
Abstract
Reporter gene-based molecular imaging can provide invaluable information on the fate of cellular therapies postimplantation. Integrating lentiviral vectors (ILVs) are commonly used for stably engineering cells; however, their potential for insertional mutagenesis poses a significant safety concern and barrier to widespread clinical adoption. In cells that slowly divide or are postmitotic in vivo, such as mesenchymal stem cells (MSCs), nonintegrating lentiviral vectors (NILVs) may be a safer alternative option because NILVs remain episomal and can provide prolonged expression profiles. Here, NILVs coexpressing fluorescence and bioluminescence imaging (BLI) reporters were engineered and used to longitudinally image the viability of human MSCs in vitro and in vivo in mice. In vitro, ILV-transduced cancer cells and MSCs maintained steady reporter gene expression over time, whereas NILV-transduced cells progressively lost signal. NILV reporter loss was accelerated in highly proliferative cancer cells compared with less proliferative MSCs. In vivo, ILV- and NILV-transduced MSCs were each detectable with BLI postintramuscular implantation, with significantly higher ILV-based signal compared with NILV-based signal. BLI signal was observed to similarly diminish over time for both cell populations, which was attributed to cell death. Despite the reduced signal intensity with NILVs and the minimal number of cells injected (40,000), live NILV-transduced MSCs were reliably visualized for up to 2 weeks. Safety is a concern for future clinical reporter gene applications. We present NILVs as a safe means of imaging reporter gene expression for slowly dividing or nondividing cells and showed effective tracking of the viability of a small number of live transplanted MSCs over time with optical imaging. Future work will evaluate improvements to episomal NILV reporter expression, explore sensitive clinically relevant reporters, and apply this approach to clinically relevant MSC applications in preclinical models. NILVs may have broad clinical applications for expression of imaging reporters and other gene products in MSC-based therapies.
Introduction
C
A powerful tool in the development of cell-based therapies has been the advancement of noninvasive cellular imaging techniques that enable the tracking of administered cells over time. The success of stem cell-based therapy depends on several different factors including the route and accuracy of cell transplantation, the functionality of transplanted cells, as well as their interaction with the host microenvironment. 10 In vivo tracking of transplanted cells is a crucial step in determining the safety and efficacy of any proposed cell therapy. Cell tracking can be accomplished by either direct or indirect means. Direct labeling requires the in vitro labeling of MSCs before transplantation. Such strategies may include the use of radionuclides for positron emission tomography (PET) or single-photon emission computed tomography (SPECT) imaging or the use of paramagnetic or ferromagnetic contrast agents for magnetic resonance imaging (MRI). 10 –12 Although these approaches have provided invaluable information, they do suffer from some drawbacks including the limited radionuclide half-life of PET and SPECT agents 12 and issues with bystander cell uptake for MRI agents. 10 Indirect labeling methods require the introduction of a reporter gene and generally also necessitate the use of a corresponding imaging reporter probe. 10,12 This approach is considered a more controllable system as only viable cells are able to express conferred genes.
Reporter gene transfer can be accomplished by a variety of means including transfection with lipoplexes, magnetofection, electroporation, and virus-based methodologies. 13 At present, no single class of gene delivery system has been developed without some limitation or potential adverse side effect. 13,14 Nonviral methods are considered safe but are substantially less efficient at gene delivery compared with viral methods. 13 Integrating lentiviral vectors (ILVs) exhibit high efficiency transduction and have the capacity to confer two or more transgenes to target cells. 1 Integrating lentiviruses can transduce both nondividing and dividing cell types 15 and enable long-lasting expression of conferred genes by integrating their vector genome into the target cell genome. These vectors have been most widely used in gene therapy and have become the vector of choice for MSC transduction. 16,17 Previous work with lentiviral transduction of MSCs has shown no alteration in MSC phenotype, in vivo homing, viability, proliferation, or differentiation potential posttransduction. 16,18,19 However, despite these positive attributes, ILV quasi-random integration has the theoretical potential to cause several undesirable effects including insertional oncogene activation, alternative splicing, aberrant transcripts, and read-through transcription of endogenous genes. 20,21 Thus, although therapeutic cells engineered with ILVs have been recently approved by the U.S. Food and Drug Administration 22 (i.e., chimeric antigen receptor T cells), ILV integration poses a significant safety concern and barrier to widespread clinical adoption. The development of nonintegrating lentiviral vectors (NILVs), also called integrase-deficient lentiviral vectors (IDLVs), provides an alternative vector for efficient and safer engineering of target cells with imaging reporter genes (and other gene products).
