
Abstract
Hepatic stellate cells (HSCs) are the primary cell type responsible for liver fibrogenesis. Transforming growth factor beta 1 (TGF-β1) and platelet-derived growth factor (PDGF) are key profibrotic cytokines that regulate HSC activation and proliferation with functional convergence. Dual RNA interference against their receptors may achieve therapeutic effects. A novel RNAi strategy based on HSC-specific GFAP promoter-driven and lentiviral-expressed artificial microRNAs (amiRNAs) was devised that consists of an microRNA-30a backbone and effective shRNAs against mouse Pdgfrβ and Tgfbr2. Then, its antifibrotic efficacy was tested in primary and cultured HSCs and in mice affected with carbon tetrachloride–induced hepatic fibrosis. The study shows that amiRNA-mediated Pdgfrβ and Tgfbr2 co-silencing inhibits HSC activation and proliferation. After recombinant lentiviral particles were delivered into the liver via tail-vein injection, therapeutic amiRNAs were preferentially expressed in HSCs and efficiently co-knocked down in situ Tgfbr2 and Pdgfrβ expression, which correlates with downregulated expression of target or effector genes of their signaling, which include Pai-1, P70S6K, and D-cyclins. amiRNA-based HSC-specific co-silencing of Tgfbr2 and Pdgfrβ significantly suppressed hepatic expression of fibrotic markers α-Sma and Col1a1, extracellular matrix regulators Mmps and Timp1, and phenotypically ameliorated liver fibrosis, as indicated by reductions in serum alanine aminotransferase activity, collagen deposition, and α-Sma-positive staining. The findings provide proof of concept for the use of amiRNA-mediated co-silencing of two profibrogenic pathways in liver fibrosis treatment and highlight the therapeutic potential of concatenated amiRNAs for gene therapy.
Introduction
H
TGF-β1 is a well defined profibrogenic cytokine that promotes HSC activation. TGF-β1 activates HSCs through the formation of a complex on plasma membranes with type II (TGFβR2 or TβRII) and type I (TβRI) serine/threonine kinase receptors, which subsequently recruits and phosphorylates the mothers against decapentaplegic homologs (SMADs). 7 The active SMADs complex then translocates into the nucleus and mediates transcription of downstream target genes that are involved in regulation of HSC transdifferentiation and apoptosis, ECM protein production, and degradation. When bound by TGF-β1, TβRII is activated and trans-phosphorylates the type I receptor and activates its kinase, therefore acting as the initiator and key component of the canonical TGF-β/SMAD signaling pathway. 7 Following activation, the proliferation of activated HSCs is modulated by several growth factors and cytokines, including PDGF, the most potent mitogen for activated HSCs. PDGF is composed of four polypeptide chains (A, B, C, and D). PDGF-A and -B are secreted as homodimers or heterodimers and are involved in autocrine and paracrine stimulation of enhanced mitogenicity and increased ECM production of activated HSCs through the PDGF receptor, the tyrosine kinase of homodimer, or heterodimer of α and/or β subunits.
As the most potent mitogenic cytokine of activated HSCs, PDGF-BB mainly binds to PDGFR-β, which activates the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) and phosphatidylinostital 3-kinase (PI3K)/Akt/P70S6K signaling pathways to promote HSC proliferation. 8 The TGF-β1 and PDGF signaling pathways are among key networks that regulate the gene expression signature of HSCs and parallel-enhanced expression of their ligand/receptor pairs, TGF-β1/TβRII and PDGF/PDGFR-β, in activated HSCs and in the fibrotic liver, as PDGF and PDGFR-β expression correlates with necrotic inflammation and fibrosis in fibrotic liver and poor prognosis of cirrhosis. 9 –13 Crosstalk between TGF-β1 and PDGF pathways and their reciprocal modulation are also implicated in liver fibrogenesis. TGF-β1 could enhance the mitogenic effect of PDGF-BB by transactivating the expression of PDGF-B and PDGFR-β genes via SMAD in activated HSCs. 14 –16 However, PDGF upregulates the expression of TGF-β1 and its receptors in mesenchymal cells, including HSCs, and the PDGFR-β tyrosine kinase inhibitor can downregulate the expression of both TβRI and TβRII in HSCs. 17,18
Targeted therapeutic strategies against experimental liver fibrosis with a humanized antibody, dominant-negative soluble receptor, and compound inhibitor have been used to antagonize receptor ligand interactions and/or intracellular components of TGF-β1 and PDGF pathways. 5,6 Combinational antifibrotic treatment with different therapeutic targets has been reported to be feasible and efficacious. For example, treatment with a tyrosine kinase inhibitor (TKI) nilotinib or PTK/ZK attenuates liver fibrosis through multiple mechanisms, including TGF-β1 and PDGF pathway inhibition. 18,19 Dual-pathway blockade with PDGFR-β inhibitor imatinib and TβRI inhibitor galunisertib ameliorates radiation-induced pulmonary fibrosis. 20 However, most TKIs have poor specific selectivity and cause systematic adverse effects, including severe hepatotoxicity, which hinders their application for the management of hepatic fibrosis. 21
RNA interference (RNAi) targeting the TGF-β1 and PDGF pathways has been shown to be a useful strategy for treating liver fibrosis. Small interfering RNA (siRNA) or short hairpin RNA (shRNA)-based RNAi strategies that target specific individual components of the TGF-β1 and PDGF pathways, such as TGF-β1, TβRII, and their downstream effectors, as well as PDGFR-β, attenuates the fibrotic phenotype both in HSCs and in rodent models. 22 –27 However, studies have demonstrated that virally overexpressed shRNA in the liver can trigger cytotoxicity, leading to organ failure and even death, in experimental animals. 28 –30
Since exogenous shRNAs and endogenous microRNAs share common RNAi machinery, sustained high-level shRNA expression may cause saturation of key components of RNAi machinery. These include nuclear karyopherin exportin-5 (Xpo-5), a limiting factor in the microRNA (miRNA) pathway that mediates the nuclear export and stabilization of shRNA/miRNA, and Argonaute 2 (Ago-2), the rate-limiting determinant of RNAi efficacy, toxicity, as well as persistence that acts as the catalytic engine for mammalian RNAi, resulting in downregulation of tissue-derived miRNA that impedes normal cellular function and is thereby associated with morbidity. 28,29,31 –33 The risk of overloading an endogenous small RNA pathway can be minimized by optimizing shRNA sequence and dose. A shRNA-based gene therapy study for Huntington's disease (HD) in mice reported that both the active shRNA against the HD gene and the control shRNA could induce neurotoxicity in the brain. However, shRNA-mediated neurotoxicity was significantly attenuated without compromising HD gene silencing efficacy when the active sequences that were toxic in the context of shRNAs were placed into artificial microRNA (amiRNA) expression system, a novel promising RNAi technology increasingly used for targeted gene silencing therapy. 34
The basic principle of the amiRNA approach is to replace the sequence of mature miRNA in its pre-miRNA stem-loop with the designed shRNA that targets a specific gene of interest, which permits the artificial hairpin embedded in the natural miRNA backbone to be processed into effective miRNA using the same cellular miRNA biogenesis machinery as natural miRNAs. 35 The flanking regions derived from human miRNAs, such as miR-30a, miR-31, and miR-223, have been widely used as scaffold for the incorporation of recombinant shRNAs, among which the stem-loop backbone of human miR-30a is the most widely utilized for amiRNA vector construction. 36 The natural miR-30a itself is expressed in HSCs and could protect against liver fibrosis by attenuating TGF-β1 signaling. 37 AmiRNA strategy presents several advantages over traditional siRNA- or shRNA-based RNAi approaches, as it is highly efficacious, easier to control, and has fewer off-target side effects.
