
Abstract
Clinical gene therapy has made important advances over the last decade. Among neurological diseases, severe genetic neurodegenerative conditions have been the focus of initial clinical applications. Gene therapy has also addressed complex neurodegenerative diseases, particularly Parkinson's disease, with encouraging results in human patients, demonstrating that specific targeting of central nervous system (CNS) cells is a relevant strategy for severe pathologies and that efficient access to the CNS with viral vectors is an achievable goal. The purpose of this review is to summarize the gene therapy clinical applications that have been conducted for neurodegenerative diseases. Limitations and hurdles to obtain and demonstrate benefit in patients, and the new developments that should allow new clinical applications with high beneficial potential are discussed.
Introduction
G
Gene therapy has made important advances over the last decade. After initial drawbacks and disappointing results, as commonly observed during the development of new medical approaches, breakthroughs in clinical gene therapy trials have emerged in a variety of monogenic diseases, including blood disorders (primary immunodeficiencies, hemoglobinopathies, and hemophilia), ocular diseases, cancer, and neurological diseases.
Gene therapy is particularly attractive for central nervous system (CNS) diseases. However, it raises specific limitations and hurdles, and neurological disorders are among the most difficult pathologies to cure.
First, the brain is a complex organ system in which disease processes induce a large spectrum of pathological states that can have broad consequences on neural development, plasticity, and function, as well as on metabolism. The complexity of the pathophysiology of neurodegenerative diseases and of cell interactions is often imperfectly understood, limiting the development of therapeutic approaches.
Second, brain access is limited, protected by physical barriers such as the blood–brain barrier (BBB) that prevents the delivery of therapeutic agents to the CNS and particularly the use of systemic treatments. These physical constraints and the lack of specificity of pharmacological approaches explain why most drugs and neurosurgical procedures have not proven effective in the treatment of CNS disorders.
Third, because of its complexity and limited access, not only are therapeutic targets difficult to identify and therapeutic agents uneasy to deliver, but also evaluation of clinical outcome after treatment is a complex issue. Evolution of the neurodegenerative process is not a linear process, and a well-characterized natural history of the disease is crucial to understand the clinical outcome under treatment. In that regard, the design of clinical trials and the availability of surrogate markers and sophisticated technologies such as brain imaging (magnetic resonance imaging [MRI], diffusion tensor imaging [DTI], or positron emission tomography [PET]) play a major role, particularly in Phase I/II initial proof-of-concept studies involving a limited number of patients.
Among brain diseases that have led to clinical trials, severe genetic neurodegenerative conditions have been the focus of numerous studies due to their devastating consequences, the absence of pharmacological treatment, and the rationale to propose gene delivery to restore missing function and stop neurodegeneration. Beside monogenic disorders, gene therapy has also addressed complex neurodegenerative diseases, particularly Parkinson's disease (PD), with encouraging results, demonstrating that specific targeting of CNS cells is a relevant strategy for severe neurological conditions and that access to the CNS is no longer the Holy Grail.
This review aims to summarize the gene therapy clinical applications that have been conducted so far for neurodegenerative diseases and to highlight new developments that should rapidly allow new clinical applications with high beneficial potential. It also considers limitations and hurdles that are still faced to obtain and demonstrate significant benefit in patients and how these can and will be overcome.
Viral Vectors for CNS Gene Therapy
Adeno-associated virus (AAVs) and lentiviral vectors (LVs) have emerged as the vectors of choice for the gene therapy of neurodegenerative diseases.
AAVs
AAVs are 4.7 kb non-enveloped single-stranded DNA parvoviruses. Recombinant AAVs (rAAVs) are the most commonly used gene therapy vectors for the CNS because of their safety, nonpathogenic nature, and ability to transduce neurons in vivo efficiently. AAV tropism is serotype dependent, depending on interaction with specific receptors on target cells. 1 Serotypes 1, 2, 5, 8, 9, and rhesus (rh).10 have been the most studied for CNS applications. 2 –5 Their specific effectiveness depends on the brain region, the target cell type, and the species. This underlines the importance of preclinical work in large-animal models such as dogs, cats, primates, and pigs to design and optimize therapeutic protocols for human patients. 6 rAAVs have demonstrated long-term gene expression in vivo in primates and also in human patients, with no evidence of neuroinflammation. 7
AAVs infect cells by interacting with specific receptors that differ between serotypes. For this reason, AAV tropism is serotype dependent. 1 AAVs can trigger innate or adaptive immune responses against the vector and the transgene. 8,9 In humans, preexisting immunity is most prevalent against AAV serotypes 2 and 5. The significant lower prevalence of serotypes such as AAVrh.10 supports their use in clinical trials. 10,11 The intensity of the immune response depends on the targeted tissue and the dose of vector that is delivered. Intraparenchymal injection of limited doses of vectors is likely limited compared to high vector dose and systemic delivery. 12
Certain serotypes of AAV also have the potential ability to cross the BBB and enter the CNS, 13 –15 making AAV vectors attractive tools for the therapy of neurodegenerative diseases. Preclinical proofs of concept has been made, particularly with AAV9, 15 –17 allowing their possible use for clinical application.
The greatest limitation of AAV-based vectors remains their large-scale production, a major issue toward the development of AAV vectors for systemic administration in human patients. Important progress has been made in the development of alternative methods using a cell suspension system toward large-scale good manufacturing practice compliance, allowing for an approved gene therapy product. 18 –20
LVs
LVs are enveloped single-stranded RNA viruses. Lentivectors have been developed from primate lentiviruses such as the human immunodeficiency virus type 1 (HIV-1) and non-primate lentiviruses, in particular the equine infectious anemia virus (EIAV). 21 –24 These vectors, pseudotyped with the envelope of the vesicular stomatitis virus G (VSVG), have a broad cell tropism, including neuronal and glial cells, 25 and LVs are used for in vivo CNS gene therapy. Because lentiviruses enter through the nucleus of the cell through a nuclear pore and do not need cell division and nuclear membrane disruption, they can efficiently infect quiescent cells. 26,27
The ability of LVs to integrate into the host's genome, leading to stable transgene expression DNA, is useful for ex vivo gene therapy applications. Stem cells, particularly hematopoietic stem cells (HSCs), can be stably transduced using lentivectors, allowing for potentially permanent, indefinitely persisting expression within the host cell, despite repeated cell division. 28 However, integration occurs at random sites, and these multiple integrations can promote insertional mutagenesis, as previously observed with retroviruses. 29,30 This risk has to be considered for both ex vivo and in vivo gene therapy. Even the long-term follow-up of gene therapy trials has not shown any insertional mutagenesis associated adverse event. 31 –33
Gene Delivery Methods for CNS Gene Therapy
CNS gene therapy relies on two types of approaches that have been repeatedly used to treat CNS diseases in a number of severe conditions in human patients (Fig. 1 and Table 1).
