
Abstract
CRISPR/Cas9-mediated programmed cell death protein 1 (PD-1) disruption in chimeric antigen receptor (CAR) T cells could be an appealing choice to improve the therapeutic efficacy of CAR T cells in an immunosuppressive tumor microenvironment. In most of the reported cases, Cas9 was delivered into T cells by way of electroporation with RNA or protein. However, transient expression of Cas9 by transfection with a plasmid encoding its gene is apparently simpler, as it avoids the steps of in vitro transcription of DNA or protein production. This study tried nucleofection into human primary T cells of plasmids encoding both CRISPR/Cas9 for disrupting the PD-1 gene and the piggyBac transposon system for expressing CD133-specific CAR in one reaction. Based on drug selection, CD133-specific CAR T cells were obtained in which, on average, 91.5% of the PD-1 gene sites were disrupted, but almost no Cas9 gene expression was found in the final engineered CAR T cells. The PD-1-deficient CD133-specific CAR T cells showed similar levels of cytokine secretion and improved proliferation and cytotoxicity in vitro, and enhanced inhibition of tumor growth in an orthotopic mouse model of glioma, compared to conventional CD133-CAR T cells. The described method could be useful for the production of PD-1-deficient CAR T cells for cancer immunotherapy.
Introduction
C
Genome editing has so far been reported mainly for CD19-specific CAR T cells. However, CAR T cells targeting solid tumors might benefit even more from genome editing, with the aim of counteracting the inhibition caused by components of an immunosuppressive microenvironment.
CD133 is a well-known marker for cancer stem cells in many solid tumor types, including glioma, lung, colon, and ovarian cancer. 13 CD133-specific CAR T cells or natural killer (NK) cells have already been reported to kill patient-derived glioma stem cells or ovarian cancer stem cells. 14,15 A 4.5-month-long partial response from CD133-specific CAR T-cell treatment has been achieved in a patient with advanced cholangiocarcinoma. 16 PD-1 gene targeting by CRISPR/Cas9 might help to improve CD133-specific CAR T cells.
The CRISPR/Cas9 system requires two essential components: the nuclease Cas9 and a chimeric single-guide RNA (sgRNA). With the guidance of the sgRNA, Cas9 binds to the target site and produces double-strand breaks, which are repaired by non-homologous end joining, causing unexpected gene mutations. 17 In previous studies aiming at PD-1 disruption in CAR T cells, Cas9-encoding mRNAs or Cas9 proteins were delivered into T cells by electroporation. 10 –12 However, for clinical use, the synthesis of RNA or protein would increase the number of manufacturing steps compared to the use of traditional gene delivery vehicles such as plasmid DNA. In addition, RNA or protein delivery has the disadvantage that the delivered elements are not stably integrated into the genome of the recipient cell. This means that for generating genome-edited CAR T cells, the T cells have to be modified at least twice (e.g., by using a combination of electroporation and virus transduction). Therefore, a simplified and efficient gene delivery method would be highly desirable for this purpose. Successful delivery of all elements into T cells using plasmids would be an attractive strategy, facilitating manufacture and reducing the modification steps. 18
This study developed a simplified protocol for generating PD-1-deficient CD133-specific CAR T cells. Plasmids (not RNAs or proteins) encoding all elements for both genome editing (CRISPR/Cas9 system) and CAR gene delivery and expression (piggyBac transposon system) were nucleofected together into primary T cells. The efficiency of gene modification was evaluated, and the functionality of the PD-1-deficient CD133-specific CAR T cells was analyzed subsequently in vitro and in vivo.
Materials and Methods
Plasmids
The plasmids used in this study have been described previously. The CD133-CAR piggyBac transposon vector and the Super piggyBac transposase plasmid for expression of the CD133-specific CAR were prepared according to Zhu et al. 14 pST1374-Cas9-GFP and pGL3-U6-hPD-1-sgRNA(1 + 2) for PD-1 disruption in T cells were prepared following the protocols of Su et al. 19
Primary cells and cell lines
Frozen peripheral blood mononuclear cells (PBMCs) from healthy donors were purchased from ALLCELLS (PB005F). The tumor cell lines used in this study were generated based on a previous report with modifications. 20 Briefly, the glioma cell line U251 was purchased from the Cell Bank of the Chinese Academy of Sciences. Based on this cell line, a CD133-overexpressing line (U251 CD133-OE) was generated by nucleofection with a piggyBac transposon vector encoding the human CD133 protein and the transposase plasmid, followed by drug selection. The stable transfectant was nucleofected again with a piggyBac transposon vector encoding firefly luciferase and the transposase plasmid, followed by drug selection to obtain a luciferase-expressing tumor cell line (U251 CD133-OE luc).
Gene editing and expansion of T cells
PBMCs were thawed and rested for 1.5–2 h. Then, 2 × 107 cells were re-suspended in 100 μL of buffer from the Human T cell Nucleofector® kit (Lonza; VPA-1002) and used for one nucleofection reaction; 5 μg of pST1374-Cas9-GFP, 5 μg of pGL3-U6-hPD-1-sgRNA(1 + 2), 5 μg of CD133-CAR piggyBac transposon vector, and 5 μg of Super piggyBac transposase plasmid were added. For preparation of the conventional CAR T cells, only the piggyBac transposon and transposase were used. Electroporation was performed with a Nucleofector™ II/2b device (Lonza) using the U-014 program. After nucleofection, the cells were immediately transferred into 3 mL of prewarmed T-cell medium in a 12-well plate.
To stimulate the T cells, on day 0, Dynabeads™ Human T-Expander CD3/CD28 (ThermoFisher Scientific; 11141D) were added at a ratio of three beads per cell, and the cell culture was supplemented with 300 IU/mL of interleukin (IL)-2. On day 14, allogeneic PBMCs from five donors were irradiated with 40 Gy using an X-ray biological irradiator (Rad Source Technologies, Inc.; RS2000PRO) and added to the T-cell culture at a ratio of 10:1. In addition, the cell culture was supplemented with 50 ng/mL of anti-CD3 antibody (Miltenyi; 130-093-387) and 300 IU/mL of IL-2 for the second round of stimulation and expansion. Two days after each stimulation, the T cells were washed and further cultured in T-cell medium (RPMI 1640 + 10% fetal bovine serum, penicillin–streptomycin, and GlutaMAX) supplemented with 300 IU/mL of IL-2 for expansion. The medium was refreshed every 2–3 days. Seven days after the first stimulation, the beads were removed, and CD133-CAR-expressing T cells were selected with 1 μg/mL of puromycin. Transfected T cells were analyzed on day 27 (13 days after the second stimulation). Non-transfected T cells were generated as described above but without nucleofection and drug selection.