The siRNA method can only generate transient and short-term gene silence effects. The intracellular half-life of delivered siRNA is relatively short due to the degradation by RNase A-like nuclease, and their concentration drops upon cell division; it may also be difficult to deliver siRNAs efficiently to specific target cells, especially in primary cell types. 38,39 While shRNA-based gene therapy often causes tissue toxicity due to overloading the RNAi machinery, amiRNAs as siRNA shuttles could provide improved safety and may be better suited for gene therapy. 34,40,41 AmiRNA shuttles may provide superior silencing by simulating natural miRNA machinery more closely. Additionally, amiRNA processing by Drosha, which is bypassed by shRNAs, may improve entry into the RNAi pathway. 42
Recent studies have shown that Dicer is imprecise in processing commonly used shRNA stem-loop designs, which increases the risk of aberrant guide- and passenger-strand mediated off-target effects. However, the natural loop configuration of amiRNA complies with the “loop-counting” rule that ensures precise Dicer cleavage. 43 Promoters used in shRNA interference plasmids mainly consist of RNA polymerase III (Pol-III) response promoters without tissue specificity. However, amiRNA constructs may be under the control of Pol-III or Pol-II response promoters and therefore may be more amenable to Pol-II-mediated transcription than shRNAs that have limited spacing flexibility for Pol-II-based expression. 44,45 This advantage could allow for the use of an miRNA backbone combined with Pol-II (but not the Pol-III promoter) to provide an effective and safe dose of active miRNA. 46 It also allows for cellular- and tissue-targeted expression of inhibitory RNAs using cell-specific promoters. 36 The glial fibrillary acidic protein (GFAP) is a HSC marker with high and specific expression, the GFAP promoter could efficiently drive targeted expression of exogenous genes in HSCs. 23,47,48
The amiRNA strategy is advantageous for point-mutated oncogenes and diseases caused by point mutation gain of function, as designed amiRNAs could specifically target point-mutated mRNA while sparing wild-type counterparts. 49 Several natural miRNAs are grouped to form clusters in the genome. As such, the generation of mature miRNAs could be achieved through generation of a single polycistronic transcript from which all other miRNAs could be naturally processed. The multi hairpin of concatenated amiRNAs approach was recently developed to improve the knockdown of a single target or to silence either two or three different mRNA targets. 42,50 –52 This strategy is particularly useful for improving silencing efficacy toward single or combined targets and for overcoming attenuation of knockdown caused by target mutations.
This study proposed an RNAi strategy based on amiRNA with multiple targets as a novel gene therapy method for the treatment of hepatic fibrosis. Lentiviral-expressed concatenated amiRNAs were designed against mouse TβrII and Pdgfrβ genes under the control of a HSC-specific GFAP promoter. Then, the antifibrogenic effects of the dual-gene silencing by amiRNA was investigated in vitro and in vivo in well established models for HSC transdifferentiation and liver fibrosis.
Methods
Cells and animals
The murine HSC line JS1 was provided by the Division of Digestive Diseases at the Zhongshan Hospital of Fudan University. Human embryonic kidney 293T cells were purchased from the Cell Bank of Shanghai Institutes of Life Sciences at the Chinese Academy of Science. All cells were cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone) with 10% fetal bovine serum (FBS; Nelso Biotech). The SPF grade BALB/c mice and Sprague–Dawley (SD) rats were obtained from the Shanghai Laboratory Animal Center of Chinese Academy of Sciences Co. Ltd. and were housed and maintained in the Laboratory Animal Center at the Shanghai Medical College of Fudan University. The Animal Ethics Committee of Shanghai Medical College Fudan University approved the protocols for animal experiments (permit no. 20130227-068). All animals received human care.
shRNAs
Interfering shRNAs sequences targeting murine Pdgfrβ or Tgfbr2 genes were designed using the Sigma–Aldrich shRNAs database and were synthetized by the Shanghai HuaGene Biotech Company. The targeting sequences for Pdgfrβ are: 5′-GTGGACTCCGATACTTACT-3′ (shP1), 5′-GTACGGTGGTGTGTTCATATC-3′ (shP2), and 5′-GTGAGAGGAAGCGTATCTATA-3′ (shP3). The targeting sequences for Tgfbr2 are: 5′-TCGGCAGCTGTACATTGACTTT3′ (shT1), 5′-CCAGATCGTGTGTGAGACTTT-3′ (shT2), and 5′-CTCAGGAAATGAGATTGATTT-3′ (shT3). The two unrelated negative control shRNAs (shNC1, 5′- CCTAAGGTTAAGTCGCCCTCG-3′; shNC2, 5′-TTCTCCGAACGTGTCACGT-3′) do not target any genes and are commonly used in RNAi studies. They were provided by the Shanghai HuaGene Biotech Company.
The shRNAs were inserted into retroviral vector pMOK.1-puro, and positive clones were confirmed by sequencing. The retroviral particles containing shRNAs against Pdgfrβ or Tgfbr2 and shNC were packaged into 293T cells and transfected into JS1 cells, respectively. The stably transfected JS1 cells were screened by neomycin resistance analysis, and the interfering efficiency was measured by analyzing Pdgfrβ and TβrII mRNA and protein expression.
Construction of pLenti-amiRNAs vectors for Pdgfrβ and TβrII silencing
amiRNAs were designed for Pdgfrβ and Tgfbr2 co-silencing. Their components included the structural backbone of miR30-shRNA, as reported previously, and two concatenated pre-miR30-shRNA cassettes embedding respective valid interfering shRNAs (amiR-PT) or negative control shRNAs (amiR-NC) as cargos. 23,44,53 Their positioning was not taken into consideration, as it was previously reported that the relative position of an amiRNA in the multi-amiRNA transcript does not affect its individual RNAi activity. 54 The miR30 backbone was chosen as the shRNA loading vehicle, since processing the larger pre-miRNA transcript to its functional miRNA has been well characterized in vitro and in vivo, and human miR30 is now the most widely utilized for amiRNA vector construction. 36,55 These full-length amiRNAs, amiR-PT1 (shP1 and shT2), amiR-PT2 (shP2 and shT3), and the negative control amiR-NC (shNC1 and shNC2) were synthetized by the Shanghai HuaGene Biotech Company. Their full-length sequences are shown in Supplementary Table S1.
The secondary structure of these amiRNAs was predicted using the RNAstructure software. 56 The lentiviral microRNA expression vector pLenti-CMV-microRNA-EGFP-T2A-Puro was purchased from the Obio Technology Co. Ltd. The human GFAP gene promoter was used as the driver for HSC-specific expression of amiR-PTs. The 5′-flanking region of GFAP gene (−2216 to +45 relative to the transcriptional start site) was amplified according to a previously published method, and the original CMV promoter of the vector was replaced with the GFAP promoter between the MluI and PstI cleavage sites. 48
Since the majority of human miRNA loci are located within intronic regions and are transcribed by Pol-II as part of their hosting transcription units, the active amiR-PT and the negative control amiR-NC were inserted into the intronic cloning site with cleavage of NdeI and BamHI of the enhanced green fluorescent protein (EGFP) gene, which is closely downstream of human GFAP promoter. 57 The EGFP fluorescent reporter enables precise tracking of lentiviral transduction and amiRNA expression and allows analysis or purification of only those cells that productively express the amiRNA. The positive clones were confirmed by direct sequencing. Then, the lentiviral amiRNA expression vectors containing functional components of GFAP promoter, artificial microRNA30-sh-Pdgfrb-sh-Tgfbr2, and EGFP-puro were constructed and named as pLenti-amiR-PT1, pLenti-amiR-PT2, and pLenti-amiR-NC, respectively. The new lentiviral amiRNA expression vectors and packaging vectors were co-transfected into 293T cells with Lipofectamine 2000 (Invitrogen). The culture supernatants were collected, concentrated, and used as a virus stock. The QuickTiter kit (Cell Biolabs) was used to measure the titer of viral particles according to the manufacturer's instructions.
Quantitative reverse transcription polymerase chain reaction
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was used to quantify the transcriptional expression of coding genes for Pdgfrβ and Tgfbr2, their signaling downstream targets or effectors (Smad7, Pai-1, C-myc, Pi3k, P70s6k, Cyclind1, and Cyclind3), fibrotic markers (α-Sma, Col1a1, Timp1, Mmp2, Mmp11, and Mmp13), and β-actin as inner control in cultured cells or liver tissues. Total RNA was extracted using TRIzol reagent (Invitrogen; 15596-026) and subjected to reverse transcription using the PrimeScript RT reagent Kit (Takara; RR037A) and relative quantitative PCRs using the SYBR Premix Ex Tap kit (Takara; DRR420A). The ΔΔCt method was applied for relatively quantitative analysis of gene expression, which was performed in triplicate for each sample, and statistical analysis was carried out based on three independent experiments.