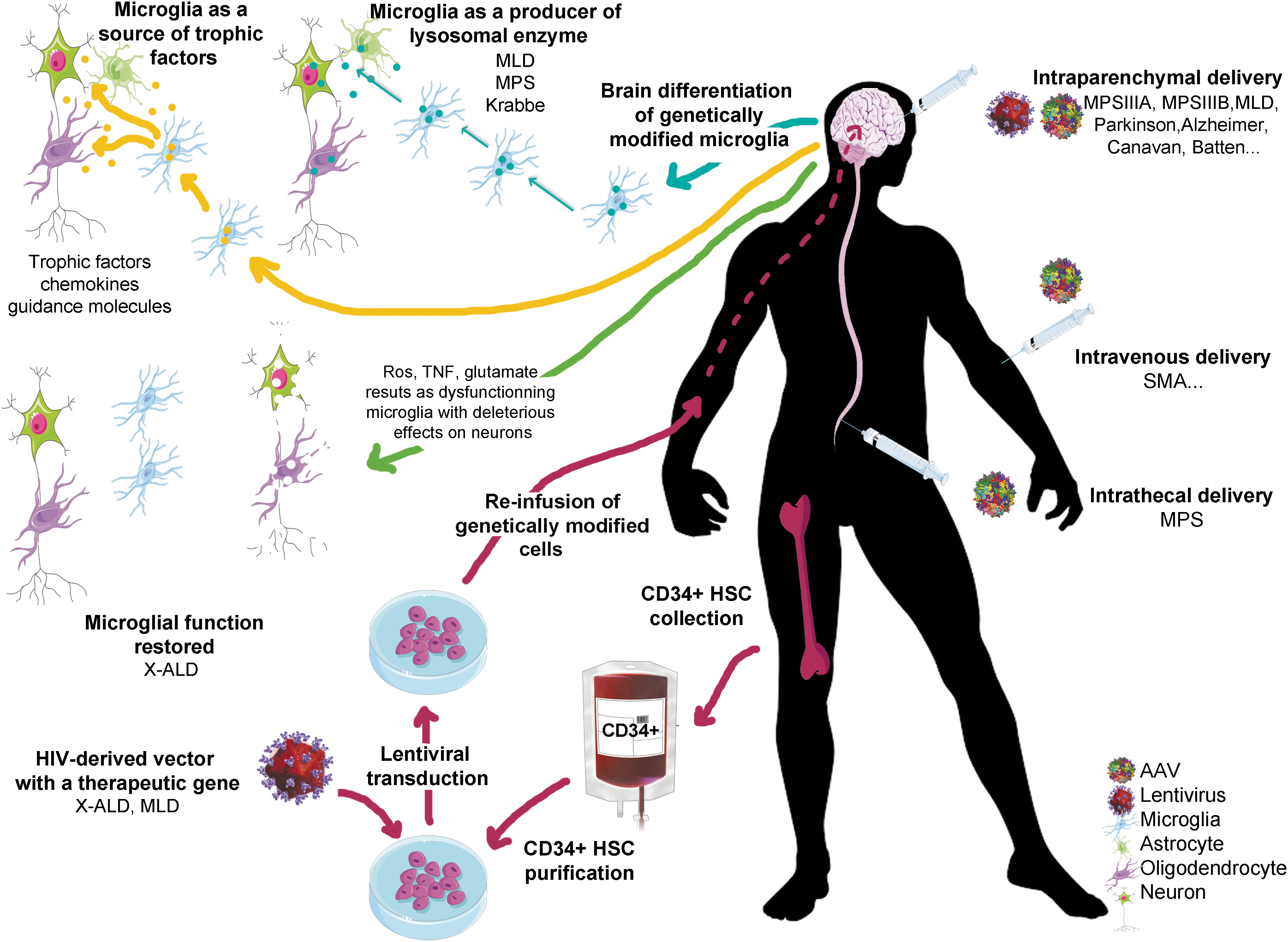
In vivo and ex vivo gene therapy strategies for the treatment of central nervous system (CNS) diseases. In vivo gene therapy (right) involves the direct infusion of vectors (lentiviral vectors or adeno-associated virus vectors) encoding the therapeutic gene. Three ways of administration are presented: direct intraparenchymal delivery, intravenous delivery, and intrathecal delivery. On the left side, ex vivo gene therapy is based on the transplantation of genetically modified hematopoietic stem cells (HSCs) to treat CNS diseases. After harvesting hematopoietic cells from the bone marrow of the patient, the CD34+ cell fraction containing HSCs is purified and transduced with a human immunodeficiency virus–derived vector carrying the therapeutic gene. Genetically modified cells are reinfused into the patient and reconstitute the bone-marrow stem-cell compartment. A fraction of stem cells or myeloid progenitors is able to migrate to the CNS, cross the blood–brain barrier, and locally differentiate into genetically modified microglia-like cells. Microglia can be genetically corrected (as in X linked-adrenoleukodystrophy) to restore normal function, leading to beneficial consequences for neurons. In the case of lysosomal diseases, microglia is been used as a source of secreted enzyme to correct neighboring deficient cells. Microglia could also be used to secrete therapeutic factor such as trophic factors or chemokines. Ros, reactive oxygen species; TNF, tumor necrosis factor.
List of clinical trials
The CAG promoter designation includes both the CBA and CB promoter.
MPS, mucopolysaccharidosis; MLD, metachromatic leukodystrophy; ALD, X-linked adrenoleukodystrophy; SMA, spinal muscular atrophy; IP, intraparenchymal; IV, intravenous; IT, intrathecal; O, ongoing; NR, not recruiting; C, completed; VG, vector genome.
