
Abstract
Retinitis pigmentosa type 43 (RP43) is a blinding disease caused by mutations in the gene for rod phosphodiesterase 6 alpha (PDE6A). The disease process begins with a dysfunction of rod photoreceptors, subsequently followed by a currently untreatable progressive degeneration of the entire outer retina. Aiming at a curative approach via PDE6A gene supplementation, a novel adeno-associated viral (AAV) vector was developed for expression of the human PDE6A cDNA under control of the human rhodopsin promotor (rAAV8.PDE6A). This study assessed the therapeutic efficacy of rAAV8.PDE6A in the Pde6a nmf363/nmf363-mutant mouse model of RP43. All mice included in this study were treated with sub-retinal injections of the vector at 2 weeks after birth. The therapeutic effect was monitored at 1 month and 6 months post injection. Biological function of the transgene was assessed in vivo by means of electroretinography. The degree of morphological rescue was investigated both in vivo using optical coherence tomography and ex vivo by immunohistological staining. It was found that the novel rAAV8.PDE6A vector resulted in a stable and efficient expression of PDE6A protein in rod photoreceptors of Pde6a nmf363/nmf363 mice following treatment at both the short- and long-term time points. The treatment led to a substantial morphological preservation of outer nuclear layer thickness, rod outer segment structure, and prolonged survival of cone photoreceptors for at least 6 months. Additionally, the ERG analysis confirmed a restoration of retinal function in a group of treated mice. Taken together, this study provides successful proof-of-concept for the cross-species efficacy of the rAAV8.PDE6A vector developed for use in human patients. Importantly, the data show stable expression and rescue effects for a prolonged period of time, raising hope for future translational studies based on this approach.
Introduction
R
Mutations in the rod-specific cyclic-guanosine monophosphate (cGMP) phosphodiesterase (PDE6) are the third common cause of autosomal recessive retinitis pigmentosa (arRP). In rod photoreceptors, PDE6 is the main cGMP-hydrolyzing enzyme regulating the intracellular cGMP concentration. 6,7 PDE6 is a heterotetrameric complex composed of two catalytic domains (PDE6A and PDE6B) and two inhibitory subunits (PDE6G). 8 –10 Mutations affecting PDE6 functionality have been found in 2–4% of patients with arRP 11 and were confirmed to lead to progressive rod degeneration. In areas where rod cells are completely lost, cones undergo degeneration secondary to rods, although they are not affected directly by the genetic defect (bystander effect). As human visual performance largely relies on cones, there is great variability in the subjective age of onset, progression, retinal appearance, and final visual outcome.
In human patients, inherited diseases often show a variable phenotype due to the wide range of disease-causing mutations. While murine knockout models commonly best match the situation in patients with complete loss of function, mice with specific point mutations may be better suited for the assessment of the corresponding human phenotypes. Regularly, differences in disease severity are linked to the available fraction of the protein translated from the affected gene. It was recently shown that a number of human disease–causing mutations in the PDE6A gene also lead to a degeneration of rod photoreceptors in respective murine models. However, the time course is more rapid, and degeneration takes place within the first weeks of life. 12 In addition, modest differences were found in the kinetics of rod degeneration between the Pde6a missense mutations investigated (R562W, D670G, and V685M). In particular, the Pde6a nmf363/nmf363 mouse line, carrying a point mutation in D670G amino acid substitution and thus for simplicity referred to in the remaining text as D670G, displayed a relatively mild phenotype. 12,13 It was possible to link these differences to the degree of remaining activity of PDE6.
Currently, no curative treatment for RP exists. In related retinal disorders, gene therapy has made tremendous progress, 14 and thus a promising role in this approach is in the preservation and possible restoration of visual function in RP as well. Proof-of-concept studies in small- and large-animal models of RP have already successfully demonstrated that gene supplementation of Pde6a using adeno-associated virus (AAV) vectors is a promising therapeutic approach for this disease group. 15 –17 In 2012, the RD-CURE Consortium consisting of clinicians and researchers of the University Eye Hospital Tübingen, the Ludwig-Maximilians-University Munich, and the Columbia University New York was initiated. The partners within RD‐CURE work on the different steps necessary for the clinical translation of gene therapy in retinal degenerations. This preclinical study was conducted within the framework of RD-CURE.
For use in clinical trials, a novel AAV vector was developed containing the human PDE6A cDNA (rAAV8.PDE6A). The present study set out to evaluate the trans-species efficacy of rAAV8.PDE6A on the basis of the Pde6a D670G preclinical mouse model. The study shows that the new vector was able to drive human PDE6A protein expression in murine rod photoreceptors, thereby improving the retinal phenotype of the D670G mouse model. This rescue effect of PDE6A gene supplementation persisted up to the end of the observation interval 6 months after injection.
Materials and Methods
Ethics statement
All procedures concerning animals adhered to the ARVO statement for the use of animals in ophthalmic and vision research and were reviewed and approved by a competent board appointed by the respective German government agency (Regierungspraesidium Tuebingen).