To detect quantitatively expression levels quantitatively of primary transcripts of the transduced amiRNAs (amiR-PT1, amiR-PT2, and amiR-NC) in transfected cells and transgenic mice, using the same reversely transcribed total RNA samples as above, qPCR was also performed to measure the relative expression of the primary transcript of the amiRNAs with the same pair of primers that are located at their shared miR-30a backbone. The sequences of PCR primers are listed in Supplementary Table S2.
Western blot
Western blot was used to measure protein expression using primary antibodies against Col1a (Santa Cruz Biotech; sc-59772), Pdgfrβ (Abcam; ab32570), TβrII (Abcam; ab186838), α-Sma (Abcam; ab5694), and β-Actin (Abmart; M20010). Goat anti-rabbit and goat anti-mouse immunoglobulin Gs (IgGs; Jackson Immuno Research Company; FZ111-165-003 and FZ115-165-003) were used as secondary antibodies. The gray level of protein bands was measured with Image J software, and protein bands relative to the inner control β-Actin were used to analyze expression. Protein expression analysis was performed in triplicate for each sample, and statistical analysis was carried out based on three independent experiments.
Cell proliferation analysis
The murine HSC line JS1 with Pdgfrβ and TβrII co-silencing and control were achieved by stable transfection with amiR-PTs or amiR-NC lentivirus and neomycin resistance screening, and validated by analysis of mRNAs and protein of Pdgfrβ and TβrII. Cells were inoculated onto a 96-well plate. Each well contained 2,000 cells, and 10 repeats were used for every treatment. Over a continuous 5-day period, cellular proliferation was detected using a cell counting kit-8 (CCK-8; Dojindo; CK04-3000T). The effect on HSC proliferation of Pdgfrβ and TβrII co-silencing was evaluated by analyzing cell growth curves according to the daily absorbance of cells (OD450).
Isolation and purification of rat and mouse primary HSCs
To investigate the effect of Pdgfrβ and TβrII co-silencing on HSC activation, primary HSCs of adult normal male SD rats (200–250 g) were isolated and purified using an improved method of in situ pronase–collagenase perfusion followed by density gradient centrifugation according to the manufacturer's protocol. 58 The pre-perfusion liquid (D-Hanks liquid free of Ca2+ and Mg2+) flowed into liver tissue via continuous portal vein pumping and flowed out from the inferior vena cave. When the blood in liver tissue was washed clean, perfusion in situ of pronase (Sigma–Aldrich; P8811; 0.4 mg/mL) and collagenase (Invitrogen; 17104019; 0.4 mg/mL) solution was carried out successively at 37°C for 5 min. Then, the liver was excised, dispersed, and incubated in digest solution (containing 0.2 mg/mL pronase and 2 mg/mL DNaseI) at 37°C for 5 min with gentle stirring. Adding cooled DMEM (containing 10% FBS) stopped the digestion, and single-cell suspension from liver tissue was then prepared by filtering through a mesh.
After several washings with DMEM, the cell pellet was re-suspended with 5 mL of 15% OptiPrep (Axis-shield). The upside of the cellular layer was carefully loaded with 5 mL of 11.5% OptiPrep. Then, 2 mL of DMEM was added on the upper layer. Cellular suspension was subjected to density gradient centrifugation at 1,400 g for 17 min at 4°C. The cellular fraction located in the middle of DMEM and 11.5% OptiPrep were HSCs, and their viability was determined using the Trypan blue exclusion test. Lipid/retinoid droplets of cells were stained with Nile red reagent (GMS12196; Genmed Scientific) and diluted with PBS for 10 min at 37°C under dark conditions. After staining, cells were observed under a fluorescence microscope. Mouse primary HSCs were also prepared to validate the cellular effect of dual-gene silencing. The remaining mouse liver tissue was homogenized and incubated in digest solution, followed by density gradient centrifugation as described above. Primary mouse HSCs were then collected and subjected to Western blot analysis.
amiRNA-mediated Pdgfrβ and TβrII co-silencing in a murine liver fibrotic model induced with carbon tetrachloride
Eighteen normal male BALB/c mice (4 weeks old; ∼16 g each) were randomly divided into three groups: (1) Pdgfrβ and TβrII dual-knockdown mice infected with pLenti-amiR-PT1 lentiviral particles (amiR-PT1; n = 7), (2) the negative control group infected with pLenti-amiR-NC lentiviral particles (amiR-NC; n = 7); (3) and the normal control mice without carbon tetrachloride (CCl4) administration and amiRNA treatment (normal; n = 4). Lentiviral particles were transfected into mouse livers at a titer of 2 × 107 TU each via tail-vein injection every 5 days. In addition, liver fibrosis was induced via intraperitoneal injection of CCl4 (diluted with olive oil 1:4) at 5 mL/kg body weight and 2.5 mL/kg for subsequent injections every 3 days. Normal control mice received olive oil and 0.9% NaCl injections. After 4 weeks of treatment, animals were humanely killed. Serum was prepared from a centrifuge of collected blood samples at 3,000 g for 10 min at 4°C. Then, serum alanine aminotransferase (ALT) activity was measure using a biochemistry analyzer.
Three blocks of liver tissue samples (0.5 cm thick) were separated from the center section of left lobe of the liver. One block was fixed in 4% paraformaldehyde solution for the paraffin section, the second was frozen rapidly in the liquid nitrogen for the frozen section, and the third was used for total RNA extraction. The reaming liver tissue was homogenized for primary HSC preparation. The independent and parallel gene therapy experiments used the same protocol. However, for the second amiRNA, pLenti-amiR-PT2, 14 normal male mice were randomly divided into three groups as follows: (1) treated mice with Pdgfrβ and TβrII co-silencing via transduction of pLenti-amiR-PT1 (amiR-PT2; n = 4), (2) treated mice with transduction of the negative control pLenti-amiR-NC (amiR-NC; n = 4), and (3) the normal untreated control mice (normal; n = 4).
Masson's trichrome staining and immunohistochemistry
Mouse live tissue was separated from the middle section of the left liver lobe, fixed in 5% paraformaldehyde for approximately 2 days, and embedded into paraffin blocks. The liver tissue slides (5 μm thick) were stained using Masson's trichrome staining kit (Sigma–Aldrich; HT15-1KT) for collagen or subjected to immunohistochemistry for α-Sma staining according to standard protocols. Collagen deposition was evaluated by adding the total blue areas of one field (200 × Masson's trichrome staining picture) calculated using Image-Pro Plus software. This program was also used to measure integrated optical density (IOD) of α-Sma-positive areas. The final liver fibrosis level of one sample was determined as the mean of six random different fields for each section.
Immunofluorescence assay
Liver tissue frozen sections were fixed in 5% paraformaldehyde for 30 min at 37°C, followed by blocking, rinse, incubation with primary antibodies against Pdgfrβ and TβrII and DAPI (Beyotime; C1002), and incubation with fluorescence-labeled secondary antibody (Cy3-labeled goat anti-rabbit IgG; Beyotime; A0516) according to standard protocols. The sections were observed under fluorescence microscope, and pictures were taken at 200 × and 600 × magnification.
Statistical analysis
Data are presented as means ± standard deviation of at least three independent experiments for every assay. The statistical significance was analyzed using Student's t-test for independent samples. p-Values of <0.05 were considered of statistical significance.