In vivo
delivery of recombinant viral vectors
This the first and the most frequently developed strategy (Table 1). Therapeutic vectors are usually delivered locally, in specific parenchymal brain regions, through neurosurgical stereotactical injections. This is particularly adapted to diseases involving specific brain regions (such as PD 34 and Huntington's disease [HD] 35 ) or to lysosomal storage diseases (LSDs) in which the therapeutic protein is secreted and can be recaptured by other cells (cross-correction), allowing for diffusion of the therapeutic protein. 6 The capacity of certain AAV serotypes to be transported to area distal from the injection sites may also increase the therapeutic benefit. 36,37
It is likely that intraparenchymal delivery is better immunotolerated than intrathecal or Intravenous (IV) injection, since brain tissue is relatively immunoprivileged, and intraparenchymal delivery requires administration of low doses of vector. 8,38,39
More recently, new in vivo protocols have emerged using intrathecal administrations that have demonstrated efficiency in preclinical studies. A study for Batten disease is ongoing based on AAV9-CLN6 delivery (clinical trial NTC02725580; Table 1). Systemic injection, theoretically the easiest and safest way to inject therapeutic vectors, was previously limited by the incapacity of AAV vectors to cross the BBB. Two clinical trials are ongoing using AAV9 for mucopolysaccharidosis type III (MPSIIIA) and for spinal muscular atrophy (SMA; Table 1). Even though IV delivery of rAAV vectors will likely be associated with complex issues related to immunity, off-target expression, and technical limitations (high amount of vectors to be administered), this is an important step in the field of CNS gene therapy applications.
Ex vivo gene therapy for CNS diseases
This approach relies on ex vivo genetic manipulation of HSCs purified from the blood or the bone marrow with LVs, followed by autologous transplantation of transduced cells. 40 –44 This leads to the renewal of microglia compartment expressing the transgenic therapeutic proteins after hematopoietic reconstitution. Such strategy is well designed to treat neurological disorders in which microglia defect is the primary lesion, such as X-linked adrenoleukodystrophy (X-ALD). 31,45 This is also an efficient way to deliver secreted proteins into the brain, thus using microglia as a cell factory to produce and broadly deliver therapeutic proteins. 32,41
Recent Achievements and Ongoing Clinical Programs
In the past years, several clinical trials have been launched for neurodegenerative diseases. These clinical trials are based—as discussed above—on ex vivo gene therapy relying on the capacity of hematopoietic progenitors to cross the BBB, or on direct viral delivery, mostly through intraparenchymal and more recently through intrathecal or IV delivery.
In vivo delivery
LSDs
LSDs are encompassing more than 50 individual inherited metabolic diseases that result from the deficiency of a lysosomal function leading to the progressive storage of undigested or partially digested materials and consequently cellular and metabolic dysfunction. 46 More than 70% of LSDs have a CNS component, mostly characterized by neurodegeneration of more than one brain region and widespread neuroinflammation. 47 LSDs are for several aspects the ideal candidates for gene therapy approaches. Most LSDs are autosomal recessive monogenic disorders, and the molecular basis underlying the protein deficiency is well understood. Due to their severity, LSDs are good candidates for such approaches based on the benefit/risk ratio, and several proofs of concept have been established, not only in mouse models but also in large animals. Lysosomal enzymes have the capacity to be secreted, and thanks to their manose-6-phosphate residue, they can be taken up distantly from the cell that produced them and can be functional in the lysosome. The gene therapy approach is of particular interest because corrected cells may express supraphysiologic levels of enzyme that can be a source of newly synthetized enzyme to correct cells distantly and especially cells that are poorly transduced by viral vectors. 48,49 Interestingly, lysosomal enzymes may be transported through the cerebrospinal fluid (CSF) within the ventricles or via axonal transport. 36
Twenty-eight clinical trials have been launched, based on local delivery of AAV vectors for LSD (Table 1). Most (19) are based on intraparenchymal injection. More recently, one was started based on intrathecal (Batten disease) and one on IV injection (MPSIIIA).
Metachromatic leukodystrophy
The most severe form of metachromatic leukodystrophy (MLD) is the late infantile form in which first signs usually appear in the second year of life and patients die before the age of 7 years. 50 These patients have a deficiency in arylsulfatase A (ARSA), leading to accumulation of sulfatides (a main component of the myelin sheath), and demyelination of the CNS and peripheral nervous system and, consequently, severe motor and cognitive defects. 50 Preclinical studies demonstrated that intracerebral delivery of AAV5- and AAVrh10-ARSA efficiently and stably corrected an ARSA-deficient mouse model. 48,51,52 Large-animal studies demonstrated wide brain diffuse and the possibility of detecting ARSA not only in neurons but also in oligodendrocytes. 39,53 A Phase I/II clinical study (clinical trial NCT01801709; Table 1) to assess both the safety and efficiency of ARSA gene transfer in the brain of children affected with early-onset forms of MLD is still ongoing in France. This study is based on the intracerebral administration of AAVrh.10-CAG-hARSA in 12 sites in the white matter of both brain hemispheres with simultaneous delivery (through imaging guided tracks with two deposits per tracks), as in a nonhuman primate (NHP) study. 39 The first two patients received a low dose (1 × 10e12 vg), while the following patients received a higher dose of 4 × 10e12 vg. No results are available yet.
MPSIIIA
Sanfilippo syndrome type A, also known as MPS type IIIA, is caused by an autosomal recessive defect in the gene encoding a lysosomal heparin-N-sulfamidase: the N-sulfoglycosamine sulfohydrolase (SGSH). While the SGSH enzyme is ubiquitously expressed in tissues, the related clinical manifestations of its deficiency are mainly neurological. Early symptoms are usually observed during the first 2 years of life, with a progressive deterioration of cognitive and motor abilities in the first decade and death occurring in the second decade. Affected children require specific care in early childhood and progressively develop profound mental retardation with minimal somatic manifestations. 54,55 Two clinical trials have been launched in the past years for this disease.