ILVs contain an integrase that inserts a reverse-transcribed viral DNA cassette into the host genome. 23 Although there are several means of generating NILVs, the most common method is through mutation of the integrase catalytic site, thereby disrupting catalytic integration while maintaining other functions such as reverse transcription and nuclear import. 24 NILVs employ the innate propensity of ILVs to form episomal DNA circles as an intermediate during infection. 25 NILV-transduced cells will harbor these circular nonreplicating nuclear episomes but, without the capability of replication, the NILV episomes will be lost during rapid division, thereby limiting their use for long-term expression in highly proliferative cells. However, in postmitotic cell populations, or cells with limited in vivo proliferation such as MSCs, NILV expression may persist for extended periods, offering a potentially safer approach with drastically reduced integration and minimization of undesirable cellular events. 26 –28
In this study we generated ILVs and NILVs expressing fluorescence and bioluminescence imaging (BLI) reporters and compared the longitudinal expression of each vector in rapidly proliferating cells (cancer) and slowly proliferating cells (MSCs). We then compared the longitudinal BLI signal of ILV- and NILV-tagged human MSCs in mice to determine performance and to highlight any potential limitations. To our knowledge this study represents the first use of NILVs to safely and noninvasively image the location and viability of an MSC population in vivo.
Materials and Methods
Generation of lentiviral plasmids
Third-generation lentiviral packaging plasmids pMDLg/pRRE and pRSV-Rev, as well as a vesicular stomatitis virus glycoprotein (VSV-G) envelope-expressing plasmid, pMD2.G, were obtained from Addgene (Cambridge, MA) (gifts from D. Trono; Addgene plasmids #12251, #12253, and #12259, respectively).
29
A third-generation lentiviral transfer vector, pUltra-Chili-Luc, was also obtained from Addgene (gift from M. Moore; Addgene plasmid #48688). Mutagenesis of the pMDLg/pRRE plasmid was performed with an In-Fusion HD cloning kit single-fragment insertion protocol (Takara Bio USA, Inc, Mountain View, CA) and mutagenic primer pairs (vector, 5′-GTAAAAACAGTACATACA
To generate our dual-modality reporter gene transfer vector (Fig. 1A), we substituted the ubiquitin C promoter on pUltra-Chili-Luc with a human elongation factor 1α promoter (pEF1α) and firefly luciferase was substituted with a codon-optimized version of firefly luciferase (luc2) via two rounds of single-fragment insertion with the In-Fusion HD cloning kit (Takara Bio USA, Inc).