Results
Construction of amiRNA vectors for Pdgfrβ and TβrII co-silencing
To knockdown the expression of Pdgfrβ and TβrII in HSCs, six (shP1, shP2, shP3, shT1, shT2, and shT3) shRNAs were designed that specifically target Pdgfrβ and TβrII genes. They were inserted into retroviral vectors packaged in 293T cells, and integrated into the murine HSCs (JS1) through stable transfection. The interference efficiency of shRNAs in JS1 cells is shown in Fig. 1. Interference with each of the three shRNAs for Pdgfrβ all resulted in significant reduction in its transcript and protein levels. Two of the three shRNAs for TβrII (shT2 and shT3) remarkably reduced the expression of its mRNA and protein, suggesting that these designed shRNAs can efficiently knock down target gene expression in transfected HSCs. Based on these valid interfering shRNAs for Pdgfrβ or TβrII, three full-length amiRNAs that contain the structural backbone of miR30-shRNA and two functional concatenated pre-miR30-shRNA cassettes embedding respective valid-interfering shRNAs or negative control shRNAs as cargos were synthesized as follows: two therapeutic amiRNAs, amiR-PT1 (shP1 and shT2), amiR-PT2 (shP2 and shT3), and one negative control amiRNA, amiR-NC (shNC1 and shNC2). 23,36,53 The secondary structures of these amiRNAs were predicted using RNA structure software, and it was found that they are of a very similar stem-loop layout (Fig. 2A).

Screening valid short hairpin RNAs (shRNAs) for Pdgfrβ and TβrII knockdown. Three shRNAs (shP1, shP2, and shP3) against Pdgfrβ, three shRNAs (shT1, shT2, and shT3) against TβrII, and one scramble shRNA as a negative control (shNC) were stably transfected into JS1 cells through retrovirus vectors. Total RNA and protein were prepared from stably transfected JS1 cells. Differential expression analysis was performed between the cells with sh-Pdgfrβ or sh-TβrII and sh-NC.
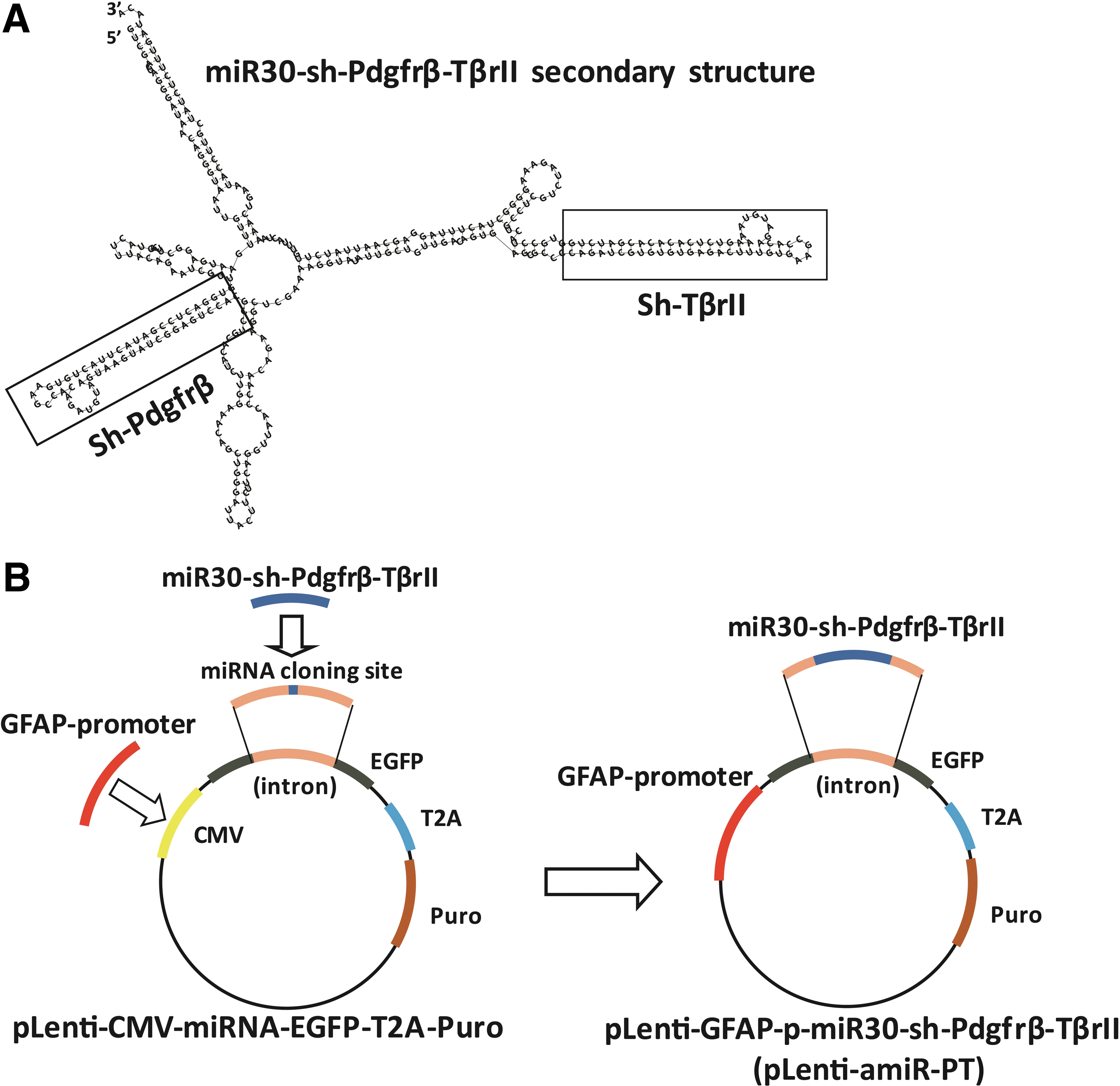
Construction of amiRNA vectors for Pdgfrβ and TβrII co-silencing. Artificial microRNA-shRNAs for Pdgfrβ and Tgfbr2 co-silencing consisted of an miR-30a backbone as the structural scaffold and two concatenated pre-miR30-shRNA cassettes embedding respective valid interfering shRNAs (amiR-PT) or the negative control shRNAs (amiR-NC) as cargos. The secondary structure of amiR-PT1 for Pdgfrβ and Tgfbr2 co-silencing, miR30-sh-Pdgfrβ-sh-TβrII, was drawn with an RNA structure.
The lentiviral microRNA expression vector pLenti-CMV-microRNA-EGFP-T2A-Puro was modified by replacing the original CMV promoter with human GFAP gene promoter, and then the synthesized amiRNAs were inserted into the cloning sites located in the intron of the EGFP gene and closely behind the GFAP promoter, such that the amiRNAs expression could be driven by the human GFAP gene promoter and demonstrated by the expression of the EGFP marker gene in the vector (Fig. 2B). These lentiviral GFAP promoter-driven amiRNA vectors, pLenti-amiR-PT1, pLenti-amiR-PT2, and pLenti-amiR-NC, were then used to co-knock down Pdgfrβ and TβrII in vitro and in vivo.
amiRNA-mediated Pdgfrβ and TβrII co-silencing suppresses HSC activation and proliferation
TGF-β plays important roles in HSC activation and ECM production, and PDGF is the strongest stimulator of HSC proliferation. Therefore, the direct effects of amiRNA silencing of their receptors on these cellular phenotypes were analyzed in both cultured and primary HSCs. In the JS1 cell lines stably transfected with pLenti-amiRNA, a substantial increase in transcriptional expression levels of transfected amiRNAs was observed compared to the control JS1 cells, especially for amiR-PT1 and amiR-PT2, indicating that the human GFAP promoter could efficiently drive the ecotopic expression of amiRNA in HSCs (Fig. 3A). A significant reduction in both mRNA and protein expression of Pdgfrβ and TβrII was also found in JS1 cells transfected with pLenti-amiR-PT1 or pLenti-amiR-PT2 compared to those transfected with pLenti-amiR-NC (Fig. 3A and B). Co-knockdown of Pdgfrβ and TβrII in JS1 cells with pLenti-amiR-PT1 or pLenti-amiR-PT2 suppressed HSC activation, as illustrated by the substantial reduction in both mRNA and protein expression of the fibrotic markers α-Sma and Col1a1 (Fig. 3A and B). Proliferation analyses of JS1 cells showed that growth rates for cells stably transfected with pLenti-amiR-PT1 or pLenti-amiR-PT2 were significantly lower compared to the control (Fig. 3C).