The first study led by Lysogene (FR0013233475—LYS; Table 1) was a non-comparative, open-label, Phase I/II clinical study (clinical trial NCT02053064), with the intracerebral delivery of an AAVrh10 encoding both the SGSH and the SUMF1 cDNAs. This approach is based on the observation that direct-to-brain administration is best for such diseases because of the profound CNS component and presence of the BBB limiting the passage of therapies administered via peripheral routes. Four patients with MPSIIIA aged 6 years and 7 months, 6 years and 5 months, 5 years and 10 months, and 2 years and 8 months received treatment in 2011 and 2012. The study shows that intracerebral administration of AAVrh.10 carrying human SGSH and SUMF1 cDNAs was safe and well tolerated. The neurosurgery itself was uneventful. Safety data collected show good tolerance, a lack of adverse events related to the injected product, no increase of infectious events, and no biological sign of toxicity related to the investigational therapy. At 1 year analysis, cognitive benefit was suggested by neurocognitive evaluation in the child who received the vector at the age of 32 months, while the benefit on neurocognitive evaluations was more limited in the three older patients. All patients showed improvement in behavioral disorders, hyperactivity, and sleep disorders. 56 A 5-year follow-up study of the four children treated is ongoing to pursue the evaluation of the safety and tolerability data. Safety data continue to demonstrate no treatment-related adverse events. Lysogene is currently planning a pivotal trial in 2018 in the United States and Europe with a second-generation more potent gene therapy technology. The dose and the route of administration have been improved, in particular by replacing the PGK promoter with a more robust CAG promoter. The refined construct has shown to have an increase in mean enzyme expression of approximately 75% and is therefore expected to yield improved efficacy.
An open-label, dose escalation Phase I/II gene transfer was initiated by Abeona Therapeutics (clinical trial NCT02716246) based on the IV delivery of a scAAV9.U1A-hSGSH (Table 1). Four patients were included (three low doses: 5 × 10e12 vg/kg; one high dose: 1 × 10e13 vg/kg), with ages ranging from 2.5 to 6.9 years. Enrollment in the high-dose group is still ongoing and should encompass nine patients in total. Immunosuppressive therapy using prednisolone was administered from day 1 through to at least day 60. A sustained decrease of liver and spleen volume has been observed in two patients evaluated at 6 months, and a decrease of urinary and CSF accumulation of heparan sulfate at day 30. The 6-month post-treatment evaluation of the first two patients showed some improved cognitive capacities, with stabilization of scores in several Muller subdomains and on the Vineland test, as well as improved ability to complete items on the Leiter-R nonverbal IQ assessment (oral communication ASGCT 2017, abstract 292). These preliminary data suggest biological activity in the liver, spleen, and CNS. A 12-month follow-up is planned.
MPSIIIB
San Fillippo type B syndrome is a LSD that causes progressive deterioration of cognitive abilities after 2–4 years and leads to the death of the patient. MPSIIIB is caused by the accumulation of partially degraded heparin sulfate oligosaccharides consecutive to the deficiency of alpha-N-acetylglucosaminidase (NAGLU). 57 Two clinical trials have been launched for this disease.
The first was an open-label, single-arm, monocentric Phase I/II clinical study (clinical trial ISRCTN19853672) based on the intracerebral delivery of an AAV5 encoding the NAGLU cDNAs. Included patients were aged between 18 months and 4 years, and exclusion criteria were severe brain atrophy, loss of independent walking, and any medication designed to modify the natural course of the disease during the 6 months prior to treatment. A dose of 4 × 10e12 vg/patient at 16 deposits (white matter of both cerebral and cerebellar hemispheres) was injected during a 2 h delivery period, and immunosuppressive therapy was associated based on tacrolimus and mycophenolate mofetil. Importantly, the procedure was well tolerated in all patients. During the 24-month follow-up after intracerebral delivery of AAV5 encoding NAGLU, clear cognitive benefits were noted, despite differences between the treated children. 58
An open-label, dose-escalation, Phase I/II gene transfer was initiated by Abeona Therapeutics based on the IV delivery of a scAAV9-CMV-NAGLU (Table 1) but without any other details.
Canavan disease
Canavan disease is a childhood genetic leukodystrophy in which the enzyme aspartoacyclose (ASPA) is defective. This deficiency leads to an increase of its substrate, N-acetyl aspartate (NAA), in the CNS, resulting in impairment of normal myelination and spongiform degeneration of the brain. The clinical trial enrolled 21 patients. AAV2/ASPA was injected into six sites with a volume of 150 μL, which represents 1 × 10e9 vg at a rate of 2 μL/min using a borosilicate catheter linked to Hamilton syringes. 59 The first available data correspond to the study of immune tolerance and the evidence of minimal systemic signs of inflammation and immune response. 8 After a 10-year follow-up, brain NAA concentrations, which were the primary outcome measure, showed a decrease after treatment and were closed to NAA concentrations in the non-Canavan reference range. 60 Together with these molecular findings, evidence of a regional effect of the gene therapy has been showed by MRI with normalization of T1 in the splenium of the corpus callosum, suggesting increased myelination and/or decreased water content, as well as stabilization of brain atrophy, although variability among subjects in the timing and rate of change of pretreatment disease progression complicated the analysis. One notable clinical feature was the relative decrease in seizure frequency in patients who received gene therapy, 60 which was also reported in the tremor rat model of Canavan disease after AAV2-ASPA gene therapy. 61
Batten disease
Late infantile neuronal ceroid lipofuscinosis (LINCL; also known as Batten disease) is caused by a deficiency in tripeptidyl peptidase I (TPP-1), a proteolytic enzyme encoded by the CNL2 gene and more rarely to the CLN6 protein, which encodes a transmembrane protein of the endoplasmic reticulum. 62 Direct stereotactical injections of AAV2 or AAV5 vectors carrying a human CLN2 transgene into the mouse model of the disease resulted in significant improvement of the disease course. 63 After AAV2 vector injection, long-term expression of TPP-1 activity was achieved in rats and African green monkeys. 64 To mediate better levels and spread of TPP-1 protein activity, efficiency of AAVrh.10 vector encoding the CLN2 gene was performed. Administration at postnatal day 2 led to a remarkably persistent widespread expression concomitant with improvement of a number of CNS function tests and significant survival advantage compared to administration at 3 or 7 weeks. 65 Ten patients with LINCL have been enrolled in a Phase I/II clinical trial (clinical trial NCT00151216). Patients received a total amount of 2.5 × 10e12 vg divided into 12 locations (six sites and two deposits per site). 66 One patient died 49 days post surgery after an epileptic seizure on day 14 but no evidence of CNS inflammation. At the 6-month follow-up, treated patients showed a slower decline assessed with the modified Hamburg neurological rating scale, but unfortunately disease progression could not be blocked. 67 Due to the significant superiority of AAVrh.10 vector in the mouse model, a study of the efficiency and tolerance in NHPs was performed, and a clinical trial (clinical trial NCT01161576 and 01414985) with AAVrh.10 vector encoding CLN2 started in 2011. The first group of six patients received 9 × 10e11 vg of AAVrh10-CAAG-hCLN2 vector, and the second group of 10 patients received 2.85 × 10e11 vg. No results are yet available.
