
Abstract
Thalassemia is a recessive monogenic hematological disease associated with reduced amounts of functional hemoglobin caused by mutations/deletions in at least one of the globin genes. This disease has attracted significant attention throughout the years in terms of genetic diagnosis and developments in gene and cell therapy. Here, recent progress is reviewed in the genetic diagnosis and development of therapeutics for thalassemia, particularly β-thalassemia, in China and around the world.
Introduction
T
In the early 1980s, a large-scale survey involving 900,000 people across 28 provinces was conducted to determine the prevalence of hemoglobinopathies and thalassemia in China. 2 Results of this survey showed a high population frequency of thalassemia in the southern regions of China, with the highest incidences seen in the Guangxi Zhuang autonomous region and Guangdong Province (including Hainan Province). 2 –9
Thalassemia is caused by mutation or deletion of the α- or β-globin genes, resulting in reduced amounts or absence of globin chains and, in turn, decreased levels of functional hemoglobin (Hb; Fig. 1). Imbalances in the α/β ratio of the Hb heterodimer can produce the various symptoms associated with thalassemia. Most α-thalassemias are caused by deletion of the α-globin gene, whereas β-thalassemia is often caused by point mutations in the β-globin gene. More than 800 variants in the β-globin gene have been described that cause β-thalassemia. 10 Some mutations lead to complete loss of β-globin production, called β0 thalassemia, whereas other mutations lead to a markedly reduced rate of β-globin production, called β+ thalassemia.
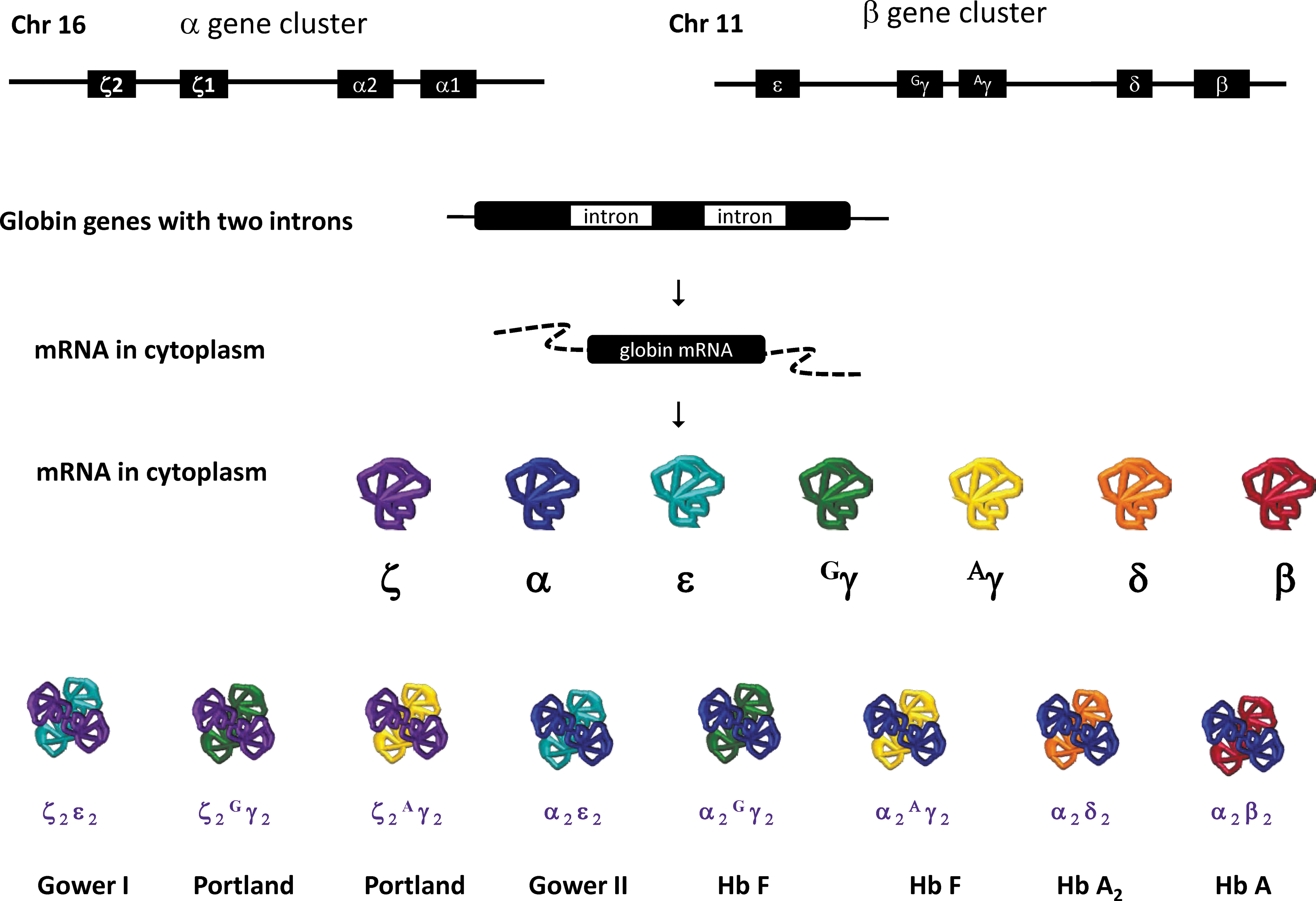
Schematic of globin locus and the respective hemoglobins generated at different developmental stages. The α-like globin cluster locus on chromosome 16 harbors four genes that encode the α-like subunits of hemoglobin: ζ1-globin, ζ2-globin, α1-globin (HBA1), and α2-globin (HBA2). The β-globin locus on chromosome 11 harbors five genes that encode the β-like subunits of hemoglobin: ɛ-globin (HBE1), Gγ (HBG2), Aγ (HBG1), δ-globin (HBD), and β-globin (HBB). They are sequentially/orchestrated switched on/or off at different the stages of development. As a consequence, both α-like subunits of globin and β-like subunits of hemoglobin are transcribed followed with translation leading to the formation of various kinds of heterodimers of hemoglobins correspondingly with the different stages of development.