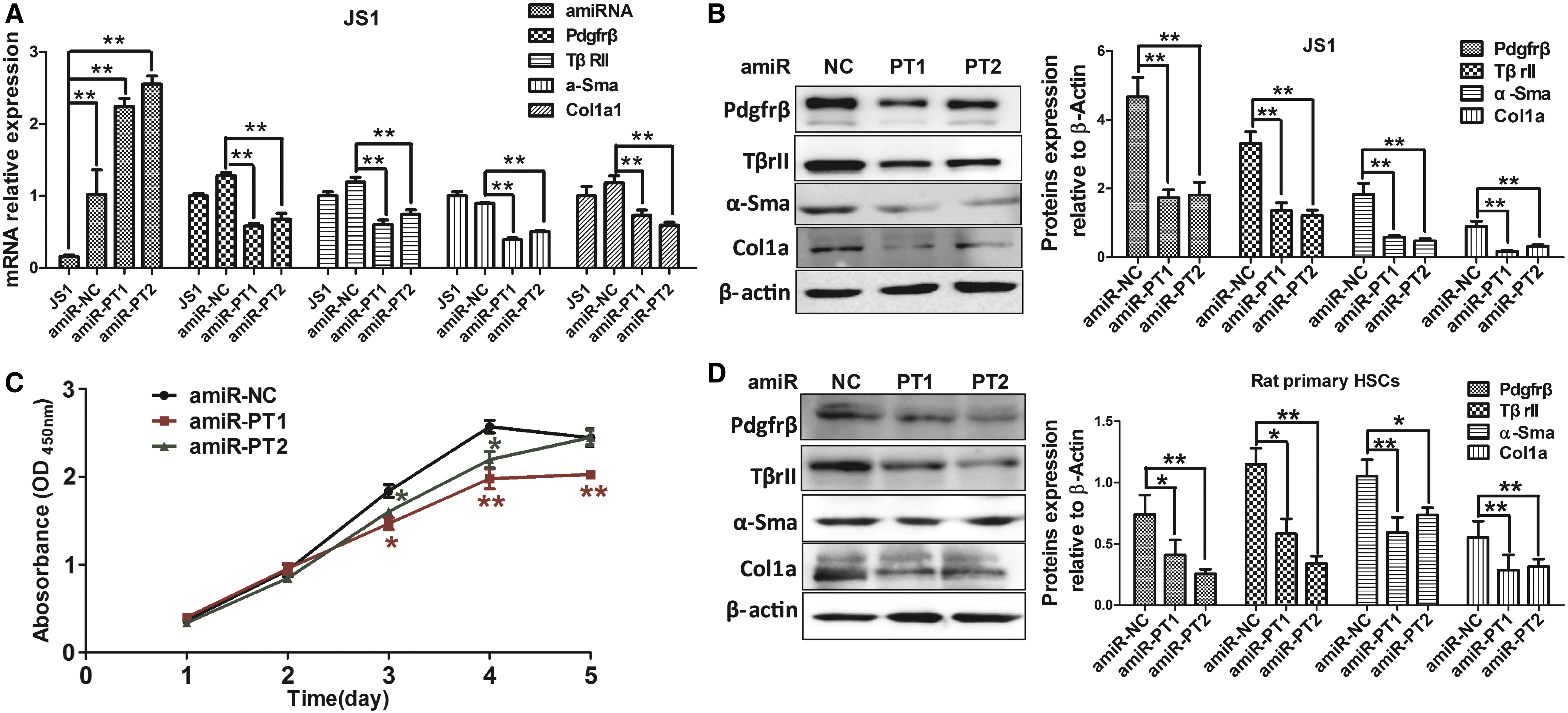
amiRNA-mediated Pdgfrβ and TβrII co-silencing attenuates HSC activation and proliferation. Co-silencing of Pdgfrβ and TβrII in HSCs was achieved by stable transfection of lentiviral-expressed amiRNAs. Transcriptional expression of transduced amiRNAs, their target genes, Pdgfrβ and TβrII, and fibrotic markers, α-Sma and Col1a1, was measured with qRT-PCR in JS1 cells across the following settings: the normal control without transfection, the negative control group transfected with amiR-NC, and the therapeutic group transfected with amiR-PT1 or amiR-PT2
To validate the phenotypic effects of Pdgfrβ and TβrII knockdown on HSC activation, fresh rat primary HSCs were prepared and culture-induced activation of primary HSCs was characterized by immunofluorescence staining of Desmin and α-Sma (Supplementary Fig. S1). The purity of isolated primary HSCs, as demonstrated by the cell population with positive Nile red staining for lipid droplets in the quiescent HSCs, was approximately 91%. In accordance with the results of cell line assays, following transfection of pLenti-amiRNA into fresh rat primary HSCs, Pdgfrβ, TβrII, α-Sma, and Col1a1 expression in cells with dual-knockdown with amiR-PT1 or amiR-PT2 were consistently and significantly reduced compared to the control (Fig. 3D). In vitro experiments suggested that amiRNA-based Pdgfrβ and TβrII dual-knockdown could suppress cellular fibrotic phenotypes, such as HSC activation and proliferation.
amiRNA-mediated Pdgfrβ and TβrII co-silencing ameliorates experimental hepatic fibrosis in mice
The antifibrotic effects of amiRNAs silencing of Pdgfrβ and TβrII were further evaluated in the liver fibrosis mouse model, which was validated by analysis of the expression of fibrosis marker proteins Col1a1 and α-Sma. Expression and distribution of EGFP, the proxy marker for the transduced amiRNAs and of their targets Pdgfrβ and TβrII, in liver tissue were assayed with immunofluorescence staining. Pdgfrβ and TβrII expression in isolated primary mouse HSCs were measured using Western blot.
Consistent with our previous report, the lentiviral vector with the GFAP promoter could successfully deliver exogenous genes—amiRNAs in this study—into the liver and drive their targeted expression in HSCs. 48 EGFP-positive cells with green fluorescence and Pdgfrβ or TβrII-positive cells with red fluorescence were dominantly distributed in the portal area where HSCs are expected to reside (Fig. 4). Most of the green fluorescent EGFP-positive cells transduced with pLenti-amiR-PT1, shown by yellow arrows in Fig. 4A and C, manifested much weaker red florescence than their adjacent cells. In the control group of pLenti-amiR-NC transduction, concomitant strong green fluorescence and high red fluorescence in EGFP-positive cells were observed (Fig. 4B and D).

Expression and distribution of interfering amiRNA and its targets, Pdgfrβ and TβrII, in the mouse liver. The interfering Lenti-amiR-PT1 and negative control Lenti-amiR-NC were transduced into mouse livers by intraperitoneal injection. Frozen sections of mouse liver tissues were prepared and underwent double-staining immunofluorescence to detect protein expression of Pdgfrβ and TβrII. Expression and distribution of amiRNAs were surrogated using the green fluorescence of enhanced green fluorescent protein. Immunofluorescence assays illustrate the expression and distribution for Pdgfrβ
In accordance with the in vitro results, quantitative analyses of amiR-PT1 transcripts in liver tissue showed that amiRNA was efficiently delivered and expressed (Fig. 5A). Quantitative analyses of cellular expression in mouse primary HSCs of Pdgfrβ and TβrII proteins also showed that pLenti-amiR-PT1 transduction consistently significantly reduced in target protein expression (Fig. 5B). These data demonstrate that the lentiviral-expressed GFAP promoter-driven amiRNA can efficiently and specifically co-knock down the expression of Pdgfrβ and TβrII in HSCs in vivo.

amiRNA-mediated Pdgfrβ and TβrII co-silencing ameliorates carbon tetrachloride (CCl4)-induced liver fibrosis in mice. Three groups of male BALB/c mice were used in the in vivo experiments as follows: interfering amiRNA therapy (amiR-PT1; n = 7) and negative control (amiR-NC; n = 7) in fibrotic mice with CCl4 administration; and normal control mice without CCl4 administration and amiRNA treatment (normal; n = 4). Pdgfrβ and TβrII co-silencing caused by amiRNA was determined by analysis of hepatic expression amiRNA, Pdgfrβ, and TβrII. The hepatic injury and fibrogenesis caused by CCl4 injury was evaluated by measuring serum ALT activity, collagen deposition, and hepatic expression of α-Sma and fibrotic markers.