Animals
D670G mice (C57BL/6J-Pde6a nmf363/nmf363) carry an A to G missense mutation in exon 15 of the Pde6a gene, causing an amino acid change from aspartic acid to glycine (D670G) in the PDE6A protein. 13 They were obtained from the Neuroscience Mutagenesis Facility (NMF) at the Jackson Laboratory (Bar Harbor, ME). D670G and C57BL6/J wild-type control animals were housed under standard white cyclic lighting. They had free access to food and water, and were used irrespective of gender. For the gene therapeutic study, 16 Pde6a D670G mice were treated. A cohort of 12 was used for the short-term study, and a cohort of four was used for the long-term study.
Cloning and production of rAAV vectors
Standard cloning techniques were used for vector assembly. All sequence manipulations were verified by sequencing. The expression cassette consists of a 0.8 kb rod photoreceptor-specific human rhodopsin (hRHO) promoter 18 and the full-length (2.58 kb) human PDE6A cDNA, which was amplified from human retinal cDNA (primer forward 5′-AAAGCGGCCGCCACCATGGGCGAGGTGACAGCA G-3′ and reverse 5′-GGAGCGGCCGCTTACTGGATGCAGCAGGAC-3′). Additionally, the expression cassette contains a 0.54 kb woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) with mutated WXF-open reading frame 19 and a 0.2 kb bovine growth hormone polyadenylation signal (BGHpA). In a second step, the described expression cassette and the pGL2.0 backbone were assembled to the final pGL2.0-hRHO-hPDE6A-mWPRE-KanR plasmid using a NEBuilder® cloning kit (New England Biolabs, Ipswich, MA). The plasmid backbone contains randomized, synthetic DNA fragments (Eurofins Genomics, Huntsville, AL), the ITRs from the pSub201 cis-plasmid, 20 a kanamycin resistance gene (KanR), and the pUC18 (Clontech) origin of replication. Production of plasmid DNA and AAV vectors was performed according to standard manufacturing procedure by Aldevron (Fargo, ND) or previously described processes 21 at the Laboratoire de Thérapie Génique, UMR 1089 (Nantes, France), respectively. AAV serotype 2/8 was used for packaging of the viral genome.
Sub-retinal rAAV injections
Sub-retinal applications of the rAAV vector were conducted, as previously described. 22 Briefly, mice were anesthetized by subcutaneous injection of ketamine (66.7 mg/kg) and xylazine (11.7 mg/kg), and their pupils were dilated with tropicamide eye drops (Mydriaticum Stulln; Pharma Stulln GmbH, Stulln, Germany). Young mice (14 days old) received a single sub-retinal injection of 1 μL of rAAV particles (2.2 × 109 total vector genomes) at the dorsal part. The injection was performed free hand under a surgical microscope (Carl Zeiss, Oberkochen, Germany). Special care was taken to avoid damage of the lens. As previously published, 22,23 in vivo imaging was performed (described in next section) to validate the treatment area and bleb formation.
In vivo imaging techniques
Both confocal scanning laser ophthalmoscopy (cSLO) and spectral domain optical coherence tomography (sdOCT) were performed immediately after sub-retinal injections to aim for a quality control including the treatment area as well as the integrity of retinal morphology (e.g., the status of retinal degeneration). 24 –26 A second in vivo measurement in the same individual was conducted to determine the short-term treatment efficacy at 4 weeks post injection (PI). For the long-term study, an additional time point was conducted at 6 months PI. In cases of severe damage such as a full retinal detachment or lens opacity, mice would be excluded from further analysis. This was, however, not the case in the present study.
Imaging was performed by a Spectralis TM HRA + OCT (Heidelberg Engineering, Dossenheim, Germany), 24 –26 and the corresponding imaging software package Eye Explorer (HEYEX v5.3.3.0, Heidelberg, Germany). Scans were acquired at a speed of 40,000 scans per second and each two-dimensional B-scan containing up to 1536 A-scans. For analysis, the data were exported as 24 bit color image files and processed in CorelDraw X5 (Corel Corp., Ottawa, Canada). Fundus structures were analyzed using cSLO, as previously described. 27
Electroretinography
Electroretinography (ERG) was performed using a Ganzfeld bowl, a direct current amplifier, and a PC-based control and recording unit (Multiliner Vision; VIASYS Healthcare, Conshohocken, PA), as described previously. 28 Prior to ERG measurements, mice were dark-adapted overnight. On the day of examination, mice were anesthetized, as described above for sub-retinal injections, and the pupils were dilated with tropicamide eye drops (Mydriaticum Stulln; Pharma Stulln). Single flash series as well as flicker frequency series at 3.0 cd·s/m2 ISCEV standard flash intensity were obtained under dark-adapted (without background illumination, 0 cd/m2) and light-adapted conditions (with a background illumination of 30 cd/m2 turned on 10 min previous to the first recording).