Flow cytometric analysis
Cells were washed with FACS buffer (phosphate-buffered saline [PBS] containing 0.5% bovine serum albumin and 2 mM of ethylenediaminetetraacetic acid) and stained in the same buffer. Antibodies were used at the concentrations according to the manufacturers' instructions. The cells were analyzed by flow cytometry on a CytoFLEX instrument (Beckman Coulter). The data were analyzed by CytExpert (Beckman Coulter) or FlowJo (Tree Star).
To determine CD133-CAR-positive cells after transfection, cells were collected 13 days after the second stimulation, washed and stained with mouse anti-c-Myc antibody (clone 9B11; Cell Signaling; 2276S), followed by a secondary rabbit anti-mouse immunoglobulin G F(ab′)2 antibody (Jackson Immunotech). CAR T-cell phenotype characterization was performed using anti-CD95 (BD; 562616), anti-CD45RO (BD; 555493), and anti-CD62L (BD; 559772).
To detect the expression of PD-L1 and CD133 on tumor cells, the cells were incubated with human FcR blocking reagent (Miltenyi; 130-059-901) for 10 min at 4°C. Then, the following antibodies were used: PE mouse anti-human CD274 (BD; 557924), CD133/1 (AC133)-PE (Miltenyi; 130-098-826). The corresponding isotype control antibodies were used as controls.
PD-1 disruption in T cells was analyzed based on the expression of PD-1 on the cell surface: 106 conventional CD133-CAR or PD-1-deficient CD133-CAR T cells were co-cultured with 105 irradiated (70 Gy) U251-CD133 OE cells in 24-well plates in 2 mL of T-cell medium per well with 100 IU/mL of IL-2. Three days later, PE mouse anti-human CD279 (BD, 560795) and APC-H7 mouse anti-human CD3 (BD, 560176) were used to detect the PD-1 expression on CD3-positive T cells.
To determine the expression of Cas9 protein in T cells, indirect evidence was collected by flow cytometry based on green fluorescent protein (GFP) signals 1 day and 8 days after nucleofection. GFP was co-expressed with Cas9 as a fusion protein.
To determine the number of human T cells in mouse blood, red blood cell lysis buffer (Beyotime; C3702) was added for removing the red cells. The human T cells were stained with the following antibodies: PE-Cy™ 7 mouse anti-human CD45 (BD; 557748) and APC anti-human CD3 (4A Biotech; FHA003-050). Finally, the cells were fixed with 4% formalin solution, washed extensively using PBS, and analyzed by flow cytometry.
Efficiency of PD-1 gene disruption
PD-1 gene disruption was assessed, as previously described. 19
Cytotoxicity assay
U251 CD133-OE luc cells (104) were seeded into 96-well plates. Conventional or PD-1-deficient CD133-specific CAR T cells were added according to different effector-to-target cell ratios. The cells were cultured in 200 μL of T-cell medium without cytokines for 24 h. All cells were collected and transferred to a 96-well white microplate (PerkinElmer); 0.75 mg/mL of D-luciferin K+ salt (PerkinElmer; 122799) was added, and the signals were read immediately using an EnSpire Multimode plate reader (PerkinElmer). The percentage of target cell death was calculated according to the following formula: Lysis (%) = (1 – signalco-culture well/signaltumor alone well) × 100.
Cytokine secretion assay
Conventional or PD-1-deficient CD133-specific CAR T cells (2 × 105) were co-cultured with 105 U251 CD133-OE cells at an effector-to-target cell ratio of 2:1 in a 96-well plate in 200 μL of T-cell medium without cytokines for 24 h. The supernatant was collected, and the concentrations of cytokines in the medium including interferon (IFN)-γ, IL-2, tumor necrosis factor (TNF)-α, and granulocyte-macrophage colony-stimulating factor (GM-CSF) were measured with the AlphaLISA kits from PerkinElmer (IL-2, AL221C; IFN-γ, AL217C; TNF-α, AL208C; and GM-CSF, AL216C) according to the manufacturer's instructions.
T-cell proliferation assay
T cells were labeled with 0.5 μM of carboxyfluorescein diacetate succinimidyl ester (CFSE; eBioscience; 65-0850-84) following the manufacturer's protocol. Target cells were irradiated with 70 Gy using an X-ray irradiator (Rad Source Technologies, Inc.; RS2000PRO). CFSE-labeled T cells (106) were co-cultured in a 24-well plate with 5 × 105 irradiated target cells in 1 mL of T-cell medium supplemented with 300 IU/mL of IL-2 per well. At the indicated time points, T-cell proliferation was analyzed by flow cytometry.
Antitumor function in vivo
All animal experiments in this study were approved by the Shanghai Administrative Committee for Laboratory Animals. The orthotopic mouse glioma model used has been described previously.
14
For intracranial tumor implantation, 2 × 105 luciferase-expressing U251 CD133-OE luc cells were injected into the brain of 6- to 8-week-old female NPG mice (NOD-Prkdcscid Il2rgnull; Beijing Vitalstar). To monitor tumor growth, the bioluminescence imaging (BLI) signals were measured using an IVIS spectrum imaging system (PerkinElmer) twice per week. For these measurements, the mice were anesthetized and received 150 mg/kg of
Bio-safety assay
Female NOG (NOD.Cg-PrkdcscidIl2rgtm1Sug/JicCrl) immunodeficient mice (Beijing Vital River) at 4–6 weeks of age were inoculated with 5 × 106 conventional or PD-1-deficient CD133-specific CAR T cells in 200 μL of PBS solution by intravenous injection. After inoculation, the animals were checked daily for mobility, food and water consumption, body weight gain/loss, ruffled fur, and any other abnormal effects. Death and any other observed clinical signs were recorded. At days 7 and 30 post inoculation, peripheral blood samples of all mice were collected, and the remaining human T cells were measured by flow cytometry. At study termination, the mice were sacrificed and the liver, spleen, lung, and bones were harvested, fixed in 4% formalin solution, and further analyzed histopathologically by hematoxylin and eosin staining.
Statistical analysis
All the calculations were performed using GraphPad Prism v6.0 (GraphPad Software, Inc.). p-Values of <0.05 was considered statistically significant.