Therefore, next, the therapeutic effects of amiRNA-based Pdgfrβ and TβrII dual knockdown were investigated in situ in the liver fibrosis mouse model. The degree of CCl4-induced live tissue injury was evaluated by measuring serum ALT enzyme activity. The results showed that ALT activity in mice with pLenti-amiR-PT1 transduction was significantly reduced compared to the control group (Fig. 5C).
Masson and immunohistochemical staining of collagens and α-Sma were used to assess liver fibrosis progression. Masson staining showed that hepatic fibrosis degree, as illustrated by the percentage of collagen, was mitigated in the treatment group with therapeutic pLenti-amiR-PT1 interference (p = 0.036; Fig. 5D). The IOD of positive α-Sma areas for the therapeutic group was also significantly decreased compared to the control (p = 0.031; Fig. 5E).
The transcriptional expression in liver tissue of a panel of typical fibrogenic marker genes was further profiled with qRT-PCR, and it was found that all tested fibrotic genes were significantly downregulated in the pLenti-amiR-PT1 group compared to the pLenti-amiR-NC group as follows: Col1a1 (p = 0.015), tissue inhibitor of metalloproteinase 1 (Timp1; p = 0.052), and members of the matrix metallopeptidase (MMP) family—Mmp2 (p = 0.015), Mmp11 (p = 0.023), and Mmp13 (p = 0.045; Fig. 5F).
In the parallel independent experiment with the second amiRNA (amiR-PT2), similar therapeutic effects of amiR-PT2 interference were consistently observed. In situ Pdgfrβ and TβrII dual knockdown by pLenti-amiR-PT2 transduction reduced expression of collagenase (p = 0.036) and α-Sma (p = 0.042; Supplementary Fig. S2A and B). These suggest that amiRNA-based Pdgfrβ and TβrII dual knockdown alleviates experimental liver fibrosis in mice.
amiRNA-mediated Pdgfrβ and TβrII co-silencing inhibits expression of target or effector genes in vitro and in vivo
To decipher the underlying biological mechanism for the antifibrotic therapeutic effect of amiRNA against Pdgfrβ and TβrII, transcriptional expression was measured in JS1 cells and in mice fibrotic liver tissue for two panels of well-known target or effector genes implicated in the development and progression of liver fibrosis as follows: Col1a1, Smad7, Pai-1 (plasminogen activator inhibitor type 1), and c-Myc of the Tgf-β signal pathway, and Pi3k, p70s6k, Cyclind1, and Cyclind3 of the Pdgf signal pathway. It was found that Pdgfrβ and TβrII dual knockdown in JS1 cells transfected with pLenti-amiR-PT1 correlated with an overall reduction in transcriptional expression of target genes of Tgf-β/Smad pathway and effector genes of the Pdgf pathway, among which Col1a1, Pai-1, and p70s6k were significantly downregulated (Fig. 6A).
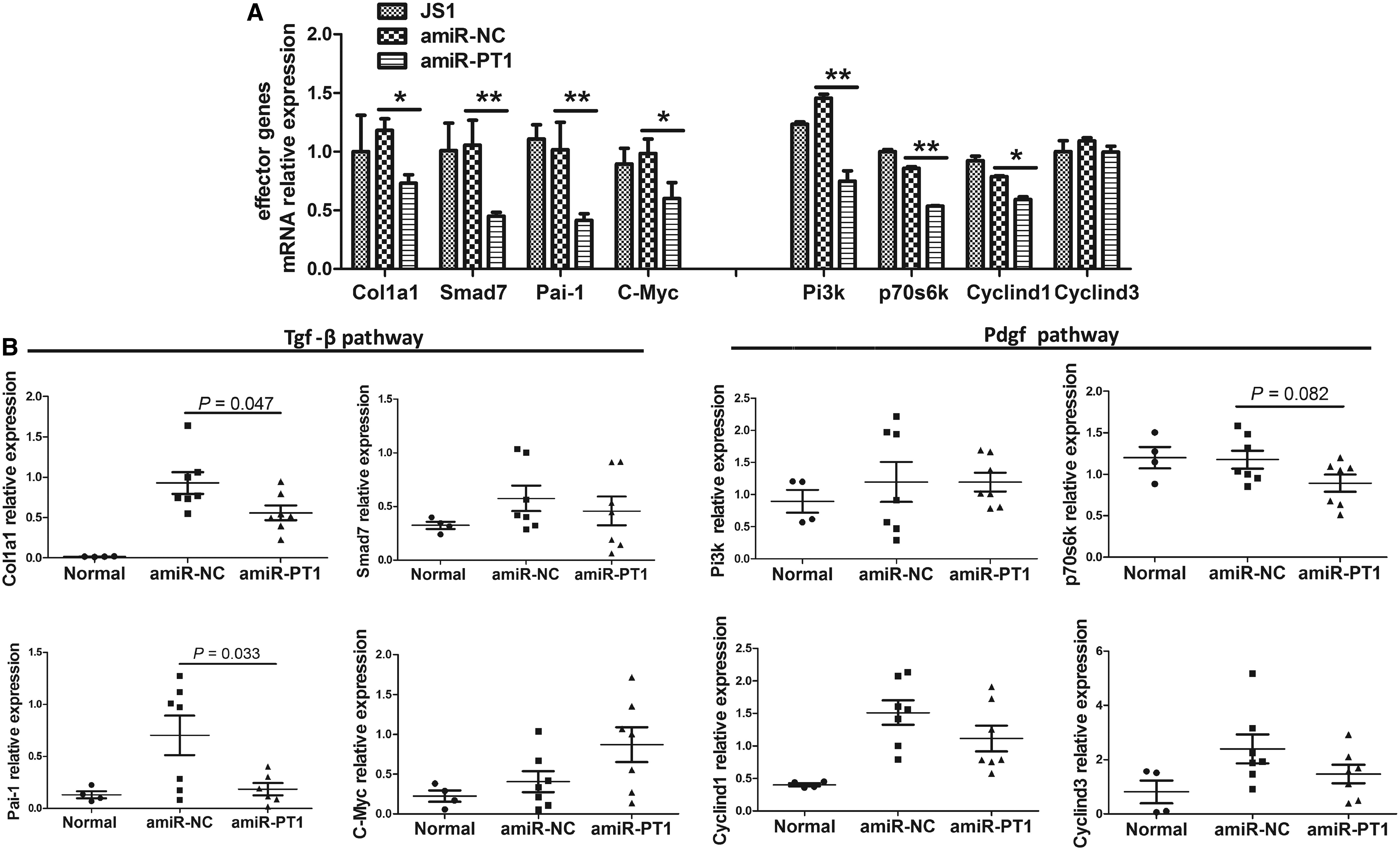
amiRNA-mediated Pdgfrβ and TβrII co-silencing suppresses the expression of their target or effector genes involved in liver fibrosis. The transcriptional expression of target or effector genes of the Pdgf and Tgf-β pathways was analyzed with qRT-PCR in JS1 cells stably transduced with interfering amiR-PT1 and negative control amiR-NC
Consistent with the in vitro data, Pdgfrβ and TβrII dual knockdown with amiR-PT1 in the mouse liver in vivo also suppressed expression of Col1a1 (p = 0.047), Pai-1 (p = 0.033), and p70s6k (p = 0.082) compared with the amiR-NC transduction group (Fig. 6B). In vitro co-silencing of Pdgfrβ and TβrII with amiR-PT2 resulted in downregulation of their target or effector genes in JS1 cells. Dual knockdown in vivo with amiR-PT2 also downregulated the expression of Col1a1 (p = 0.084), Pai-1 (p = 0.052), and p70s6k (p = 0.016) in the mouse fibrotic liver (Supplementary Fig. S2C and E). Taken together, amiRNA-based Pdgfrβ and TβrII dual knockdown may downregulate the target or effector genes of their signaling pathways, which mechanistically accounts, at least partly, for the antifibrotic effects of therapeutic amiRNAs.