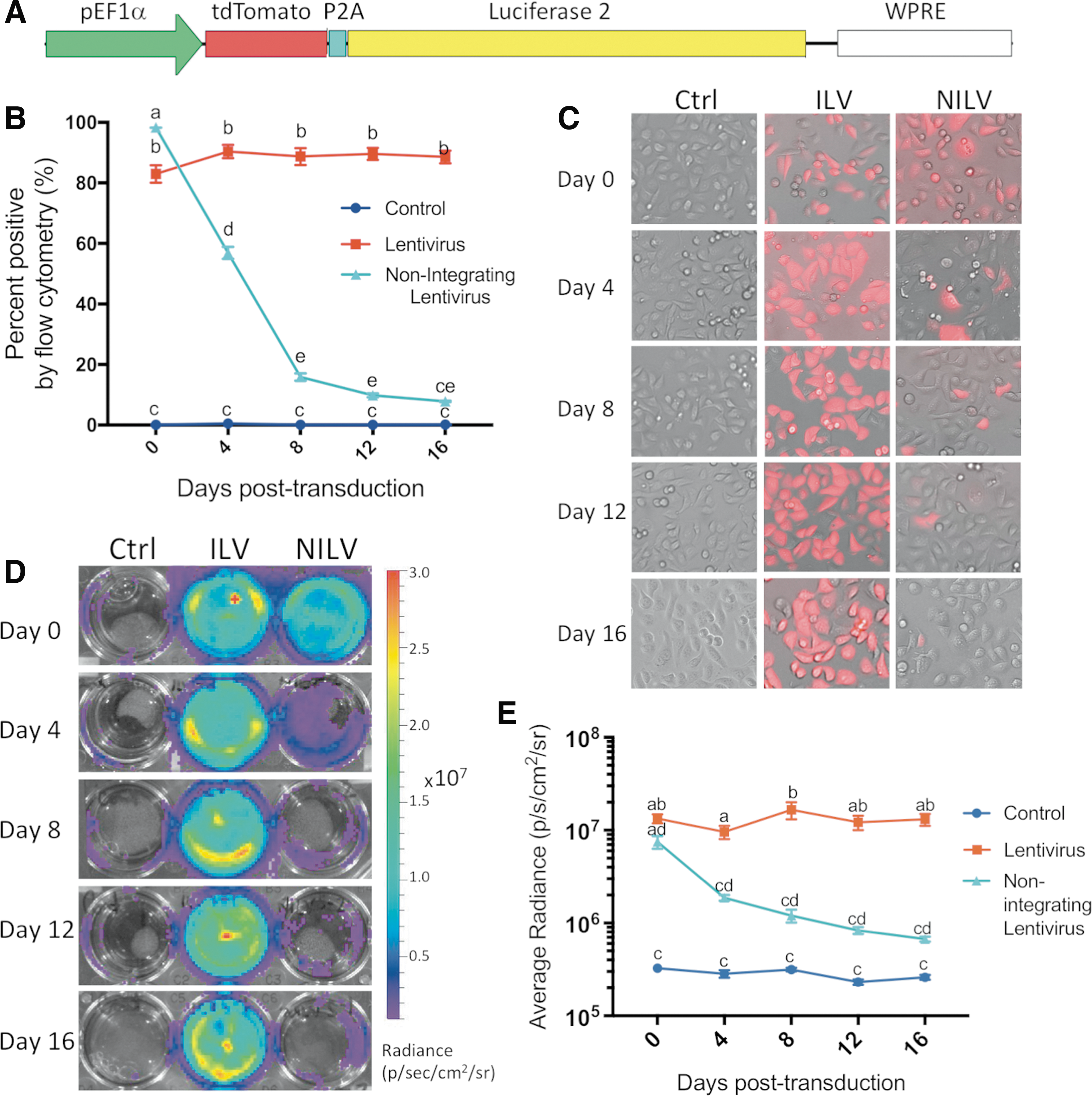
In vitro analysis of reporter gene function in integrating lentiviral vector (ILV)- and nonintegrating lentiviral vector (NILV)-transduced highly proliferative MDA-MB-231 cancer cells over time.
Lentiviral production
To generate pEF1α-driven, tdT/luc2-expressing self-inactivating ILVs, packaging, envelope, and transfer plasmids were cotransfected into 293T cells, using Lipofectamine 3000 according to the manufacturer's lentiviral production protocol (Thermo Fisher Scientific, Waltham, MA). Similarly, NILVs were produced in the exact same manner except that the D64V/D116N pMDLg/pRRE plasmid was substituted for the pMDLg/pRRE packaging plasmid. Vector supernatants were harvested 24 and 48 h after transfection and filtered through a 0.45-μm filter. As NILV production is less efficient, produced NILV was concentrated with a Lenti-X concentrator according to the manufacturer's instructions (Takara Bio USA, Inc). ILV and NILV preparations were aliquoted and frozen at −80°C until use. ILVs and NILVs were transduced at various volumes into 10 5 H1299 cells, using Polybrene (8 μg/mL; Sigma-Aldrich, St. Louis, MO) and the functional viral titer (in infectious units [IFU]/mL) was determined by flow cytometry on a BD FACSCanto system (BD Biosciences, San Jose, CA). The average ILV production was quantified as 7.8 × 10 6 ± 1.3 × 10 6 IFU/mL, and the average production of concentrated NILV was calculated as 1.5 × 10 6 ± 1.4 × 10 6 IFU/mL.
Cells
Human breast cancer cell culture and lentiviral transduction
Human MDA-MB-231 breast cancer cells (HTB-26; American Type Culture Collection [ATCC], Manassas, VA) were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM; Wisent, Saint-Jean-Baptiste, Canada) supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), and streptomycin (100 μg/mL). Cells were grown until 90% confluent in a humidified chamber at 37°C with 5% CO2. For MDA-MB-231 cell transductions, cells were seeded in 6-well plates in complete medium and transduced at a multiplicity of infection (MOI) of 5, using Polybrene at 8 μg/mL. The MOI was chosen on the basis of a previously conducted MOI optimization experiment, in which an MOI of 5 was shown to consistently result in >90% transduction (data not shown). Cells underwent a medium change 24 h posttransduction and were washed 48 h posttransduction with three changes of phosphate-buffered saline (PBS). Thereafter, as described below, MDA-MB-231 cells were assessed by flow cytometry and seeded for in vitro analysis. The day of the first flow cytometric analysis was set as day 0.