Immunohistochemical analysis
Upon completion of the in vivo recordings, mice were sacrificed at 4 weeks PI (short-term experiments) or 6 months PI (long-term experiment). The eyes were enucleated, fixed in 4% paraformaldehyde, and processed for immunohistological analysis. Vertical cryo-sections were stained with a specific rabbit polyclonal antibody directed against the human PDE6A (1:500; Novus Biologicals, Cambridge, United Kingdom; cat. no.: #NBP1–87312), anti-cone arrestin (1:500), 29 or anti-cGMP (1:2,000; provided by Dr. Harry Steinbusch, Maastricht University, Maastricht, Netherlands). 30 The immunosignal was detected with a Cy3-tagged donkey anti-rabbit IgG secondary antibody. Cell nuclei were stained with Hoechst33342 or DAPI. Confocal images from the immunostained cryo-sections were collected using a Leica SP8 confocal laser scanning microscope or Zeiss Imager Apotome microscope. Statistical analysis was performed using GraphPad Prism software (GraphPad Software, La Jolla, CA). To compare two groups, an unpaired Student's t-test was applied. Unless otherwise stated, all values are given as the mean ± standard error of the mean.
Results
PDE6A and cGMP expression levels in the D670G retina
As differences in disease severity are commonly linked to the fraction of the protein available, first expression of PDE6A was assessed in wild-type retina as well as in the V685M and D670G mutants (Supplementary Fig. S1). In the healthy wild type, on postnatal day (P) 13, PDE6A was found to be strongly expressed in photoreceptor outer segments (Supplementary Fig. S1A). In contrast, in the V685M mutant, there was virtually no PDE6A expression (Supplementary Fig. S1B), while the D670G mutant showed some protein expression in what appeared to be relatively short outer segments (Supplementary Fig. S1C).
To evaluate if the fraction of PDE6A protein produced by mutants may be physiologically active, cGMP levels in the same animals were also assessed on P13. Because of the eminent function of PDE6 for photoreceptor cGMP hydrolysis, PDE6A expression is expected to be inversely correlated to photoreceptor cGMP accumulation. While in both wild-type (Supplementary Fig. S1D) and D670G-mutant retinas (Supplementary Fig. S1F) no or very few cGMP-positive photoreceptors were found, many photoreceptors in V685M retinas showed strong cGMP immunoreactivity (Supplementary Fig. S1E). This finding indicates, in line with other studies, 12,13 that the D670G mutation allows for a biologically relevant degree of residual PDE6 function, whereas no biologically relevant protein function was found in the V685M mutant.
Induction of transgene expression
To assess whether the rAAV.PDE6A treatment was successful, expression of the human transgene and preservation of photoreceptors were analyzed using immunohistochemistry at 4 weeks PI and 6 months PI (Fig. 1). As in previous works, 31,32 this study relied on the comparison of the treated eye (TE) to the contralateral untreated eye (UE). In the UE, only one to two rows of photoreceptors were remaining, and PDE6A expression was absent at 4 weeks PI and 6 months PI, as previously described (Fig. 1A). 12,16,17 Therefore, treated areas in the TE could be clearly identified by the presence of PDE6A transgene and more than two remaining photoreceptor rows. Immunolabeling with antibodies specific for PDE6A protein (green) clearly showed the presence of virally encoded human PDE6A protein in the treated but not in the untreated part of the retina. The mean gray value of fluorescence intensity was significantly higher in treated than in untreated areas at 4 weeks PI (38.83 ± 2.76 vs. 2.16 ± 0.24 units; p < 0.0001) and 6 months PI (29.38 ± 4.14 vs. 1.08 ± 0.17 units; p < 0.001). Furthermore, the availability of the missing PDE6A protein preserved retinal morphology in treated areas, whereas the degeneration continued to progress in untreated areas. Outer nuclear layer (ONL) thickness in treated parts was significantly more preserved than in untreated parts at 4 weeks PI (19.62 ± 1.83 μm vs. 6.73 ± 0.54 μm; p < 0.0001) and 6 months PI (21.53 ± 1.87 μm vs. 5.92 ± 0.48 μm; p < 0.001). At the end of the observation period 6 months PI, it was found that PDE6A expression had persisted in treated areas, and so did the protective effect on the retina (Fig. 1B). Importantly, a distinct preservation of transgene expression and retinal layers was observed up to 6 months PI. When comparing the time points 4 weeks PI and 6 months PI, no obvious difference in the measured parameters fluorescence intensity (38.83 ± 2.76 units vs. 29.38 ± 4.14 units; p = 0.10) or ONL thickness (19.62 ± 1.84 μm vs. 21.53 ± 1.81 μm; p = 0.58) could be observed.