Molecular Diagnosis of Thalassemia in China
The first molecular diagnosis of thalassemia, which was also the first for any human disease, was carried out by Kan's group in 1976. Liquid DNA–DNA hybridization was measured in vitro, and the number of α-globin genes in DNA prepared from cultured amniotic fluid cells was quantified by hybridization with a radioactive DNA probe complementary to the α-globin gene. Results from this study showed whether the fetus a carried an α-thalassemia gene. 11
In the early 1980s, DNA dot-blot hybridization for prenatal diagnosis of α-thalassemia was conducted by Zeng and Huang. 12 This was the first example of molecular diagnosis of α-thalassemia in China, and the efficiency of molecular diagnosis represented a significant improvement over that achieved with liquid hybridization.
Southern blot analysis after digestion of DNA samples with restriction endonuclease was subsequently applied for molecular diagnosis of thalassemias, 12 –14 including β-thalassemia. 15,16
Shortly after polymerase chain reaction (PCR) was invented in 1985, 17 it was used for rapid diagnosis and carrier testing of several inherited disorders. PCR-based techniques now provide a rapid and relatively simple method for diagnosis of thalassemia and are used in an increasing number of countries. The techniques have proven to be reliable and extremely accurate.
In the late 1980s, PCR was used by the authors' group to diagnose a variety of genetic diseases, including thalassemia. PCR-allele-specific oligonucleotide (ASO) probe techniques showed that the most common mutations associated with β-thalassemia are a frameshift at CD41/42, IVS-II-nt.654 C > T and a nonsense mutation at CD 17 A>T, −28 A>G. 18 Multiplex allele-specific amplification (MAS-PCR) for prenatal diagnosis of β-thalassemia can simultaneously detect these common mutations. About 50 fetuses at risk for severe β-thalassemia were prenatally diagnosed using this method. 19 To date, these two methods are the most frequently used for genetic diagnosis of thalassemia.