Results
Development of PD-1-deficient CD133-specific CAR T cells
To generate PD-1-deficient CD133-specific CAR T cells, a protocol was developed based on two key points: (1) all the exogenous genes were delivered into T cells by nucleofection with plasmids; and (2) the cells with PD-1 disruption were enriched according to their CAR expression by puromycin selection. The enrichment of PD-1-deficient CD133-specific CAR T cells had been designed according to the following two hypotheses: (1) multiple plasmids could enter one cell simultaneously; and (2) stable transposon-mediated integration of exogenous DNA could be assumed to occur less frequently than CRISPR/Cas9-mediated genome editing. The important elements in the plasmids encoding Cas9 and CAR are shown in Fig. 1A. GFP and c-Myc tag have been included in the constructs for the detection of Cas9 and CAR, respectively, and the puromycin resistance gene was co-expressed with the CAR through a T2A peptide for the selection of CAR-positive cells, as shown in Fig. 1A. The process is summarized in Fig. 1B. Briefly, PBMCs were isolated from blood. The four plasmids (coding for Cas9, the sgRNA, the transposase, and the transposon) were nucleofected into human T cells in one reaction. One day later, transfected T cells were activated by anti-CD3/CD28 beads and expanded in complete medium supplemented with IL-2. One week after activation, puromycin was added to enrich CAR-expressing T cells. On day 14, the T cells were activated again with anti-CD3 antibody and feeder cells, and IL-2 was supplemented to support expansion for another 13 days.
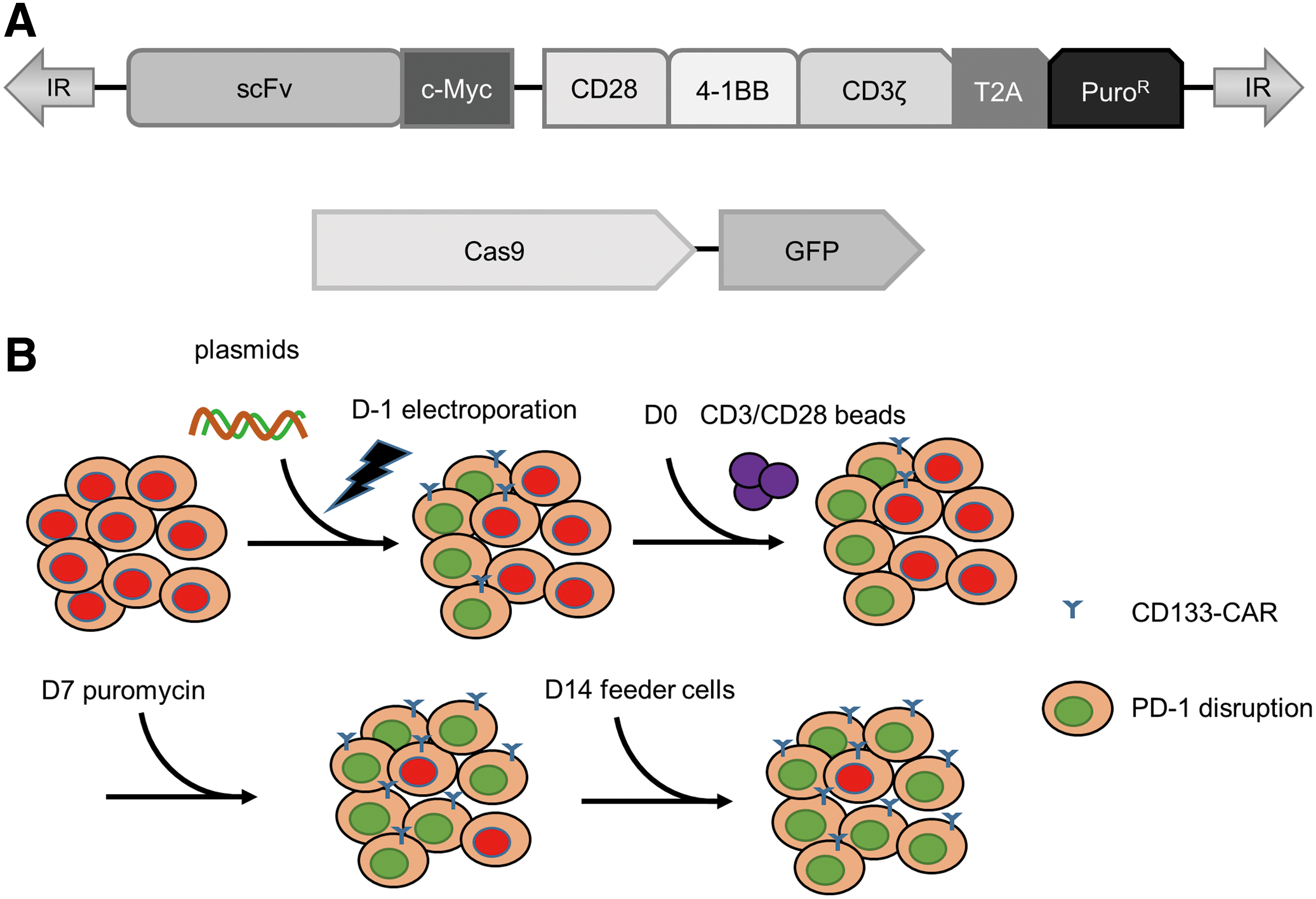
Generation of programmed cell death protein 1 (PD-1)-deficient CD133-specific chimeric antigen receptor (CAR) T cells.
To determine whether the expansion folds of cells could be adequate for future clinical usage, a proliferation assay of cells from three individual donors was performed, and the cell numbers were recorded every 2–3 days. The proliferation rate varied depending on different donors (Fig. 2A). The average fold change of total cell number was 1.00 for the early stage (days 0–7 in the first round of expansion), 1.99 for the 7 days of puromycin selection, and 39.35 for the second round of expansion with puromycin. In total, an average 80-fold increase of cell number was achieved after two rounds of expansion. In addition, a big cell loss before stimulation was observed, as nucleofection was toxic to T cells, and part of cells in PBMC even died without nucleofection (Fig. 2B).