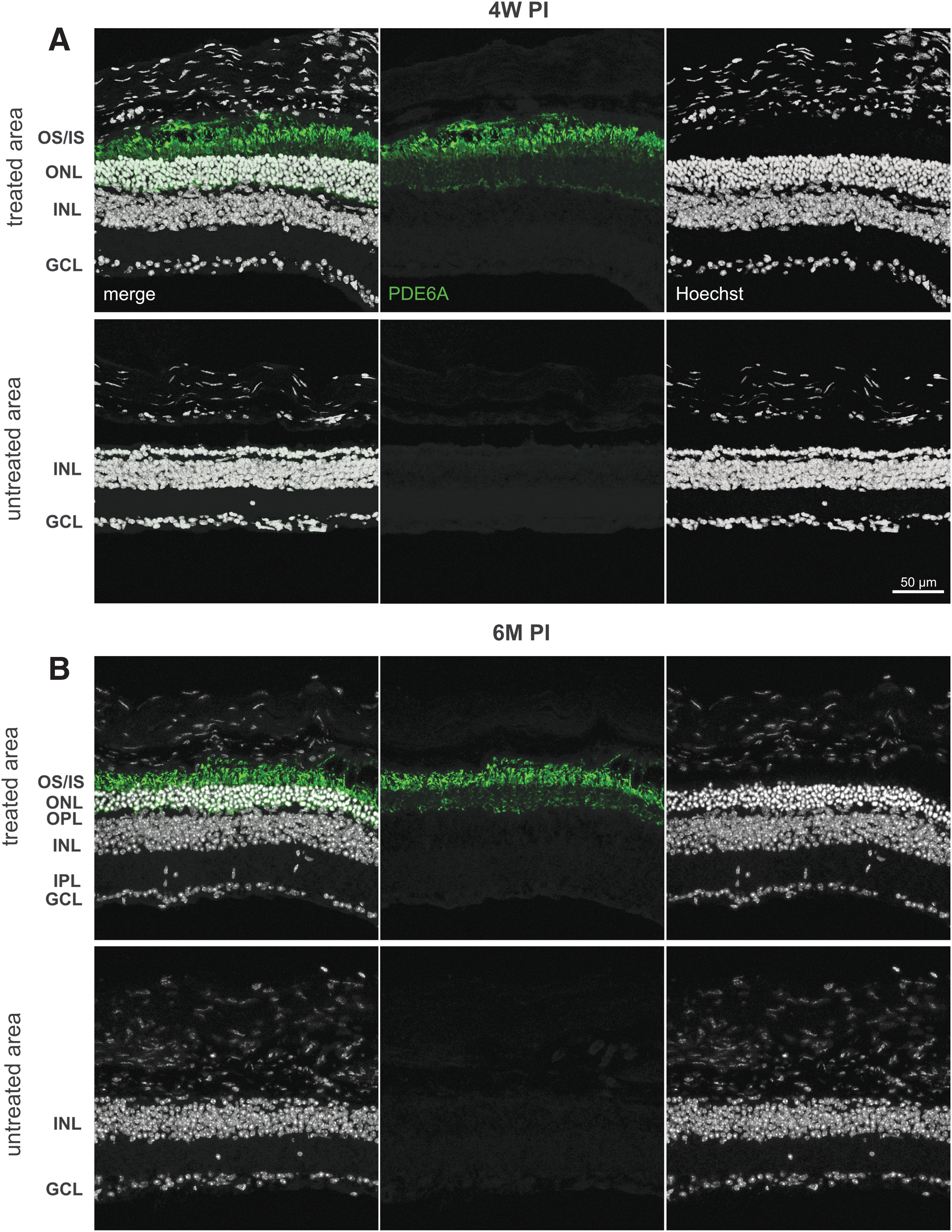
Preservation of photoreceptor integrity in D670G mice after rAAV8.PDE6A treatment: Histomorphological data. Immunohistochemical staining of representative treated and untreated retinal areas at 4 weeks
Preservation of outer retinal morphology
At 1 month of age, the retina of D670G mice is already considerably degenerated, marked by a very thin ONL with only a few photoreceptor rows (Fig. 2A, top). 12 However, a novel finding was that the apparent lack of the formation of regular outer segments also led to a detachment of the retina from the underlying retinal pigment epithelium (RPE; Fig. 2A, center and bottom, ventral part). This in vivo finding was confirmed by ex vivo data (see comparison Fig. 2B vs. 2C).