Real-time PCR is an extension of PCR methodology. Using two different Taqman probes coupled with the 5′ nuclease activity of Taq DNA polymerase, allele-specific detection of amplification (allelic discrimination) can be achieved and consequently used to obtain genotype information. 20,21
In the past two decades, methods based on real-time PCR have been developed, and molecular diagnosis of thalassemia by real-time PCR is now widely used in China. For example, Huang et al. developed a novel real-time PCR assay based on multicolor melting curve analysis (MMCA) methodology that uses multicolor probes in a melting curve analysis. 22 This method has been used to diagnose deletional and non-deletional mutations of both α-thalassemia 23 and β-thalassemia. 24 Almost all of the major α- or β- thalassemia mutations can be identified simultaneously using this approach. Thalassemia detection kits based on MMCA are now available and have been approved by the CFDA after double-blind testing of thousands of thalassemia samples.
High-resolution melting analysis (HRM) based on real-time PCR and melting curve analysis is faster, simpler, and less expensive than alternative approaches requiring separations or labeled probes. Therefore, HRM is suitable for rapid screening of thalassemia hotspot mutations. In the authors' lab, duplex and triplex amplicon genotyping was developed to identify the abovementioned common β-thalassemia mutations simultaneously in patients or carriers. The entire procedure for mutation detection can be completed within 30 min. 25 Moreover, approaches based on the HRM technique that can distinguish common non-deletional mutations in α-thalassemia and common β-thalassemia mutations have been established in several labs in China. 26 –28
Next-generation sequencing (NGS) techniques provide high speed and throughput, such that genome sequencing projects that took several years with the Sanger technique can now be completed in a matter of weeks. 29 Recently, NGS has been widely used for the rapid diagnosis of various genetic diseases, including thalassemia. For example, Xu et al. designed an NGS panel targeting the coding regions of globin genes and four modifier genes. In molecular screening analysis of 10,111 couples, they detected 4,180 individuals who carried a total of 4,840 mutant alleles and identified 186 couples who were at risk of having affected offspring. The advantages of NGS were reflected by the finding that 12.1% of the pathogenic or likely pathogenic variants were undetectable by traditional methods. 30
The discovery of cell-free fetal DNA (cfDNA) in maternal plasma in 1997 provided new possibilities for noninvasive prenatal testing (NIPT). 31 NGS allowed quantification of millions of DNA molecules in a single sample, thus greatly increasing the sensitivity of molecular tests using cfDNA. Xiong et al. developed an accurate noninvasive prenatal test using NGS for HbE and the four most common β-thalassemia mutations found in South East Asia (i.e., −28A > G, CD17A > T, CD41/42[-TTCT], and IVS-II-654C > T). Their data showed that detection of paternal mutations using NGS could be achieved with high sensitivity and specificity, without the need for invasive tests, in 50% of pregnancies at risk for α-thalassemia in cases where the father and mother carry a different mutation. 32
Molecular Manipulation of Hb Genes for Treatment of β-Thalassemia
Imbalances in the α/β ratio in the globin heterodimer can result in the various symptoms of β-thalassemia (Fig. 2). Thus, early attempts to minimize thalassemia symptoms involved restoring the α/non−α globin-chain-ratio by either increasing the amount of the globin chain resulting from a defective globin-gene, in cases of α-thalassemia or β-thalassemia; or by elevating the level of the γ-globin chain in β-thalassemia. Extensive studies have been performed to restore the α/non-α globin chain ratio using the former approach. With the development of anti-sense RNA technology, Gong et al. blocked the abnormal splicing that occurs in β654-thalassemia, one of the most common β-thalassemias found in the Chinese population (IVS-2-654 C→T), by delivering an anti-sense RNA-expressing vector specifically targeting the C to T mutation. 33 In 2008, Xie et al. used RNAi to interfere with α-globin gene expression, while simultaneously blocking the β654-aberrant splicing site in a mouse model. They achieved an improved α/β-globin chain balance, leading to a decrease in the excessive precipitation of α-globin chains around the cell membrane, and in turn a recovered phenotype. 34 Inspired by May et al., who first reported the lentiviral vector delivery of a β-globin gene to treat β-thalassemia in model mice, 35 Li et al. achieved direct-delivery of β-globin expressing genes using a lentiviral vector and observed phenotypic recovery in a mouse model. 36 Consequently, murine β654-thalassemia induced pluripotent stem cell (iPSC) lines were generated, followed by transduction with a normal human β-globin gene. The chimeric murine models were established using blastocyst injection of these modified iPSCs, 37 which could differentiate into blood cells effectively in vivo. Thalassemia symptoms were alleviated after genetic modification of the iPSCs in these chimera mice. The curative effect had dose-dependent performance associated with rising chimerism levels. The work provided a model system for prenatal therapy using iPSCs to treat thalassemia and also presented vital information for the development of future postnatal iPSC therapies in thalassemia patients.