More recently, a Phase I/II trial (clinical trial NCT02725580) was launched based on the intrathecal delivery of an AAV9-CAG-CLN6 vector at 1.5 × 10e13 vg (six patients enrolled), but no results are yet available (Table 1).
PD
PD is caused by the loss of dopaminergic neurons of substantia nigra, leading to dysregulation of interacting inhibitory and excitatory pathways. Glutamate acid decarboxylase (GAD) catalyses synthesis of GABA, the major neurotransmitter in the brain. The first clinical trial involved the injection of a mix of two AAV2 vectors, one encoding GAD65 and the other encoding GAD67 in the subthalamic nucleus (clinical trial NCT00195143). Twelve patients were enrolled, divided into three cohorts: low dose (1 × 10e11 vg), medium (3.10 × 11 vg), and high (1 × 10e12 vg). The injection encompasses 20 μL of mannitol and 45 μL of vector mix in one site unilaterally at a rate of 0.5 μL/min. No adverse events due to surgery were observed. All patients displayed an improvement on the Unified Parkinson's Disease Rating Scale (UPRDS) for motor evaluation, mostly on the side of the body contralateral to treatment. No significant differences were observed between the different groups of vector doses. 68
In the trial from Bartus et al. (clinical trials NCT00985517), six patients with moderate PD where administered bilaterally with AAV2-neurturin (CERE-120) in both the putamen and substantia nigra to counteract deficits in the axonal transport, which limit the efficacy of neurotrophic factors. The goal was to test the feasibility and tolerance of the strategy, even with the stereotactical targeting of the substantia nigra, a small structure deep within the midbrain and close to critical neuronal and vascular structures. The results of this trial showed tolerance and absence of adverse effects, therefore demonstrating that the bilateral stereotactical injection of AAV2-neurturin into these two brain regions was feasible, safe, and well-tolerated in PD patients. 69 This was completed with a recent study demonstrating the long-term safety in PD patients. Indeed, long-term follow-up in patients injected with doses ranging from 1.3 × 10e11 vg total to 2.4 × 10e12 vg total revealed no major changes in clinical assessment or imaging studies. Furthermore, the motor capacities remained stable or mildly improved relatively to baseline individuals during the whole open-label and long-term follow-up, therefore providing evidence that long-term AAV2-neurturin stereotactical delivery into the putamen or the putamen/substantia nigra is safe. 70 Four patients died from unrelated causes, and autopsies were performed on their brains. Importantly, Bartus et al. showed a mild but persistent expression of neurturin over 4 years. 71,72 These data were completely concordant with previous long-term analysis published by Bankiewicz et al. in large-animal models. 73
Dopamine is synthetized in the brain from L-tyrosine from the diet. For this process, three enzymes are essential: tyrosine hydroxylase (TH), which converts L-tyrosine into L-dopa; guanosine triphosphate cycohydrolase I (GCH); and AADC, which converts L-dopa into dopamine. In PD, the benefit of levodopa therapy decreases over time, possibly because degeneration of nigrostriatal neurons causes progressive loss of aromatic L-amino-acid decarboxylase (AADC) enzyme, which converts levodopa into dopamine. Preclinical studies in animal models showed efficient improvement of PD-related phenotype after delivery of either the three enzymes 74,75 or only AADC delivery 73,76 using viral vectors.
A clinical trial (Table 1) involved the injection of AAV2 encoding AADC in four sites of the putamen with customized infusion cannulae. Fifty microliters were injected at each site at a rate of 1 μL/min. Evidence of improvement on UPRDS criteria by 30% was described in the 10 patients, but the surgical procedure was associated with an increased risk of intracranial hemorrhage and limited headache. The observed improvement should be regarded as preliminary because of the small number of subjects; it could simply be a placebo effect. 77 Since that first clinical application, several clinical trials have been launched based on the intracerebral delivery of AAV encoding AADC transgene (Table 1). Agilis demonstrated long-term safety and therapeutic efficacy of AGIL-AADC administration for patients with AADC deficiency (oral communication ASGCT 2017, abstract 295). 78,79 Voyager therapeutics launched a clinical trial (clinical trial NCT01973543) with three doses of VY-AADC01 (7.5 × 10e11, 1.5 × 10e12, and 4.7 × 10e12vg) together with levodopa medication. No results have been published yet. However, in December 2016, Voyager announced dose-dependent improved measures of motor function and an enhanced response to levodopa after 6 and 12 months. The procedure is well tolerated, and Voyager demonstrated accurate MRI-guided delivery.
Finally, a clinical trial using a LV (Prosavin®) derived from the EIAV expressing the three key dopamine biosynthetic enzymes (tyrosine hydrolase, aromatic L-amino-acid decarboxylase, and GTP cyclohydrolase-1), with the aim of providing a continuous source of dopamine in the striatum. A Phase I, open-label, clinical study (clinical trial NCT00627588) has been initiated, including six patients at two dose levels. A 12-month follow-up has demonstrated that vector administration was safe and well tolerated at both doses, and no serious adverse events related to the product or the surgical administration were reported. A significant improvement of motor function evaluated by the UPDRS part III motor score was recorded compared to baseline. 80 Fifteen patients were enrolled, and a long-term analysis is ongoing, although no results have been published yet.