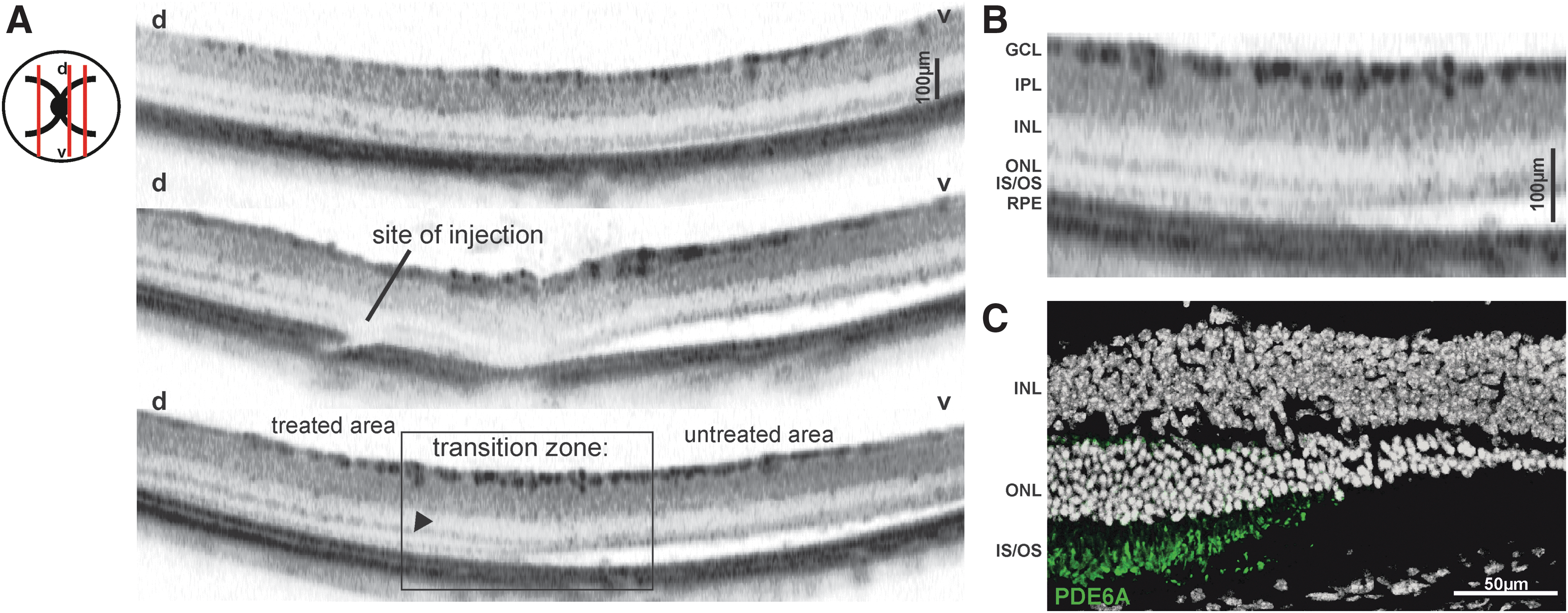
Preservation of photoreceptor integrity in D670G mice after rAAV8.PDE6A treatment: in vivo imaging. In vivo imaging of retinal layers with spectral domain optical coherence tomography at 4 weeks PI.
Strikingly, the supplementation of the PDE6A gene not only caused a preservation of the cells in the ONL, but also was able to prevent the detachment (Fig. 2A, center and bottom, dorsal part). An appreciation of the treatment effects was possible in the transition zone between treated and untreated area (Fig. 2A, bottom, magnification shown in Fig. 2B). A more detailed comparison to ex vivo immunohistological staining revealed that areas of rescue perfectly match with regions of PDE6A expression (Fig. 2C). These findings indicate that the treatment successfully prevented the development of the Pde6a-related disease phenotype, as in treated areas, less degeneration and no retinal detachment could be observed.
Restoration of retinal function
The high level of cGMP in practically all disease-causing Pde6a mutants 33 entirely prevents untreated rods from electrical signaling. Gene therapy may restore some rod functionality in treated areas, depending on the number and condition of surviving rods at the time of treatment, but the crucial outcome of therapy is rod cell survival, which is only loosely linked to rod-driven function, particularly in PDE gene mutants.
The situation is completely different for the cone system. While only rods are directly affected by the disease, cones nevertheless undergo a secondary degeneration in areas of rod loss. In all areas where rod cell survival is sufficient, cones function normally, so that cone cell survival is usually closely linked to cone-driven function.
In ERG analysis, the evaluation of treatment efficacy was based on the differences between ERG responses of TE and UE of the same individuals. A superior retinal function in TE is an indirect measure for the restoration of retinal function. As detailed above, D670G mice hold a severe deficiency in rod-mediated vision as a consequence of the defective phosphodiesterase (Supplementary Fig. S1), resulting in a lack of rod-driven components and a reduction in cone-driven ERG components (Fig. 3A, black ERG traces) in UE.