Discussion
Although liver fibrosis occurs in most types of chronic liver diseases, there is currently no clinically validated antifibrotic therapy with proven safety and efficacy for the treatment and management of patients with hepatic fibrosis. 6 Emerging antifibrotic therapies are focused on depopulating or inhibiting accumulation of fibrogenic cells and/or preventing the deposition of ECM proteins. Activated HSCs are the predominant contributor to myofibroblasts that produce and deposit ECM. Thus, HSC is considered the primary cellular target for antifibrotic therapies across all types of liver disease. 2,3 TGF-β1 and PDGF are key profibrotic cytokines with regulatory functional crosstalk. Their signaling pathways modulate HSC activation and proliferation and are well-established therapeutic targets for liver fibrosis. 6
In the present study, HSC-specific promoter-driven lentiviral-expressed amiRNAs against mouse Tgfbr2 and Pdgfrβ genes were designed. The study showed that concatenated amiRNAs could be transduced and could co-knock down the expression of Tβr and Pdgfrβ in HSCs, suppress HSC activation and proliferation, and therapeutically ameliorate CCl4-induced experimental hepatic fibrosis in mice. The findings provide a proof of concept for the use of combinational amiRNA silencing of two key profibrogenic pathways in the treatment of liver fibrosis and highlight the therapeutic potential of concatenated amiRNAs in gene therapy.
Strategies to block TGF-β1 and PDGF profibrotic pathways using RNA interference to target the cytokine receptor individually, or downstream effectors, have achieved favorable antifibrotic effects in cells and in animal models. Yang et al. investigated the in vitro antifibrotic effects of two efficient shRNAs and their concatenated cluster by targeting different sites of rat TGF-β1 mRNA, which are embedded in a miRNA30 architecture, driven by GFAP promoter. These were transiently transfected into rat HSCs HSC-T6 cells with a plasmid vector. They found that transduction of pre-miRNA-cluster mimics correlates with higher TGF-β1 silencing, reduced viability, and enhanced apoptosis of HSC-T6 cells compared to the amiRNA expression vector encoding single shRNA. 23 Fu et al. reported that transient transfection of rat HSC-T6 cells with shRNA against TβrII could efficiently knockdown its expression and inhibit HSC activation and ECM production. 24 In addition, siRNA- or amiRNA-mediated knockdown of connective tissue growth factor, a downstream effector of TGF-β1 pathway, attenuates hepatic fibrosis in the rat. 25,59
This study showed that concatenated amiRNAs targeting TβrII and Pdgfrβ exerted antifibrogenic effects in vitro and in vivo. Transduction of amiRNAs into HSC cell lines and primary HSCs significantly reduced expression of α-Sma and Col1a1 and suppressed HSC activation and proliferation. Their delivery into fibrotic liver of treated mice, in addition to silencing TβrII and Pdgfrβ in situ, reduced collagen disposition and α-Sma expression, decreased serum ALT activity, and downregulated expression of fibrotic marker genes, such as Col1a1 and matrix metallopeptidase. The phenotypic and therapeutic effects of the amiRNA silencing strategy was replicated and validated in independent in vitro and in vivo experiments with the second amiRNA designed in parallel. Therefore, amiRNA-based Tgfbr2 and Pdgfrβ co-silencing inhibited fibrotic phenotypes of HSCs in vitro and ameliorated experimental hepatic fibrosis in mice in vivo.
A principal feature of liver fibrosis is the imbalance between synthesis and proteolytic degradation of ECM proteins in the liver, which is largely due to disturbed expression of MMP, which remodels and degrades ECM and their specific inhibitors (tissue inhibitor of metalloproteinase, TIMP). Paradoxically, it was found that the antifibrotic effects of amiRNA silencing of TβrII and Pdgfrβ in mouse HSCs correlated with concomitant downregulation of both Mmps (Mmp2, Mmp11, and Mmp13) and Timp1 in the liver. MMPs and TIMPs play pivotal roles in both fibrogenesis and fibrolysis, and, while mRNA and protein expression of rodent Mmp13 (the homolog of MMP1 in human) are undetectable in healthy liver tissue, their expression is increased in activated HSCs during experimental liver fibrogenesis. 60 Mmp13 functions as the major interstitial collagenase to reduce liver fibrosis by degrading insoluble fibrillary collagens, especially type I collagen.
Systemic administration of plasmid DNA-coding Mmp13 increases expression of Mmp13 and ameliorates liver fibrosis in mice and rats. 61,62 In contrast, Mmp13 may destroy surrounding tissues to deposit newly synthesized ECM, release ECM-bound cytokines such as TGF-β1, and subsequently induce fibrogenesis. Its loss of function attenuates inflammatory reaction and suppresses HSC activation and fibrogenesis. 60,63 As a candidate marker of liver fibrosis, TIMP1 is strongly upregulated in liver tissue and serum during hepatic fibrogenesis in patients with liver disease, and its expression directly correlates with hepatic fibrosis stage. 64,65 Previous functional studies in mice with either transgenic Timp1 overexpression or antagonizing antibody also show that Timp1 promotes hepatic fibrogenesis in vivo. 66 –68 However, two recent in vivo studies in Timp1 knockout mice suggest that Timp1 is upregulated, but not essential, in hepatic fibrogenesis, and its deficiency exacerbates CCl4-induced liver injury and fibrosis. 69,70 Therefore, amiRNA-based TβrII and Pdgfrβ co-silencing in the mouse liver may result in dynamic remodeling of ECM synthesis and degradation by modulating Mmps and Timp1 expression, from relatively high levels in the diseased fibrotic liver to significantly lower levels in the treated liver with ameliorated fibrotic phenotypes.
This study also showed that amiRNA silencing of TβrII and Pdgfrβ phenotypically exerts therapeutic effects against liver fibrosis and mechanically modulates their signaling. Transduction of either amiR-PT1 or amiR-PT2 consistently resulted in significantly reduced expression of their target or effector genes, which could be typically exemplified by Col1a1, Pai-1, and p70s6k. The Col1a1 gene codes collagen type I alpha 1 chain, a well-established marker protein for HSC activation and fibrosis, and its transcriptional expression is a downstream event of TGF-β1 signaling. 71 The Pai-1 gene product is a member of the serine proteinase inhibitor (serpin) superfamily, and is the most potent cellular inhibitor of the urokinase-type plasminogen activator (uPA)/tissue-type plasminogen activator (tPA)/plasmin fibrinolytic system, which plays important roles in cellular proteolytic degradation of ECM proteins and the maintenance of tissue homeostasis. 72
The Pai-1 gene contains TGF-β response elements for SMAD binding and is significantly upregulated in fibrotic liver, and its deficiency is associated with increased activity of tPA and reduced liver fibrosis. 72 Antifibrotic treatment in a rat model of cirrhosis results in a reduction in collagen and Pai-1 synthesis and cirrhosis amelioration, and downregulating TβrII gene expression significantly suppresses TGF-β1-induced Pai-1 expression in mouse NIH3T3 cells. 73,74 The PDGF signaling pathway also activates Pai-1 transcriptional activity in primary cultured HSCs, suggesting a synergetic regulatory function of TGF-β1 and PDGF for Pai-1 gene expression. 75 As the most potent mitogen for activated HSCs, PDGF and its receptor could transduce the mitogenic MAPK/ERK and PI3K/Akt signaling pathway and activate downstream targets or effectors, such as P70S6K and D cyclins, whose expression correlates with G1/S cell cycle transition and HSCs proliferation. 8,76 P70S6K, the 70-kDa ribosomal S6 kinase, is a member of the ribosomal S6 kinase family of serine/threonine kinases and plays a crucial role in HSC proliferation, collagen expression, and cell cycle control. 77
It was found that amiRNA silencing of Pdgfrβ resulted in concomitant reduction in Pi3k, P70s6k, Cyclind1, and Cyclind3 expression. Consistent with the findings, it was reported that genetic or pharmacological inhibition of PI3K and P70S6K reduces the expression of cell cycle control proteins Cyclin D1 and Cyclin D3 and blocks HSCs proliferation. 77,78 However, not all of the analyzed target or effector genes of TβrII and Pdgfrβ showed consistent and significant reductions in transcriptional levels following amiRNA-mediated co-silencing of the two receptors in vitro or in vivo. c-Myc, which is involved in many cellular events, such as cell growth, proliferation, and differentiation, was significantly downregulated in JS1 cells transfected with amiR-PT1 or amiR-PT2, and down- or upregulated in fibrotic mouse liver tissue transduced with amiR-PT1 or amiR-PT2, respectively.