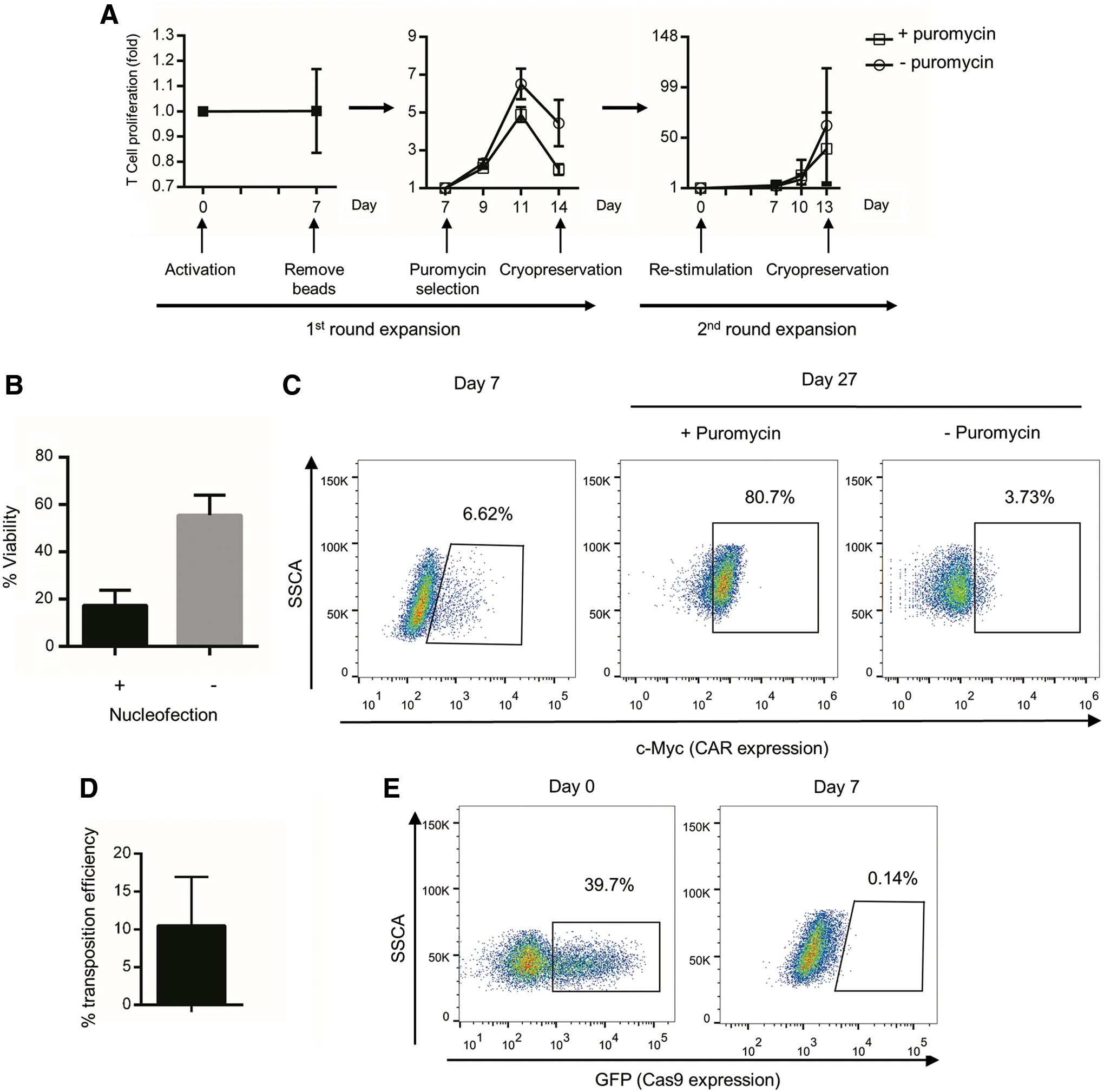
Proliferation and exogenous gene expression of PD-1-deficient CD133-specific CAR T cells.
To assess the expression of the CD133-specific CAR, T cells were analyzed by flow cytometry for expression of the c-Myc tag included in the CAR construct. As shown in Fig. 2C, CAR-positive T cells were enriched by puromycin selection significantly, and a majority of the drug-selected cells expressed the CD133-specific CAR after the second round of expansion. In addition, the transposition efficiency of transposon system we used has been evaluated according to the following formula: percentage of c-Myc positive cells/transient transfection efficiency (shown in Fig. 2D).
To determine whether the genome-edited CAR T cells express the non-human Cas9 protein, the expression of Cas9 was determined by flow cytometry by detecting the GFP co-expressed with Cas9. One day after nucleofection, the transfection efficiency for Cas9 was about 39.7% (Fig. 2E). However, 7 days later, Cas9 expression was no longer detected by flow cytometry. Therefore, a Cas9-induced immune response can probably be avoided when genome-edited CAR T cells are therapeutically used in patients.
To confirm the differentiation state of the products, CD95, CD45RO, and CD62L on the cell surface were checked by antibody staining. All the CAR T cells were CD95 positive, and more than half of them expressed CD62L and CD45RO, a surface phenotype typical of central memory T cells, 21 corresponding to an intermediate state of T-cell differentiation (Fig. 3), which has also been reported in other studies when using allogeneic PBMCs as feeder cells for the T-cell expansion. 22,23

T-cell differentiation marker expression after two rounds of expansion. CAR T cells were stained with anti-CD95
PD-1 knockout efficiency on both the DNA and protein levels
To determine the efficiency of CRISPR/Cas9-mediated genome editing in CD133-specific CAR T cells, first the PD-1 knockout efficiency was analyzed on the genome level. Genomic DNA was extracted from CD133-specific CAR T cells, and the PD-1 gene was amplified by PCR. The PCR products were incubated with the T7EN1 enzyme. As shown in Fig. 4A, the edited PD-1 genes were recognized by T7EN1 and cut to produce more than one smaller fragment. In addition, the PCR products were cloned and sequenced. As shown in Fig. 4B, insertions and deletions were found in 89.5–95% of the PD-1 gene sites, summarized for three individual donors.