Human mesenchymal stem cell culture and lentiviral transduction
Human MSCs were sourced commercially (Lonza, Basel, Switzerland) or received as a kind gift from A. Keating (University Health Network, Toronto, Canada). In the latter case, MSCs were collected from the bone marrow of healthy, young adult volunteers donated with written informed consent according to a protocol approved by University Health Network Research Ethics Board (Toronto, Canada). 30 MSCs were cultured in low-glucose DMEM (MilliporeSigma Canada Co, Oakville, Canada) supplemented with 10% FBS, and all experiments were conducted with MSCs between passages 3 and 6. Cells were propagated until 90% confluent in a humidified chamber at 37°C with 5% CO2. For MSC transductions, cells were seeded in 12-well plates in complete medium and transduced at an MOI of 75 (after optimization), using protamine sulfate (100 μg/mL; Sigma-Aldrich). Protamine sulfate was used as a substitute transduction agent as Polybrene was found to affect the viability and proliferative capability of MSCs. MSCs were transduced as described above with a medium change at 24 h and washing and flow cytometric assessment at 48 h posttransduction. Successfully transduced cells were seeded for in vitro analysis or prepared for in vivo injection as described below.
In vitro reporter gene analysis
To evaluate tandem dimer Tomato (tdTomato; tdT) expression over time, cells were assessed by both flow cytometry and fluorescence microscopy. MDA-MB-231 cells were assessed on days 0, 4, 8, 12, and 16 posttransduction and MSCs were assessed on days 0, 4, 12, and 20 posttransduction. For flow cytometric analysis, a subset of cells at each time point was washed with PBS, dissociated, and resuspended in PBS with 2% FBS. ILV- and NILV-transduced cells were compared relative to naive cells, using a BD FACSCanto system (BD Biosciences). Relative percent tdT-positive and geometric mean tdT fluorescence were measured. In addition, at each time point, adherent control, ILV-transduced, and NILV-transduced cells were imaged with an Olympus IX500 inverted fluorescence microscope (Olympus Corporation, Tokyo, Japan) equipped with an INFINITY3 camera (Lumenera Corporation, Ottawa, Canada).
At the same time points indicated above, cells were also assessed for luc2 activity over time. Equivalent cell numbers of control, ILV-transduced, and NILV-transduced cells were seeded into individual wells (10
5
cells for MDA-MB-231 experiments and 10
3
cells for MSC experiments).
Intramuscular MSC injections
Animal use and in vivo procedures were approved by the University Council on Animal Care, Animal Use Subcommittee at Western University and were conducted under all stipulated regulations and guidelines. Female nude mice were acquired at 6–8 weeks (nu/nu Foxn1, strain code 088; Charles River) and were housed in dedicated animal housing. On the day of injection, mice were anesthetized under 2% isoflurane in 100% oxygen and 4 × 10 4 ILV- or NILV-transduced MSCs in 50 μL of PBS were implanted intramuscularly into the right hind limb (n = 5 per group).
Bioluminescence imaging
BLI was performed on an IVIS Lumina XRMS in vivo imaging system (PerkinElmer). Mice were anesthetized with 2% isoflurane in 100% oxygen and imaged 0, 3, 6, 10, 14, and 17 days postinjection of MSCs. Mice received an intraperitoneal injection of 150 μL of
Tissue analysis
At endpoint, mice were sacrificed by isoflurane overdose followed by 0.9% saline perfusion. Mice were then perfused with 4% paraformaldehyde and right hind limbs were collected and further fixed overnight by submersion in 4% paraformaldehyde. Hind limb muscle was separated and cryopreserved in ascending concentrations of sucrose (10, 20, and 30%, w/v) for 24 h each. Tissue was then embedded in optimal cutting temperature (O.C.T.) compound (Sakura Finetek USA, Inc., Torrance, CA) and sectioned at 16 μm. Tissue sections were coverslipped, using DAPI (4′,6-diamidino-2-phenylindole)-containing mounting medium (ab104139; Abcam, Cambridge, UK), and assessed for tdTomato-positive MSCs with an upright Zeiss Axio Imager Z1 (Carl Zeiss, Thornwood, NY).