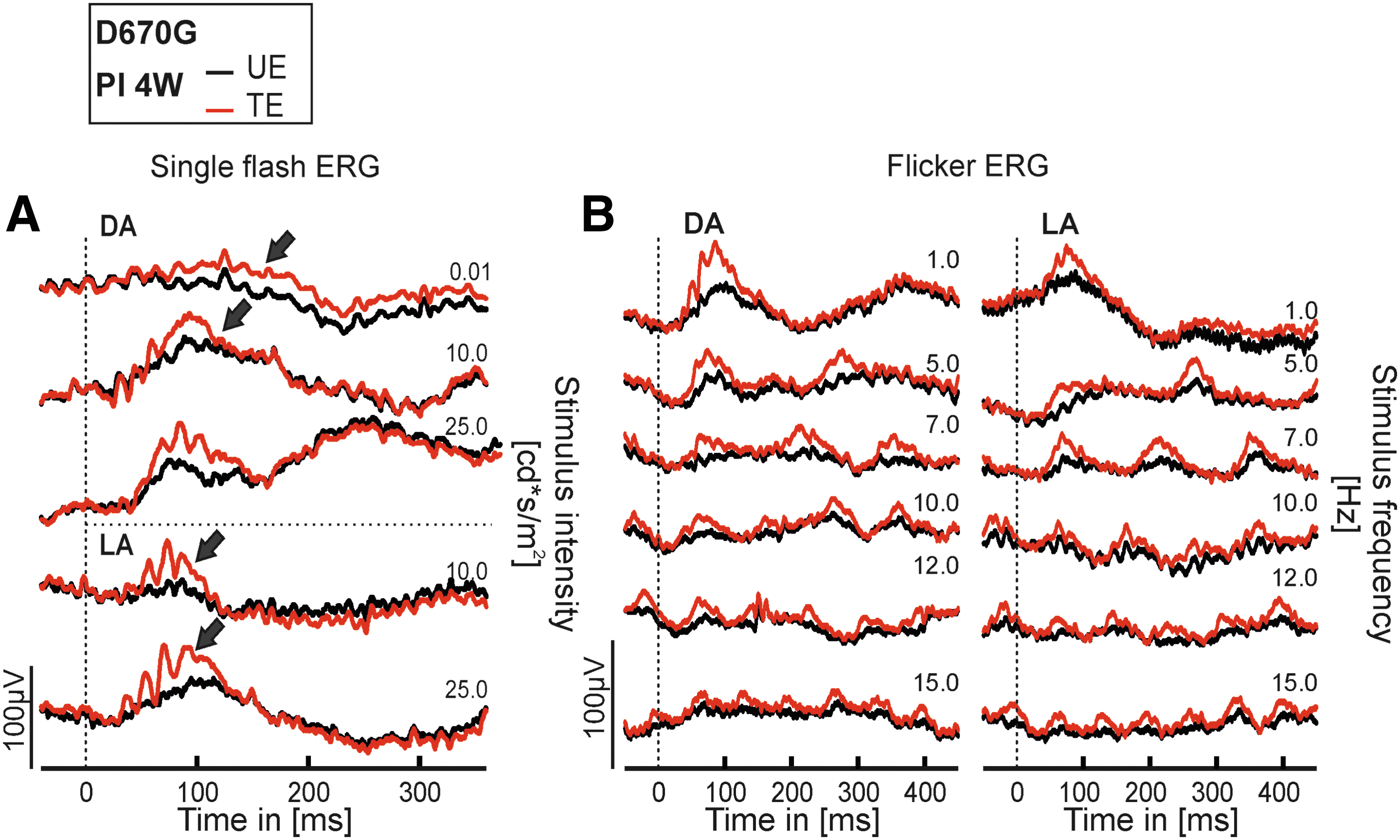
Preservation of photoreceptor integrity in D670G mice after rAAV8.PDE6A treatment: functional assessment. Electroretinographical (ERG) assessment of retinal function at 4 weeks PI. ERG data from treated eyes (red traces) and untreated eyes (black traces) of the same individual D670G-mutant mouse.
Generally, the responses shown in Fig. 3A and B are representative for treated mice. The TE of the Pde6a D670G mouse (red traces) were substantially larger than those from the UE (black traces), illustrating the functional benefit of the treatment. However, a direct effect of the treatment on rod system–driven ERG responses was not clearly detectable, neither in single flash responses at 0.01 cd·s/m2 (first trace) in Fig. 3A, nor in the flicker ERG recordings (Fig. 3B) using trains of flashes with an intensity of at 3.0 cd·s/m2 (ISCEV standard flash 34 ) in TE. Consequently, most of this outcome was due to an improvement of cone-dominated responses. The difference in dark-adapted (DA) and light-adapted (LA) flicker ERG records (Fig. 3B, left vs. right) would also apply to pure cone system–driven signals, which are larger on a dark background due to a better contrast sensitivity (Fig. 3B, left).
Durable enhancement of cone condition
The initial assessment of the rescue performance in treated D670G mice at 4 weeks PI clearly indicated a successful preservation of cone morphology and function. To assess the longevity of cone preservation, treated D670G mice were examined at 6 months PI, and the study specifically stained for a cone marker in histological sections. A representative treated and an untreated area from a retinal slice stained with cone arrestin (Arr3) and the nuclear dye Hoechst 33342 is shown in Fig. 4. While the natural course of photoreceptor degeneration in D670G mice results in thinning of the ONL to about one to two rows of photoreceptors already at 1 month of age, a healthy retina in comparison features 11–12 photoreceptor rows. 12,13 Notably, four to five rows of photoreceptors were retained in the treated area (Fig. 4A, left), indicating that the treatment led to significantly delayed photoreceptor cell loss in general and a specific long-term survival of cones in particular. In contrast, an almost complete loss of the ONL was found in the untreated region (Fig. 4A, right), and the remaining rudimentary Arr3 expression indicated both a loss of cone cells and a change in the structure of the remaining ones. In the treated regions, cone photoreceptor structures appeared relatively normal; merely a distribution of Arr3 down to the synapse region indicated that they were not entirely healthy. The decimated cones in the untreated areas had a very unnatural, almost spherical appearance, reminiscent of the “dormant cones” (Fig. 4B, right) described by Busskamp et al. 35
