gene carrier that can be extensively applied for many gene therapy applications, potentially for cancer gene therapy approaches.
Footnotes
Acknowledgments
This work was supported by Bio & Medical Technology Development Program (NRF-2013M3A9D3046431) funded by the Ministry of Science, ICT & Future Planning (MSIP), and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP; NRF-2015R1A2A2A03003553). This research was also supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grants HI14C1564 and HI16C1089).
Author Disclosure
The authors declare there are no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.