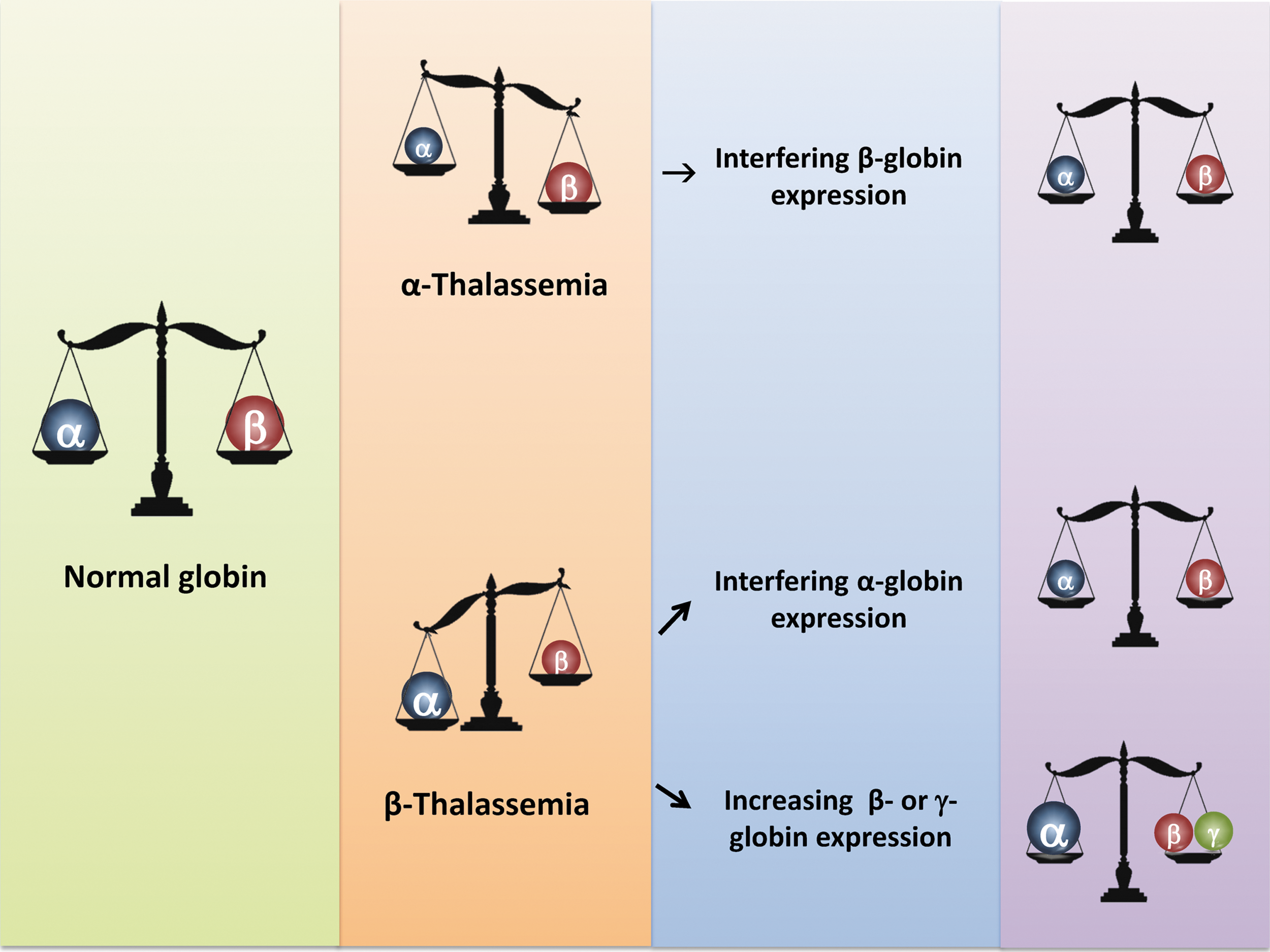
Thalassemia and principle of treatment. Left panel: normal globin heterodimers consists of α/β-globin at the ratio of 1:1. Middle panel: in thalassemia subjects, there is an imbalance of α/β-globin ratio, either >1 (for β-thalassemia), or <1 (for α-thalassemia). Right panel: treatment options for thalassemia aim to restore the α/β ratio to close to 1:1, by reducing the amount of overloaded globin subunit (β-globin for α-thalassemia and α-globin for β-thalassemia), or increasing the expression of β-like globin (in the case of β-thalassemia) to achieve the optimal α/β balance.