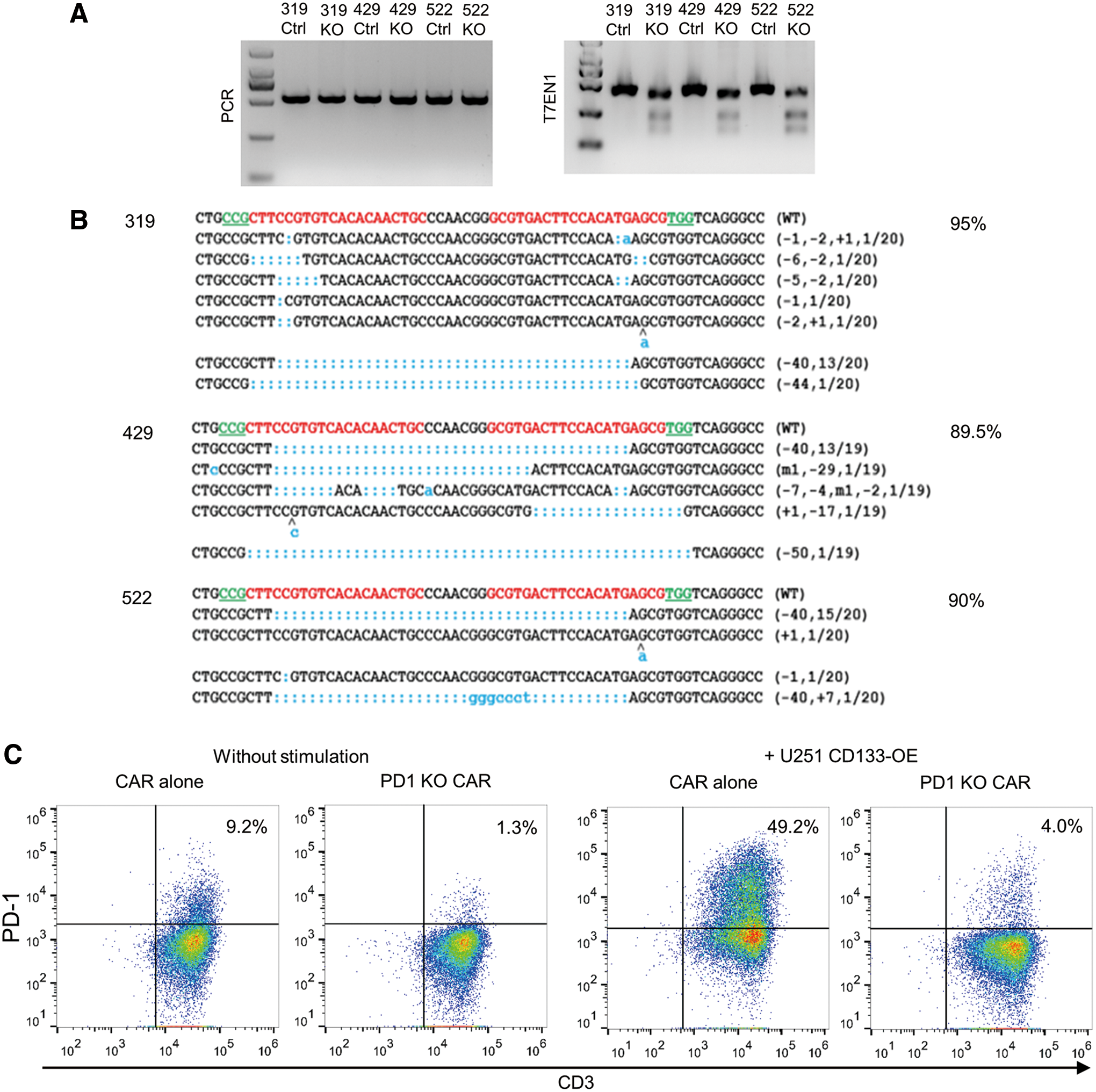
PD-1 knockout efficiency in three individual donors.
To stimulate PD-1 expression on the cell surface, conventional CD133-specific CAR T cells or PD-1-deficient CD133-specific CAR T cells were co-cultured with irradiated U251 CD133-OE cells, and PD-1 expression was thereafter measured by flow cytometry. As shown in Fig. 4C, after 3 days of co-incubation, PD-1 was upregulated on 49.2% of the conventional CD133-specific CAR T cells. In contrast, only 4.0% of the cells were positive for PD-1 in the genome-edited CAR T-cell group.
Effector functions of PD-1-deficient CD133-specific CAR T cells
First, the expression levels of both the CAR target and PD-L1 on the target cells were checked by flow cytometry. U251 CD133-OE cells were found to express both CD133 and PD-L1 highly on their surface (Fig. 5A).
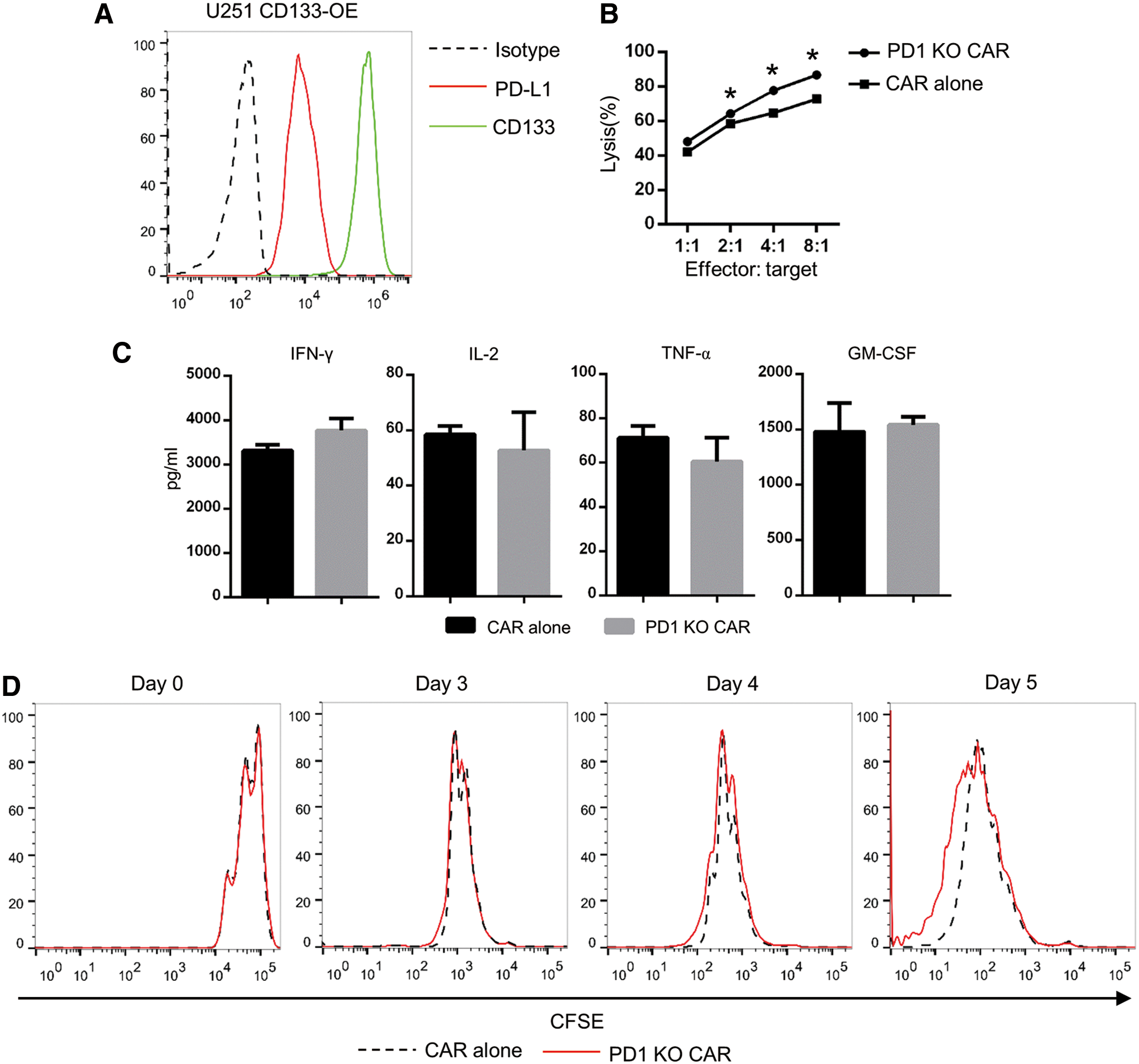
Conventional and PD-1-deficient CD133-specific CAR T cells had comparable antitumor effects in vitro.