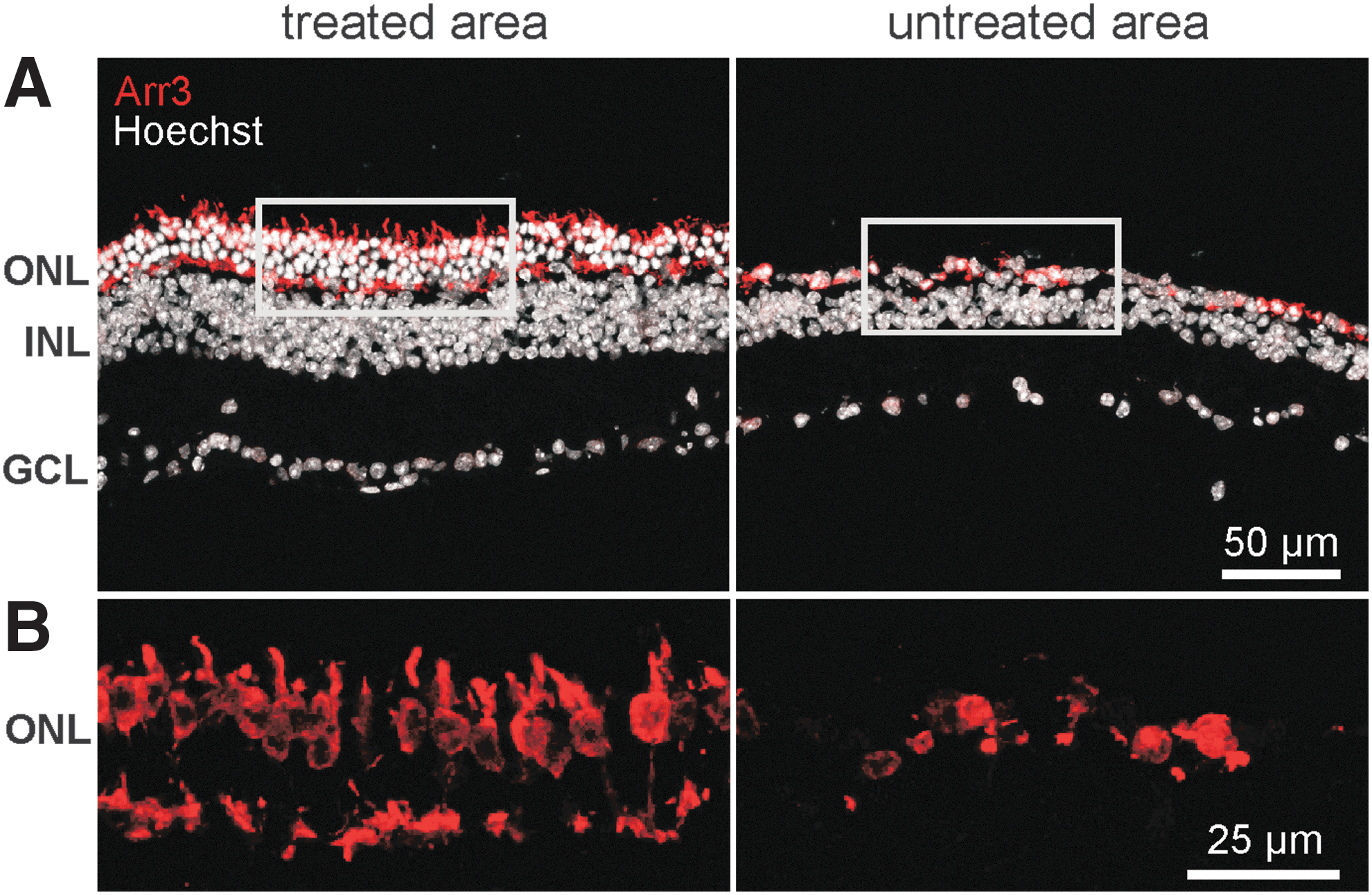
Specific histomorphological data on cone photoreceptor integrity in D670G mice after rAAV8.PDE6A treatment. Remaining cones at 6 months PI were marked with a specific antibody for cone arrestin (red).
Discussion
RP is a group of inherited blinding eye diseases that impose serious limitations upon the life of the affected individual and have profound implications for the society. As no curative treatment for RP is currently available, this study set out to implement gene replacement therapy in this group of disorders. In this work, the use of an AAV vector system for the treatment of autosomal-recessive human RP type 43 (RP43) in a respective preclinical model is described.
RP is primarily a rod disease, meaning that rod photoreceptors are directly affected by the genetic defect, while cone photoreceptors, which are most important for everyday vision, die secondarily to rods. In line with this, RP43 is caused by mutations in the gene for rod phosphodiesterase 6 alpha (PDE6A; OMIM number 613810).
It was found that differences in the severity of the human phenotype caused by disease-related mutations in the PDE6A gene corresponded well to differences in the kinetics of rod degeneration between respective murine models, 12 while the time course in the latter is in general more rapid, and the degeneration takes place within the first weeks of life. Nevertheless, it is highly probable that the phenotypic variability found among human point mutations is, like in the preclinical models, linked to the fraction of PDE6 available, which in turn governs the level of cellular cGMP and thus the operation of the cyclic nucleotide-gated (CNG) channels in phototransduction. Because for the most part they share a common pathophysiology, it is believed that murine preclinical models are very useful in the assessment of gene therapy, even if primarily designed for human patients. In this study, the trans-species approach relies on the application of a novel AAV vector for expression of the human PDE6A cDNA under control of the human rhodopsin promotor (rAAV8.PDE6A) in D670G animals.