Example of the strategies used in β-thalassemia gene and cell therapy. Autologous or human leukocyte antigen matched allogeneic cells from β-thalassemia subjects, including CD34+ hematopoietic stem cells (HSCs), bone-marrow stem cells (BMSCs), or induced pluripotent stem cells (iPSCs) derivatives will undergo functional compensation by transduction with a functional β/γ-globin gene, or in situ gene manipulation. They will then be subjected to screening and (in vitro/in vivo) induction into HSCs (except for those the source of HSCs themselves). Specific amounts of the cells will then be transfused back to the patients followed by short- and long-term observation and assessment of the clinical presentation, including HBB level, mean corpuscular volume, transfusion independency, and so on. β-thalassemia mouse models can be used to test these strategies before being applied to patients.
Induction of γ-Globin Gene Expression
β-Thalassemia symptoms could also be alleviated by increasing levels of other β-like genes. Hydroxyl urea was used to induce reactivation of γ-globin gene expression to increase Hb F levels to ameliorate anemia symptoms and achieve a more balanced α-/non-α globin chain ratio. 38,39 In 2009, Sankaran et al. revealed that the BCL11A gene repressed γ-globin expression. Thus, suppression of BCL11A expression could decrease the repressive effects on the γ-globin gene, leading to increased production of γ-globin chains that would result in a more balanced α/(β + γ) globin ratio to treat β-thalassemia. 40 In 2017, Wienert et al. reported a British β-thalassemia case that had 20% Hb F. They showed that the marked increase in Hb F seen in this case was due to a single base mutation of T to C at position −198 of the Aγ-globin gene promoter. This finding led to the use of KLF1 to bind to the aberrant promoter site, resulting in activation of γ-globin expression. 36 Moreover, Wienert et al. used CRISPR-Cas9 gene editing to edit out the T → C at −198 of the Aγ-globin gene promoter and achieved activation of γ-globin expression in HUDEP-2 erythroblast cells. 41 Recently, the Xu group used a target-based NGS assay for molecular screening and clinical genotyping of 2,252 hemoglobinopathy patients in China. 42 These screening data revealed that a common single-nucleotide polymorphism (rSNP) HBG1-rs368698783 (G/A) in the proximal Aγ-promoter can ameliorate the severity of thalassemia through epigenetic-mediated regulation of a delayed fetal-to-adult Hb switch, which could provide potential targets for treatment of β-hemoglobinopathy. 43
Functional Compensation Versus in Situ Gene Modification
Only a few approaches are available to treat β-thalassemia, such as providing a corrected β-globin gene in trans, induction of β-like globin gene expression, and in situ correction of defective genes. Over the past decades, multiple in situ gene editing techniques have been developed, including homologous recombination, zinc-finger nuclease (ZFN), transcription activator-like effector nuclease (TALENs), and CRISPR-Cas9. However, the use of most of these techniques has been limited due to issues with efficiency and off-target effects. 44 For example, methods to achieve sufficient numbers of hematopoietic stem cells (HSC) or bone-marrow stem cells (BMSC) to repopulate recipient blood are needed. In 2015, Liang et al. reported the correction of β-thalassemia mutations using the CRISPR-Cas9 gene editing technique in discarded human tripronuclear zygotes, 45 and more recently this group used CRISPR-Cas9 to correct the β-thalassemia HBB-28 (A>G) mutation in human cloned embryos. 46 The HBB-28 (A>G) mutation accounts for >10% of total β-thalassemia cases in China, and this approach could eventually be applied to treat thalassemia at the embryo stage. Further success for in situ correction of another common β-thalassemia disease, CD41/42, involved manipulation of iPSCs by both TALEN and Cas9-CRISPR techniques. 47 The authors plan to use the latter technique for future manipulation and treatment of human β-thalassemia in embryos.