SMA
SMA is a devastating neurodegenerative monogenic disorder. Children with the most severe form (SMA type I) are known as “floppy babies.” They will never sit unassisted or roll over, and they fail to maintain their head. 81 The CHOP-INTEND score measures evolution of the disease, with no patient displaying a score of >40 by the age of 6 months, and 75% of patients die or require permanent ventilation by 13.6 months. In humans, chromosome 5 contains two quite identical genes at location 5q13: SMN1 at the telomere and SMN2 at the centromere. SMN1 encodes the survival motor neuron protein (SMN), which is crucial for motor neuron survival, whereas SMN2 is only able to produce 10–20% of the full-length SMN and mostly expresses a truncated protein (SMNΔ7), which is rapidly degraded by cells.
After very promising results obtained in both mice and large-animal models with a gene therapy approach based on the delivery of scAAV9 encoding the SMN protein by intrathecal or intra-CSF delivery, 81,82 AveXis© developed a Phase I clinical trial (clinical trial NCT02122952) with a single IV administration of a scAAV9-SMN, which crosses the BBB. Fifteen patients confirmed with two copies of SMN2 gene by genetic testing were enrolled. A first cohort of three patients received 6.7 × 10e13 vg/kg of AVXS-101, and a second cohort of 12 patients received 2 × 10e14 vg/kg. After the first cutoff, all patients were alive, and only one patient from cohort 1 required permanent ventilation at 28.8 months of age. Patients in cohort 2 were free of permanent ventilation and had clear improvements in motor function: 11/12 injected patients achieved a CHOP-INTENT with >40 points, having head control and sitting with support, and 8/12 sat unassisted. Moreover, two patients could crawl, stand, and walk independently, showing a dramatic sustained impact on motor function (oral communication ASGCT 2017, abstract 291).
Alzheimer's disease
Alzheimer's disease (AD) is the most common neurodegenerative disorder, affecting 50 million people worldwide. The two 83 main neuropathological hallmarks of the disease are extracellular amyloid plaques produced from the metabolism of amyloid precursor protein (APP) and neurofibrillary tangles (NFTs) due to the accumulation of abnormal hyperphosphorylated tau protein, a microtubule assembly protein. 84 –86 Despite numerous studies in multiple animal models of the disease, there is no efficient therapy to slow down the course of the disease in humans, and this constitutes a major objective with huge medical needs.
The only gene therapy clinical trials that have been launched for AD were based on intracerebral delivery of AAV2 encoding nerve growth factor (NGF). A first report of 10 patients with mild to moderate AD who received bilateral injections (total of six deliveries) into the basal forebrain region (into the nucleus basalis of Meynert) with dose escalating from 1.2 × 10e10 vg to 1.2 × 10e11 vg; clinical trial NCT00087789) showed the safety and the tolerability of the procedure for at least 2 years post injection, and persistent bioactivity of NGF in autopsy tissues, with a trophic response to NGF and lower cognitive decline. 87 A multicentric Phase II (clinical trial NCT00876863) is ongoing on 49 patients, but results are not yet available. 88
Ex vivo gene therapy using HSCs
After HSC transplantation, a fraction of stem cells or myeloid progenitors is able to migrate to the CNS, cross the BBB, and locally differentiate into genetically modified microglia-like cells. The existence, modalities, and kinetics of CNS microglia replacement by hematopoietic-derived cells after HCT is still a matter of debate. 89,90 However, studies in mice suggest that recruitment of bone marrow–derived myeloid cells to the CNS occurs in many types of neuroinflammatory or neurodegenerative processes. 91 –93 The reconstitution of microglia-like cells after HCT can provide a local permanent source of therapeutic protein such as lysosomal enzyme or trophic factor to treat neurodegenerative diseases. It may also correct endogenous dysfunctional microglia in diseases such as adrenoleukodystrophy.
However, allogeneic HCT from healthy donors is still associated with high morbidity/mortality risk. This can be overcome by autologous transplantation of corrected cells after transduction with gene therapy vectors. Indeed, LVs have been shown to transduce HSC efficiently and allow reconstitution in vivo with LV-corrected cells. 31,32,94,95 Transduction with LVs can correct deficient microglia and its consequences on surrounding neuronal damages in adrenoleukodystrophy. In the case of LSD, the production of above-normal enzyme quantities by genetically modified cells can increase the benefit of HCT by increasing the enzyme dose delivered to the affected tissues. 41
X-ALD
X-ALD is an X-linked monogenic neurodegenerative disease. Mutations in the ABCD1 gene cause a loss of function of the ALD protein (ALDP), a peroxisomal membrane transporter. This results in impaired transport and metabolism of very long-chain fatty acids (VLCFAs) into peroxisomes. The accumulation of toxic VLCFAs leads to the breakdown of myelin. Cerebral ALD (cALD) is the most severe form, with onset usually in childhood, but also in young adults. cALD is characterized by inflammatory demyelination and neurodegeneration, leading to the loss of neurologic function and death within a few years of symptom onset. The only treatment that was shown to stop the progression of the disease is HCT from healthy donor HCT when performed at an early stage of brain disease. 96 Based on the efficacy of HCT in cALD patients, HSC gene therapy has been developed early in ALD. 96 The goal of HSC gene therapy in ALD is to halt cerebral disease progression by restoring functional ALDP expression in brain macrophages/microglia while reducing transplant-related morbidity and mortality associated with allogeneic HCT. Based on preclinical proof of efficacy, 94,95 the pioneering Phase I/II clinical trial (clinical trial NCT01896102) based on transplantation of autologous HSC corrected with a LV vector was started in 2006. Four boys with cALD received autologous HSCs transduced ex vivo with an LV containing an ABCD1 cDNA. Gene therapy resulted in functional human ALDP expression and disease stabilization, with results comparable to those obtained with HCT from a healthy donor, without the risks and complications associated with allogeneic transplant. 31 A larger multicenter study is currently ongoing and is confirming positive results (oral communication ASGCT 2016, abstract 250).