During liver injury, TGF-β and PDGF signaling can enhance fibroblast growth by upregulating c-Myc via the CDK-dependent C-terminal and middle linker dual-phosphorylated Smad2 and Smad3 (pSmad2L/C and pSmad3L/C) pathways. However, TGF-β can also inhibit HSC growth by downregulating c-Myc via the pSmad2C and pSmad3C pathways. 79 The seeming discrepancy of c-Myc expression between the in vitro and in vivo studies could partly be due to the complex modulation of the TGF-β signaling by systemic factors, such as inflammatory cytokines and hepatic cellular communication in mice that are absent in cultured HSC cell lines. Therefore, amiRNA silencing of Tgfbr2 and Pdgfrβ could, to some extent, suppress expression of the receptors and their target or effector genes, thereby inhibiting activation and proliferation of HSCs, reducing hepatic expression of fibrogenic genes and ameliorating hepatic fibrosis.
The GFAP promoter-driven and lentiviral-expressed therapeutic amiRNAs (both amiR-PT1 and amiR-PT2) consistently manifested much stronger transcriptional expression both in transfected HSCs cells in vitro and in therapeutically transduced mice liver in vivo compared to the negative control amiRNA (amiR-NC). There are two main possible explanations for this discrepancy. First, while the transcriptional levels of lentiviral-expressed amiRNAs were comparable, primary amiRNAs transcripts might be of varied cellular stability. The therapeutic and control amiRNAs shared the same backbone of pre-miR-30a and harbored two interfering shRNAs against Pdgfrβ and TβrII or two control shRNAs. The negative control shRNAs sequences in this study are commonly used in RNAi studies and were commercially provided. In the amiRNA structure prediction, highly similar secondary structures of amiR-PT1, amiR-PT2, and amiR-NC were observed (data not shown), suggesting that divergent biogenesis dynamics and cellular stability of these amiRNAs would not explain significant differences in their transcript levels.
Second, and more plausibly, co-knock down of Pdgfrβ or TβrII pathways with amiRNAs could directly or indirectly modulate their downstream factors, including transcription factors and epigenetic regulators that interplay in trans with the GFAP promoter, which could then render stronger cis-acting capability toward the loaded amiRNAs. GFAP gene expression in HSCs and brain astrocytes is strictly modulated by trans-acting factors and epigenetic regulators, among which H3K27 histone methyltransferase Enhancer of Zest Homolog 2 (Ezh2), the catalytic subunit of polycomb repressive complex 2, is an important player. Ezh2 could mediate H3K27 methylation on the GFAP promoter and directly suppress GFAP gene transcription, as endogenous Ezh2 is downregulated during primary neural progenitor cell (NPC) differentiation, which could ensure prolonged expression of this key astrogenic marker gene in NPCs. 80 Likewise, during the transdifferentiation of primary quiescent HSCs into myofibroblasts, enhanced expression of Ezh2 in HSCs promotes liver fibrosis by epigenetically silencing PPARγ expression, a pivotal negative regulator for HSC activation. 58,81 The GFAP gene manifests transdifferential stage-dependent differential expression during HSC activation. While it is highly expressed in freshly isolated primary quiescent HSCs, it is significantly downregulated in the rat-activated HSC cell line HSC-T6 and in culture-activated HSCs. 82 Its expression in vivo is inversely correlated with liver fibrosis progression in chronic hepatitis C patients. 83 Furthermore, the TGF-β1 signaling pathway could modulate the transcriptional and epigenetic regulators of the GFAP gene.
c-Myc, one of targets of the TGF-β/Smad signaling pathway, could repress GFAP expression, as its overexpression in glioblastoma multiform cells results in significant reduction of GFAP promoter activity. 84 TGF-β1 treatment significantly increases the association of EZH2 to its target gene promoter and reinforces epigenetic repression in fibroblasts from human lungs affected with idiopathic pulmonary fibrosis. It additionally significantly reduces active histone H3 and H4 acetylation at the promoter. 85 Therefore, it is reasonable that amiRNA-mediated TβrII silencing in HSCs with therapeutic amiRNAs could knock down the expression of transcription factor for the GFAP gene, such as c-Myc, and strengthen epigenetic derepression and activation of the GFAP promoter through Ezh2, eventually enhancing the driving activity of the GFAP promoter toward the embedded therapeutic amiRNAs. This positive feedback between the two functional components of the lentiviral amiRNA expression vector, amiR-PT, and its driving promoter has the potential to strengthen RNAi efficiency.
This study has some limitations and raises a few open questions to address in the future. Although we observed consistent antifibrotic effects of amiRNA-based TβrII and Pdgfrβ silencing in both primary HSCs and treated mice with CCl4-induced experimental liver fibrosis, its therapeutic effects should be validated in other models, since there are differences in pathologic features and disease mechanisms between animal models and human disease.
The therapeutic amiRNA cargo was delivered into the mouse liver via tail-vein injection by utilizing the lentiviral vector, one of the two types of virus vectors (lentiviral and adeno-associated virus [AAV] vectors) that are used in several recently approved gene therapies. 86 Lentiviral vectors transduce dividing and nondividing cells and allow persistent transgene expression through stable integration into the host genome with low genotoxicity and low immunogenicity. 39 Lentiviral expression of three amiRNAs targeting the oncogene Bcr-Abl can sufficiently suppress oncogene expression and kinase pathway activation, thereby preventing regrowth of leukemic cells both in vitro and in vivo. 87 Therefore, the lentiviral vector was used as the vehicle for the two concatenated therapeutic amiRNAs that target two profibrogenic pathways. Although the expression of amiRNA and the production of effective siRNAs against TβrII and Pdgfrβ are under the strict control of the HSC-specific GFAP promoter, the circulating delivery and pervasive distribution of the recombinant virus particles in most organs and tissues in mice complicate the controllability of gene transduction. It is therefore warranted to compare the efficacy and safety of the amiRNA-based gene therapy using other vector and delivery systems, such as the liver-trophic AAV2/8, which is an attractive candidate vector for hepatic gene transfer application due to its high transduction efficiency in liver and minimal immune response. 88
The current combinational amiRNA silencing was designed toward only two targets, since sustained high-level shRNA expression in the mouse liver could oversaturate endogenous microRNA machinery and induce hepatotoxicity and even lethality. In addition, the recommended number of concatenated amiRNAs in a multi-amiRNA expression vector should not be more than four. 28,30,50,54
In summary, the present study proposes a novel amiRNA silencing strategy targeting two profibrogenic pathways for the treatment of hepatic fibrosis. The GFAP-promoter-driven, lentiviral-expressed concatenated amiRNAs against Tβr and Pdgfrβ suppressed their signaling and reduced expression of targets and effectors, such as Pai-1, and thereby exerted antifibrogenic effects in both cultured HSCs and experimentally treated mouse livers. Since TβRII, PDGFR-β, and PAI-1 are well-defined common key regulators of tissue fibrosis and are generally implicated in fibrotic pathogenesis of multiple organs including the liver, lung, kidney, and skin, the findings provide a proof of concept for the use of dual-amiRNA interference against TβRII and PDGFR-β for the treatment of fibrosis across tissues, and highlight the therapeutic potential of concatenated amiRNAs in gene therapy. 5,72
Footnotes
Acknowledgments
The National Natural Science Foundation of China supported this study (31301050 and 81572404).
Author Disclosure
The authors have declared that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.