Autologous iPSC and Induction of HSC
The newly developed gene editing techniques to treat thalassemia involve induction of treatment cells into HSCs in vitro or in vivo before transfusion back into the patients. To date, the manipulation of these cells has not achieved satisfactory levels for clinical trials. As early as 2007, only 1 year after the first successful production of iPSCs, Hanna et al. reported the use of iPSCs to treat sickle = cell anemia in a mouse model. 48 One of the challenges for the therapeutic use of HSCs induced from iPSCs is engraftment of these cells for hematopoietic lineage repopulation in vivo, although the hemogenic endothelium derived from hPSCs may have therapeutic potential. 49 A report by Sugimura et al. showed that transduction of seven transcription factors (ERG, HOXA5, HOXA9, HOXA10, LCOR, RUNX1, and SPI1) could effectively promote hemogenic endothelium differentiation into hematopoietic stem and progenitor cells that have the capacity for long-term multi-lineage hematopoiesis in vivo. 50 Alternatively, other groups are attempting to knock out the human leukocyte antigen (HLA) to allow allogenic transfusions. 51 These results shed light on the therapeutic potential of iPSCs for treating genetic blood disorders, including thalassemia, but also highlight the need for further extensive studies of bio-safety and induction efficiency prior to their application in clinical trials.
Clinical Trials
In 2010, Cavazzana-Calvo et al. reported the first clinical trial involving transduction of the β-globin gene into CD34+ HSCs followed by transfusion back into children with severe β-thalassemia. 52 After a several year follow-up, one out of the four subjects treated had shown clinical benefit with Hb levels maintained at 9–10 g Hb/dL, as well as clonal expansion with HMGA2 gene activation. This clone gave rise to Hb levels that were one-third of normal levels 1 year after transfusion, and this effect persisted for 7 years before finally regressing without transformation. 52,53 Four β-thalassemia trials have been initiated in the United States and Europe. Of the 16 patients included in these trials, six had achieved transfusion-independent status. 53 In the meantime, a similar approach to treat severe β-thalassemia is being conducted in Nanfang Hospital, Southern Medical University, Guangzhou, China (pers. commun.). These clinical trials involved gene delivery via lentiviral vectors, instead of the commonly used adeno-associated virus vector for gene therapy, due to the superior integration of lentivirus that is more suitable for continuous production of functional compensated red blood cells. To date, no severe post-usage side effects have been reported after more than 100 clinical cases and more than 300 experimental mice models. 53 –55
Overall, as a monogenic recessive hematological disease, thalassemia is an ideal subject for developing genetic diagnostics as well as gene and cell therapies. Currently, several hurdles to developing a mature therapeutic protocol exist. For example, an efficient HSC induction system that could produce reliable in vivo expansion of functional HSCs and red cells, or suitable and well-controlled in situ gene editing and correction methods with no off-targeting events are needed. Solutions for these hurdles will lead to more successful clinical trials that will shed light on therapies for thalassemia at different developmental stages.
Footnotes
Acknowledgments
We thank Dr. Yi-tao Zeng (Shanghai Institute of Medical Genetics, Shanghai Jiao Tong University) for helpful discussions, and Guanhen Yang and Qin Cai for help with manuscript preparation. This work is supported by grants from the National Basic Research Project of China (2014CB964700 and 2014CB964701), the National Science Fund for Distinguished Young Scholars (81125003), and the Key Subjects of Shanghai (2017ZZ02019).
Author Disclosure
The authors declare they have no conflicts of interest, including financial interests.