MLD
The rationale for HCT in LSDs is based on the capacity of cells derived from the transplanted HSCs to differentiate into macrophages in the affected tissues, particularly brain macrophages/microglia that may be a permanent source of functional lysosomal enzyme. HCT may thus treat LSD patients with CNS involvement. A single intervention may provide enzyme to affected patients for their lifetime. Based on this rationale, a few thousand LSD patients have been treated with allogeneic HCT over the past decades. 97
HCT was proven to be efficient in several LSDs with CNS involvement such as MPS1 (Hurler syndrome) but is not efficient in MLD. In this disease, only a gene therapy approach based on the use of autologous HSCs and a highly efficient gene transfer using a LV vector to express human ARSA was shown in an animal model to prevent the sulfatide storage and cure the neurological disease, 98,99 indicating a critical role for enzyme overexpression in HSC-derived progeny in determining therapeutic effect. Based on these preclinical results, a Phase I/II clinical trial (clinical trial NCT01560182) of HSC gene therapy in MLD patients was started in early 2010 and is provided promising results. 100,101 Early-onset MLD patients were treated at the pre-symptomatic stage with autologous HSC transduced with an LV encoding the human ARSA cDNA after busulfan conditioning. Stable engraftment of gene-corrected HSCs was observed, leading to ARSA activity at or above normal values in circulating hematopoietic cells and in the CSF. A marked benefit was demonstrated as prevention of disease onset and/or halted progression. Patients (n = 11) showed motor and cognitive development, in contrast with the severe natural course of the disease.
Future Challenges
Encouraging results reported in Phase I/II gene therapy clinical trials for CNS diseases developed so far support the feasibility and the efficacy of AAV- and LV-based CNS gene therapy. They confirm the numerous proofs of concept that have been made for a large number of neurodegenerative diseases in small and large animals. 6,41,44 However, hurdles still need to be overcome toward demonstration of significant and sustained benefit in human patients in a growing number of indications. They include the capacity to target/restrict expression in specific CNS cell types, the efficacy of vector delivery in order to obtain an adequate expression of the transgene to reach a therapeutic effect, 102 the potential immune response that can hamper the efficacy of AAV delivery and transgene expression, and the demonstration that current strategies can provide in a single-time treatment a long-term efficacy in human patients with severe conditions.
Access to the target cells
The difficult access to particular regions of the CNS after in vivo delivery of viral vectors may result in a reduced number of cells expressing the therapeutic transgene insufficient to reach overall therapeutic levels. The route of administration and parameters of in vivo delivery protocols (injected volume, flow, and number of injections) still need to be improved to allow widespread transduction. MRI already allows the monitoring of real-time stereotactical injections of AAV particles into the brain of NHPs, 103 which is key to target specific brain regions precisely in clinical trials.
A way to improve the delivery of therapeutic substances to the brain is convection-enhanced delivery (CED). The CED procedure involves minimal surgery to implant small-diameter catheters into the brain, and this has been extensively used to target brain tumors using small pressure gradients during the injection. This technique has been used in dogs, primates, and humans, mainly for brain tumors but also for PD potentially to increase the diffusion of the vector after delivery. 104 –106
CSF delivery is a safer alternative option to intraparenchymal delivery, and several studies in NHPs showed that AAV delivery to the CSF allowed efficient transduction of localized tissues in the CNS. 107
Importantly, some AAV vectors are able to cross the BBB following IV delivery. The capacity of AAV9 to circumvent the BBB and transduce neurons within the CNS 15,108,109 has been demonstrated. Injection into neonates allowed large neuronal transduction, whereas adult delivery led mostly to astrocyte transduction. 15 AAVs with modified capsids that were engineered to cross the BBB are being continuously optimized to reach the foremost regions of the CNS with less-invasive routes of administration relative to intracranial injections, a promising approach for developing new gene therapies for neurodegenerative diseases. 13,102 To try to circumvent the limited tropism of AAVs, novel AAVs with a desired characteristic based on a selection method called CREATE (Cre-recombination based AAV targeted evolution) were recently isolated. 14 New variants were shown to have a wide transduction profile in an adult mouse system after IV delivery. 13,14
In parallel to modifying vectors, techniques have been proposed to enhance viral transduction and diffusion by improved delivery. One of them is the pre-infusion of focused ultrasound (FUS), after IV delivery of microbubbles. Oscillation of the microbubbles inside the brain capillaries by ultrasound leads to the BBB transiently opening. 110,111 In combination with AAV delivery, this technique could increase the volume of AAV transduction in the brain. 112 Another currently evaluated alternative is coupling AAVs with exosomes. This could improve AAV diffusion and limit the immune response against AAVs. Indeed, exo-AAVs are more resistant to neutralizing antibodies compared to standard AAVs, especially when administered at a low vector dose, and a significant improvement of the transduction has been reported. 113,114
HSC gene therapy based on LVs
The beneficial results obtained in the first HSC gene therapy trial based on LVs for presymptomatic MLD opens up therapeutic options for other LSD. The therapeutic benefit provided by the above-normal level of enzyme activity on critical disease manifestations after HSC gene therapy exceeded that exerted by HCT. Such a strategy could be proposed for other diseases with severe CNS involvement, such as MPSII, a disease caused by the deficiency of iduronate-2-sulfatase (IDS) for which allogeneic HCT leads to poor neurologic outcome in the transplanted patients. 115
In diseases such as X-ALD, where gene therapy aims to restore normal microglia function after hematopoietic cell reconstitution, above-normal expression of the therapeutic protein is probably not a key benefit. The rationale of gene therapy is to stop disease progression while reducing morbidity and mortality associated with allogeneic transplant thanks to the use of milder conditioning and autologous cells. The critical objective here is to achieve high rates of autologous HSC transduction with optimized LVs and transduction protocols. Encouraging results from ongoing study confirm that this goal is achievable.
New Therapeutic Targets
Gene silencing strategies to “silence” dominant genes: the example of “huntingtin”
Preclinical studies using AAV-based-shRNAs/miRNAs 116 –118 or chemically modified antisense oligonucleotides (ASOs) 119 have been done in mouse models of HD. The main hurdles in gene-silencing approaches are the off-target effects (nonspecific silencing), saturation of the cellular machinery, and immune responses against the nucleic sequence. 120 Despite this, well-tolerated lead ASOs that potently and selectively silence mutated huntingtin preserving the normal allele have been identified and tested in HD mouse models. 121 Among huntingtin-lowering drugs, the use of ASOs is the only approach that has already entered clinics (Phase I/IIa; NCT02519036); their safety and tolerability profile support its continued development. The completion of the trial and results are expected by the end of 2017. Genome editing of cells may open new avenues for further development of gene therapy clinical applications in human patients. 122
Amyotrophic lateral sclerosis (ALS) is another fatal neurodegenerative disorder with a survival of around 3–5 years after onset of the disease. Around 20% of familial cases of ALS and 1–3% of sporadic cases are due to mutations in the superoxide dismutase 1 (SOD1) gene. Recently, Stoica et al. showed extended survival and delayed hind-limb paralysis after intraventricular delivery of an AAV9 encoding a microRNA against the human SOD11 in neonatal SOD1G93A mice. 123 Borel et al. showed in the same SOD1G93A mice that systemic delivery of AAVrh10 encoding an artificial microRNA against SOD delayed onset of the disease and death of the mice. 124 Further, they showed an efficient silencing of SOD1 after intrathecal delivery of the vector in NHPs, 124 suggesting the potential therapeutic benefit of this approach in ALS patients displaying SOD1 mutations.