To determine the cytolytic capacity of the PD-1-deficient CD133-specific CAR T cells, firefly luciferase-expressing target cells (CD133-overexpressing luc+ U251 glioma cells) were co-cultured with CAR T cells for 24 h. Target cell death and viability were analyzed based on the luciferase signals. As shown in Fig. 5B, significantly stronger lytic capacity was found for the PD-1-deficient CD133-specific CAR T cells when the effector-to-target cell ratios were 8:1, 4:1, or 2:1 compared to conventional CAR T cells.
To analyze cytokine secretion, the T cells were stimulated with the target cells for 24 h. IL-2, IFN-γ, TNF-α, and GM-CSF released into the medium were measured thereafter by AlphaLISA assay. As shown in Fig. 5C, the PD-1-deficient CD133-specific CAR T cells secreted similar amounts of all four cytokines when compared with conventional CAR T cells.
To explore the expansion ability of PD-1-deficient CD133-specific CAR T cells, conventional or PD-1-deficient CD133-specific CAR T cells were co-cultured with irradiated PD-L1+ CD133+ target cells. As shown in Fig. 5D, on days 3 and 4, there was no significant difference in the proliferation rates between conventional and PD-1-deficient CD133-specific CAR T cells. However, on day 5, a proportion of the PD-1-deficient CD133-specific CAR T cells showed a higher proliferation rate than the conventional CD133-specific CAR T cells, as assessed by CFSE dilution.
In vivo antitumor activity
To determine whether the PD-1-deficient CD133-specific CAR T cells enhanced antitumor activity in vivo, an orthotopic glioma model was used in immunodeficient mice, as described previously. CD133-overexpressing U251 glioma cells were orthotopically injected into the brains of immunodeficient mice; 7 days later, non-transfected T cells, conventional CAR T cells, or PD-1-deficient CAR T cells were injected into the tumor sites, followed by two additional rounds of treatment in the next 2 weeks. Both the mice treated with conventional CD133-CAR T cells and those treated with PD-1-deficient CD133-CAR T cells showed prolonged survival. However, the mice treated with PD-1-deficient CD133-CAR T cells had a significantly longer survival time compared to those treated with the conventional CAR T cells (Fig. 6A). Tumor sizes were also assessed based on BLI signals. Using this assay, it was found that both the conventional and the PD-1-deficient CD133-CAR T cells inhibited tumor growth. However, on day 28, the mice treated with PD-1-deficient CD133-CAR T cells showed lower luciferase signals compared to those treated with conventional CAR T cells (Fig. 6B).
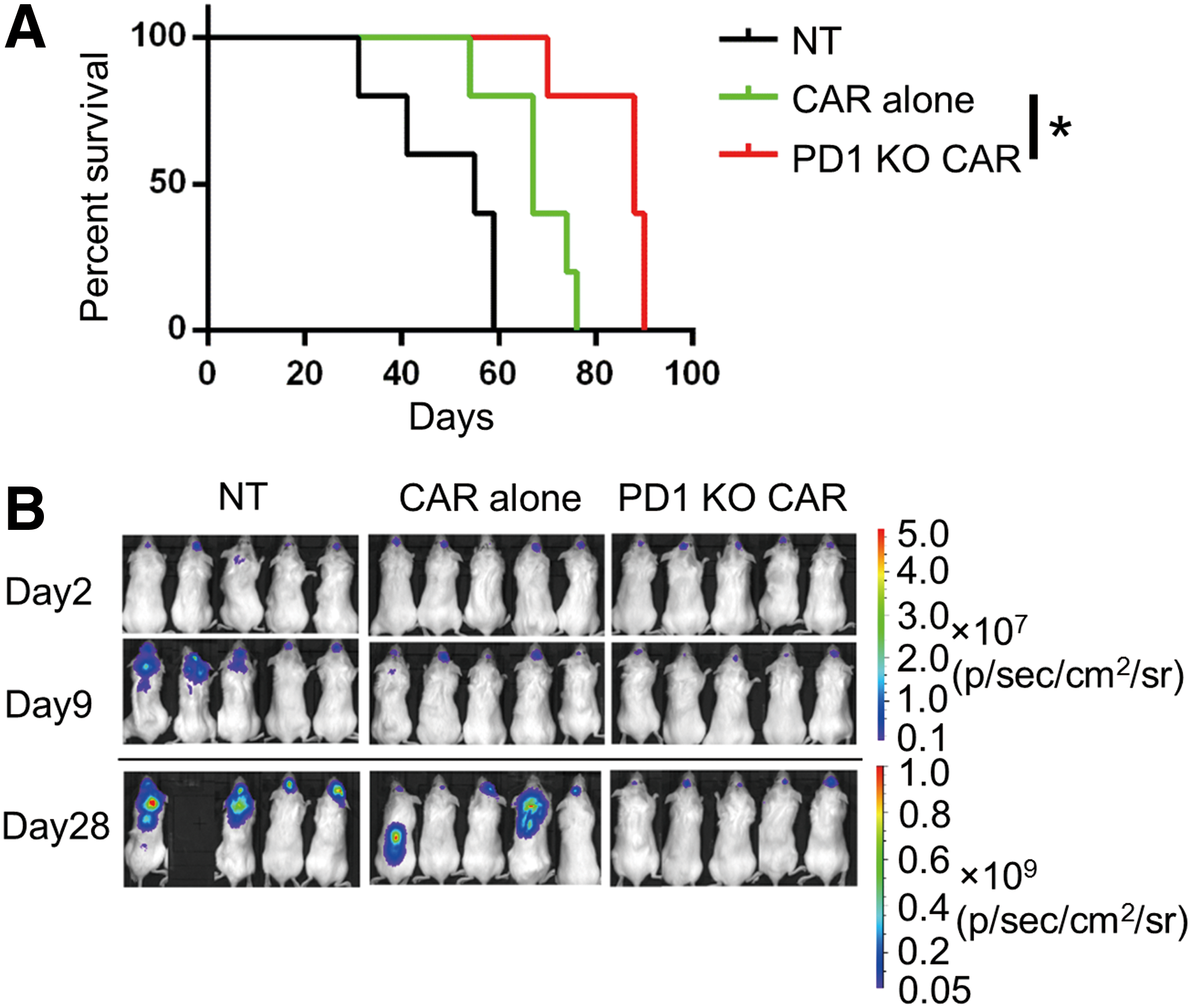
PD-1-deficient CAR T cells showed therapeutic efficacy against established, orthotopic glioma xenografts in immunodeficient mice. Firefly luciferase-expressing U251 CD133-OE cells (2 × 105) were injected into the forebrains of immunodeficient mice. On days 7, 11, and 15 of tumor growth, 2 × 106 CAR T cells or non-transfected T cells were injected via the same coordinates. Mice with >20% weight loss were euthanized.