For the assessment of therapeutic efficacy, the D670G mutant was chosen, as it mirrors a small but distinct degree of remaining PDE functionality that is most likely present in many human point mutations. In addition, the relatively mild time course of the degeneration in comparison to lines carrying more severe mutations or even the knockout situation provides for a sufficiently large window of therapeutic opportunity before cells are lost.
All mice included in this study were treated with sub-retinal injections of the vector at 2 weeks after birth. The therapeutic effect was monitored at 4 weeks PI and at 6 months PI. These time points were chosen on the basis of previous work on preclinical gene therapy and the natural course of PDE6A disease. Four weeks PI was found to be a very useful initial time point to assess the best performance of AAV-based gene therapy in mice. 31,32 Six months PI was chosen as the long-term time point due to the rather rapid retinal degeneration in PDE6 mutants 12,36 in comparison to many other disease models, where longer observation periods are usually more appropriate.
At each time point, the effect of the therapeutic transgene on retinal morphology and function was examined in vivo, and at the end of the study also histologically ex vivo. In vivo, biological function was assessed by means of ERG, while the degree of a potential morphological rescue was investigated with OCT. Ex vivo, both topography and level of expression of the PDE6A protein was investigated in immunohistological staining. The latter examination revealed a generally stable and efficient expression of PDE6A protein in rod photoreceptors of D670G mice following treatment at both at 4 weeks PI and 6 months PI, accompanied by a substantial morphological preservation of ONL thickness and rod outer segment structure. The structural preservation of photoreceptors expressing the transgene is an additional valid argument for a correct function of the PDE6A protein.
Gene therapy for channelopathies (Cnga3 and Cngb1) in a mouse model shows that about 30% of the full retina is transduced by one single injection, which was sufficient to restore respective vision-guided behavior. 31,32 Notably, a sharp boundary was found in the transition zone between the treated and untreated areas (Fig. 2), which is most likely due to the limitation of AAVs to spread beyond the sub-retinal bleb. These experiments clearly show that local treatment is sufficient for restoration of function.
For human translation, this treatment strategy offers sufficient therapeutic efficacy. However, in terms of topographical differences of photoreceptor distribution as well as the size of the affected area, an alternative treatment strategy might be to perform several sub-retinal injections to target rod photoreceptors specifically surrounding the fovea to rescue cone photoreceptors from secondary degeneration. This approach is currently being tested in larger animals and proposed for human clinical trials, but because of the small eye size it cannot be applied to mouse studies.
A novel and unexpected finding was that while initially the retina in the treated regions became briefly detached due to the sub-retinal injection, this reversed at 4 weeks PI. In contrast, the untreated regions, attached at the time point of injection, became detached at 4 weeks PI. The hypothesis is that the ongoing, rapid degeneration in Pde6a mutants leads to a malformation or even lack of regular outer segments, so that the retina becomes less tethered to the RPE and lifts off in the untreated region. Remarkably, this detachment was entirely absent in the treated areas 4 weeks PI, as marked by PDE6A expression. It is thus reasonable to assume that the improvement of photoreceptor structure by the therapy furthermore led to an enhanced condition of the retina–RPE interface.
Importantly, this led to a prolonged survival of cone photoreceptors for at least 6 months due to an amelioration of the bystander effect linked to rod cell death. In line with these findings, the ERG analysis confirmed a substantial restoration of cone function. The rescue of rod photoreceptors is due to a reduction of intracellular cGMP levels in terms of prolonged survival. However, these levels still prevent the rod system from generating ERG signals (see also Sothilingam et al. 12 ). In this study, a large degree of rescue is possible without any direct evidence for a functional rescue of the rod system in ERG outcome. In human patients, this might be different, as the protracted time course compared to mice suggests generally lower cGMP levels in diseased rod cells. Thus, the data suggest that the immunohistological outcome is a suitable biomarker for preclinical evaluation of PDE6A gene supplementation in mice.
In summary, this work highlights the value of preclinical gene therapy in Pde6a mutants and its relevance for subsequent human trials. In particular, rAAV8.PDE6A successfully rescued function and morphology in D670G-mutant mice, and the data indicate stable expression and rescue effects for at least 6 months after treatment. Importantly, the rescue of rod photoreceptors also led to extended survival of cone photoreceptors. The rescue of cone photoreceptors is of special relevance for the most important human visual functions such as visual acuity and color vision, raising hope for future translational studies based on this novel AAV vector.
Footnotes
Acknowledgments
We thank Kerstin Skokann, Gudrun Utz, and Marina Wolf for excellent technical help and Wolfgang Baehr (University of Utah) for the gift of the anti-cone arrestin antibody. This work was supported by the Tistou & Charlotte Kerstan Stiftung.
Author Disclosure
C.S., M.B., S.M., and M.W.S. are part of a patent application related to this publication. No competing financial interests exist for the remaining authors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.