Gene therapy targets for multifactorial diseases
In multifactorial neurodegenerative diseases, gene therapy strategies based on delivery of neurotrophic factors or activation of deficient metabolic pathways to slow down neurodegeneration have shown encouraging preclinical results that should rapidly lead to clinical applications. Neurotrophic factors were shown to rescue diseased cells in animal models of AD, PD, ALS, and HD, 125 and clinical trials using AAV2-GDNF and AAV2-NGF have started to be developed for AD and PD, respectively. 126 An interesting target toward clinical development is cholesterol metabolism, which plays a pivotal role in the progression of neurodegenerative diseases. Delivering the key enzyme of brain cholesterol catabolism, the cholesterol 24-hydroxylase (CYP46A1), was shown to restore cholesterol metabolism pathway and mitigate neuropathology and abnormal behavior in several animal models of AD and HD. 35,127,128 Stimulation of autophagy-dependent pathways is another example of strategy that could further lead to evaluation in human patients based on preclinical studies in animal models of PD, Lewy Body disease, AD, and spinocerebellar ataxia, in which the viral-mediated overexpression of the autophagic protein beclin-1 was shown to improve neuropathology and abnormal behavior. 129 –131
Immune response
Immune responses against the capsid and the transgene have emerged as serious obstacles for translation of AAV therapy to humans in peripheral tissues. 9,132 These are likely limited after direct injection of limited doses of viral vectors in the potentially immune-privileged brain. However, development of intra-systemic injection protocols will raise this issue, with the need to evaluate preexisting immunity, to discuss immunosuppressive protocols and to evaluate long-term follow-up.
Long-term efficacy
The long-term effectiveness of gene therapy in human patients remains to be better documented. Indeed, the long-term follow-up of clinical trial patients revealed persistent and long-lasting expression of the transgene in the human brain, 60 in line with a recent report showing persistent expression of genes for dopamine-synthesizing enzymes delivered by an AAV vector into the primate brain 15 years post gene transfer. 133 This is important confirmation, even it does not guarantee clinical improvements, as shown for PD. 134
Clinical trial design
A critical issue for CNS gene therapy is the design of clinical trials: how to identify a beneficial effect clearly in a reasonable time frame and with a limited number of patients in a Phase I/II clinical study. In many of the severe genetic neurodegenerative diseases that are the focus of gene therapy programs, the therapeutic window is narrow. Late treatment at advanced stages of diseases may not allow the desired therapeutic effect to be reached. The potential heterogeneity of the patient population and the complexity of efficacy endpoints underline the importance of the clinical study design: an adequate number of patients, inclusion criteria based on accurate characterization of the natural history of the disease, relevant biomarkers showing the mechanism of action (proof of functionality of the vector), and outcomes of disease progression. New tools, particularly imaging, will be critical to demonstrate efficacy in a limited number of patients. An example is fluorodeoxyglucose PET imaging that has been used to track dynamic biomarkers in neurodegenerative diseases. 135 –137 This will be mainly critical for novel original therapeutic strategies focusing a low number of enrolled patients, as in Phase I/II trials.
Toward the development of gene therapy products
Among the limited number of gene therapy trials that have been performed so far for severe neurodegenerative diseases, many encouraging results have been obtained in terms of feasibility, tolerance, long-term expression, and efficacy in treated patients. These first results, the current preparation of Phase II/III trials for several indications, and the improvement of vectors and delivery protocols should accelerate the development of the field in the near future.
Beside the critical issues that have been discussed above in terms of delivery, immune response, and manufacturing of gene therapy products, challenges to the progress of clinical gene therapy include regulatory and marketable difficulties. To foster the development of gene therapy products and support the entry of new innovations into the international market, a more harmonized regulation and a facilitated process for the approval of clinical trials is needed, especially for multinational trials. In Europe, a new framework for the approval of clinical trials is anticipated for the fourth quarter of 2018. 138
A major advantage of gene therapy is that it could cure a patient with one single injection. This unique single-treatment approach will require a well-defined value proposition to avoid impossible burden on global healthcare services and to establish product pricing that will provide value and be accepted by prescribers and payers. 139 Toward that goal, collaborative discussions already engaged between regulatory authorities, pharmas, and payers about innovative models to fund these revolutionary therapies will be critical. The issue of rapid access and reimbursement of novel medicinal products is crucial for patients suffering from lethal or very severe diseases with no treatment options. 138
Regardless of these important challenges, results from the first in vivo and ex vivo gene therapy clinical trials for neurodegenerative diseases are promising and demonstrate that gene therapy is a relevant strategy to slow down the progression of CNS diseases. Remarkably, the endorsement of the pioneer gene therapy product, Glybera, an AAV to treat lipoprotein lipase deficiency, by the European Agency of Medicine (EAM) 19,140 and the EAM-approved Strimvelis (GSK2696273) by Glaxo-Smith Kine, an ex vivo therapy based on stem-cell delivery for ADA-SCID patients suffering from severe immunodeficient syndrome, 141 are a crucial step toward gene-based drug development.
Footnotes
Acknowledgments
This work was supported by NeurATRIS: A Translational Research Infrastructure for Biotherapies in Neurosciences, the Fondation pour la Recherche Médicale, Bioingénierie pour la Santé 2014 “Projet DBS20140930765,” Paris Biotech Santé incubator, and the SATT (Société d'Accélération de Transfert Technologique) Ile de France Innov.
Author Disclosure
N.C. is a founder and owner of founder equity of BrainVectis Therapeutics. S.A. is an employee of BrainVectis. F.P. declares no competing interests.