Safety in immunodeficient mice
To assess the safety of the PD-1-deficient CD133-specific CAR T cells in vivo, potential uncontrolled proliferation, signs of graft-versus-host disease (GvHD), and other potential signs of acute toxicity were investigated upon intravenous injection of conventional or PD-1-deficient CD133-specific CAR T cells into immunodeficient mice. CAR T-cell numbers in the blood were detected by flow cytometry (Fig. 7A). Both conventional and PD-1-deficient CAR T cells were detectable (mean 0.5% and 0.67%, respectively) 1 week after injection, but had almost disappeared (mean 0.02% and 0.04%, respectively) by day 28 (Fig. 7B), demonstrating that there was no excessive proliferation or lymphoma formation due to the deletion of PD-1. Likewise, no GvHD signs or deaths were found. All the mice were euthanatized 28 days after the start of treatment, and the vital organs were collected and subjected to pathology analysis in which no toxicity-related abnormalities were detected (Supplementary Table S1; Supplementary Data are available online at
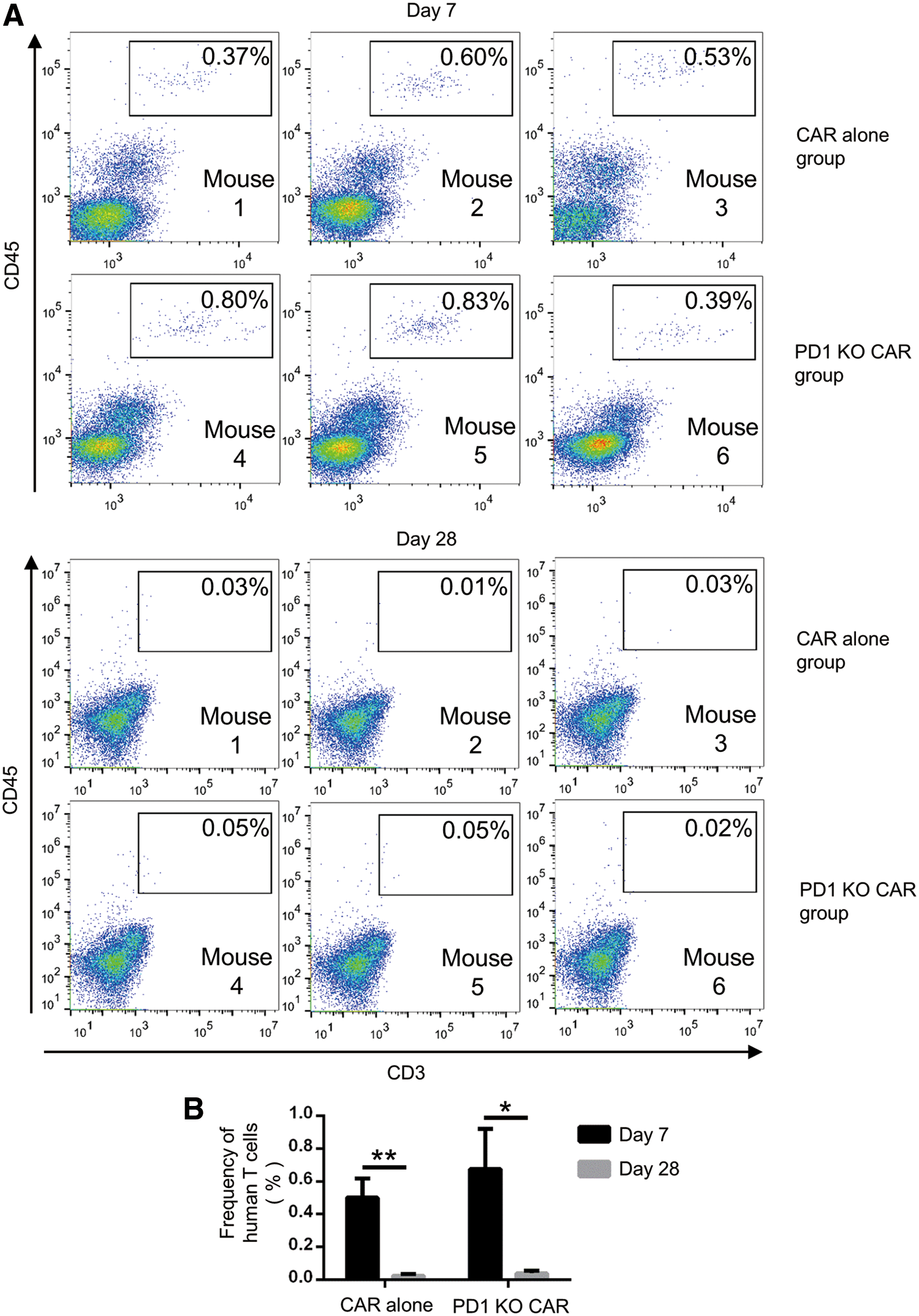
PD-1-deficient CAR T cells did not show uncontrolled proliferation in immunodeficient mice.
Discussion
This study developed a protocol in which plasmids encoding components of both the CRISPR/Cas9 and the piggyBac transposon/transposase system required for the production of genome-edited CAR T cells were nucleofected together into T cells. The CRISPR/Cas9-edited cells were selected based on stable transposon integration indicated by CAR expression. Using this protocol, CD133-specific CAR T cells with PD-1 gene knockdown were generated successfully and showed similar or enhanced in vitro functions and improved tumor inhibition in vivo compared to conventional CAR T cells. The study presents a simple but efficient method for CRISPR/Cas9-mediated genome editing in CAR T cells, and a new type of CAR T cells (i.e., PD-1-deficient CD133-specific CAR T cells), which might be therapeutically useful in treating a wide range of tumor types containing CD133+ cancer stem cells.
In previous studies, in vitro transcribed Cas9 mRNA or Cas9 protein was delivered into T cells to generate PD-1-deficient CD19-specific CAR T cells based on the combination with virus transduction. 10 –12 However, compared to RNA, protein, and virus, plasmid DNA is easier to manufacture at lower cost, especially in Good Manufacturing Practice (GMP) grade. Using DNA as gene vehicle has additional advantages. First, multiple plasmids can be electroporated simultaneously into one cell, and plasmids have sufficiently high capacity to carry multiple elements. Thus, the system will possibly enable editing of multiple genes in CAR T cells, thus generating so-called universal CAR T cells, or CAR T cells in which multiple inhibitory checkpoints are silenced. Second, transient, plasmid-mediated expression of the Cas9 gene does not pose the problem of stable Cas9 gene integration, which may cause immunogenicity when used in humans, as the Cas9 protein is not of human origin. Lastly, in the present protocol, transduction using virus is avoided by employing the piggyBac transposon system. Besides, if a viral system is chosen for delivery of the CAR gene, additional steps for transfection of CRIPSR/Cas9 system need to be performed. In contrast, the current protocol does not need too many additional steps, except mixing the plasmid-encoding CRISPR/Cas9 system together with the encoding transposon system for nucleofection. Thus, the protocol may provide an alternative method for genome-edited CAR T-cell production.
However, DNA electroporation is quite toxic for T cells, causing substantial cell death at the beginning of the manufacturing process. This, combined with the relatively low transgene efficiency of transposon transfection compared to retrovirus transduction, renders the whole manufacture process longer. Some groups working on the generation of clinical-grade CAR T cells using the transposon system have tried to optimize the expansion time (e.g., by using PBMCs or artificial antigen-presenting cells for enrichment or mini-circle plasmids for gene delivery). 18,24,25 Currently, improved electroporation devices are available, and in the future, it may be possible to reduce the toxic effects on human T cells caused by electroporation (e.g., by optimizing the electroporation conditions). Currently, the puromycin resistance gene and antibiotic selection are used for enrichment of CAR-positive T cells. For further development of CAR T cells in the clinical setting, CAR T cells may be enriched by sorting based on CAR expression, using GMP-grade equipment (e.g., CliniMACS Prodigy from Miltenyi). To obtain sufficient cells for adoptive transfer, the expansion rate should be judged. According to a previous clinical report, two batches of IL13Rα2-targeting CAR T-cells were manufactured for the treatment of a patient with recurrent multifocal glioblastoma, and regression of all intracranial and spinal tumors was observed. 3 For the 16 infusions, a total of 9.4 × 107 and 5.0 × 107 cells have been taken from the two batches of products. Considering the current protocol, to obtain 1.0 × 108 PD-1 KO CAR T cells, 1.0 × 107 PBMCs of starting materials will be required, which should not be difficult in most cases, especially when a leukapheresis device is applied to the patient instead of drawing blood directly. It has been reported that an average of 6.7 × 109 PBMCs could be acquired from one leukapheresis product. 26
In principle, PD-1 disruption will give CD133-specific CAR T cells the capacity to resist PD-1/PD-L1-induced immunosuppression. The present data show that PD-1 disruption did improve the in vitro cytotoxicity and the in vivo antitumor activity in immunodeficient mice. In addition, the study found no uncontrolled proliferation of PD-1-deficient CAR T cells or evident toxicity induced by them in immunodeficient mice. These data support the evaluation of PD-1-deficient CD133-specific CAR T cells for safety and clinical efficacy within clinical trials. Moreover, the data more generally advocate the evaluation of genome-edited CAR T cells such as PD-1-deficient CAR T cells for clinical use in the therapy of cancer patients.
How PD-1-deficient CD133-specific CAR T cells contribute to better antitumor activity in vivo is an important question and requires further studies. The authors and other groups have analyzed the in vitro functions of genome-edited T cells, including cytotoxicity, cytokine secretion, and proliferation. 11,12 Liu et al. found significant improvement in cytokine secretion for PD-1 knockout CD19-CAR T cells. 11 Rupp et al. observed enhanced cytotoxicity of PD-1 knockout CD19-CAR T cells. 12 In this study, only enhanced cytotoxicity and proliferation were found for PD-1-deficient CD133-specific CAR T cells; cytokine secretion was similar to that of conventional CAR T cells. The clear molecular and cellular effects caused by PD-1 gene disruption in CAR T cells need further exploration in the future. In addition, CARs with different intracellular signaling domains may have different abilities to resist immunosuppression. Cherkassky et al. showed that a 4-1BB CAR worked better in the immunosuppressive tumor microenvironment than a CD28 CAR. 27 In this study, a third-generation CAR was gene-edited that includes both CD28 and 4-1BB signaling domains. How different types of CAR-associated signaling domains are coordinated with the PD-1 signaling pathway will also require future studies.
Independent of CAR transduction or transfection, Su et al. observed enhanced cytokine secretion and cytotoxicity following the disruption of PD-1 in the primary T cells of patients. 19 In contrast, a study on zinc finger nuclease-mediated gene editing of PD-1 in tumor-infiltrating lymphocytes (TILs) reported a significantly increased polyfunctional cytokine profile (TNF-α, GM-CSF, and IFN-γ) but no enhanced cytotoxicity in the PD-1 genome-edited TILs. 28
In this study, simple models were used, including co-culture of CAR T cells with tumor cells in vitro and treatment of tumor-bearing immunodeficient mice with human CAR T cells, as used before. 14,20,29 Although a CD133 highly expressed U251 cell line cannot be a very suitable model to represent the patient situation, it was used to judge the CAR T cells' capability of killing tumors on a quantified level. Furthermore, though the tumor cells used expressed high levels of PD-L1, it is not clear whether the PD-1/PD-L1 pathway is the main negative regulator of CAR T cells in the models. Other immunosuppressive elements that play roles in the regulation of T-cell-mediated immune responses are not considered in the models. In the future, humanized mice, which rebuild the human immune system, may be used to study the impact of genome-edited CAR T cells.
In conclusion, a simple protocol is provided for CRISPR/Cas9-mediated genome editing in CAR T cells based on electroporation of DNA. The PD-1-deficient CD133-specific CAR T cells showed efficient antitumor function. Further research on the clinical performance and the mechanism of action of this new type of CAR T cells in the treatment of solid tumors is warranted.
Footnotes
Acknowledgments
We would like to thank the Cell Sorting Platform in SIAIS for the technical assistance. This work was supported by the National Key R&D Program (2016YFC0905901) and the local grant (2017IB020) from Yunnan Provincial Science and Technology Department.
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.