Statistical analysis
Statistical analyses were performed with GraphPad Prism version 7.0a (GraphPad Software Inc, San Diego, CA). Two-group comparisons were analyzed with an unpaired two-tailed t-test. Three-group comparisons were assessed by one-way ANOVA and Tukey's post-hoc multiple comparison test. All graphed data are presented as means and SEM. For all tests, a p value less than 0.05 was considered statistically significant, and statistical similarities and differences are denoted by letters on all graphed data, where the same letters indicate data that are not statistically significant and differing letters indicate data that are statistically different with a p value that is at least less than 0.05.
Results
Longitudinal ILV- and NILV-driven reporter expression in cancer cells
We first compared reporter expression with ILVs and NILVs over time in MDA-MB-231 breast cancer cells, a highly proliferative cell line (doubling time of approximately 20 h according to Schneider and colleagues 31 ). These experiments were performed (n = 3, biological replicates) to confirm maintenance of ILV expression and loss of NILV expression as previously reported, 26,32 as well as to compare longitudinal reporter expression in slowly dividing transduced MSCs. As expected, longitudinal flow cytometry and fluorescence microscopy analysis confirmed that ILV-transduced MDA-MB-231 cells maintained steady tdT fluorescence with an average of 88.0 ± 2.9% tdT-positive cells across all assessed days (Fig. 1B and C). Similarly, stable bioluminescent signal was evident at an average radiance of 1.30 × 10 7 ± 2.52 × 10 6 photons/s/cm2/sr in 10 5 ILV-tagged MDA-MB-231 cells across all time points (Fig. 1D and E). Immediately after NILV transduction (day 0) efficient gene transfer was evident in MDA-MB-231 cells (98.3 ± 0.1% tdT+ by flow cytometry; Fig. 1B) and robust luc2 activity, albeit significantly lower (p < 0.05) than with ILVs, was noted with an average radiance of 7.49 × 10 6 ± 2.02 × 10 6 photons/s/cm2/sr per 10 5 cells (Fig. 1E). The percentage of NILV-transduced MDA-MB-231 cells (56.9 ± 3.4%) was significantly lower than ILV-transduced cells (90.4 ± 3.9%; p < 0.05) by 4 days posttransduction, and further reduced to 9.9 ± 1.1% by day 12 compared with ILV-tagged cells (89.6 ± 3.5%; p < 0.05) (Fig. 1B). By day 16 (the last time point evaluated), 7.867 ± 0.4% of MDA-MB-231 cells remained tdT-positive, suggesting either residual NILV integration or the retention of nonproliferative cells, as seen in previous studies. 28,33 –35 Similarly, our BLI analysis revealed a significant 75.0% reduction in NILV-driven luc2 activity at 4 days posttransduction compared with day 0, and a 91.1% reduction by 16 days (Fig. 1D and E). Collectively, NILVs efficiently transduced MDA-MB-231 cells and conferred dual reporter gene expression, and the rapid loss of reporter expression suggests that in the majority of cells NILVs remained episomal.
Longitudinal ILV- and NILV-driven reporter expression in MSCs
To assess our ability to efficiently label MSCs we first used ILVs to identify the ideal MOI by transducing MSCs from various donors (n = 6, biological replicates) at MOIs of 0, 10, 25, 50, 75, and 100. Successful gene transfer was determined by tdT expression, where any fluorescence above that of naive untransduced control cells was deemed positive transduction. Transduction efficiency was observed to peak between 52% and 98% in various MSC populations, and in all cases no significant increase in transduction or decrease in viability was observed at an MOI greater than 50 (Fig. 2A). In all further MSC in vitro assessments an intermediate MOI of 75 was used and only MSC populations that showed >60% transduction at an MOI of 75 were employed (six of eight tested populations qualified). We next compared the transduction efficiency of ILV versus NILV at an MOI of 75 in MSCs. Flow cytometric analysis on day 0 (immediately after the end of the transduction) revealed that the percentages of MSCs transduced with NILV (79.2 ± 14.0%) and ILV (80.2 ± 15.9%) were not statistically different (p = 0.11; Fig. 2B). Despite equivalent transduction, differential expression of tdT was evident as the geometric mean fluorescence of ILV-transduced MSCs was 5.6 ± 3.6-fold brighter than those transduced with NILVs (p < 0.001; Fig. 2C and D). Similarly, BLI analysis revealed a 22.2 ± 10.3-fold difference in total enzyme-based luc2 signal between ILV- and NILV-tagged MSCs (p < 0.0001; Fig. 2E and F).
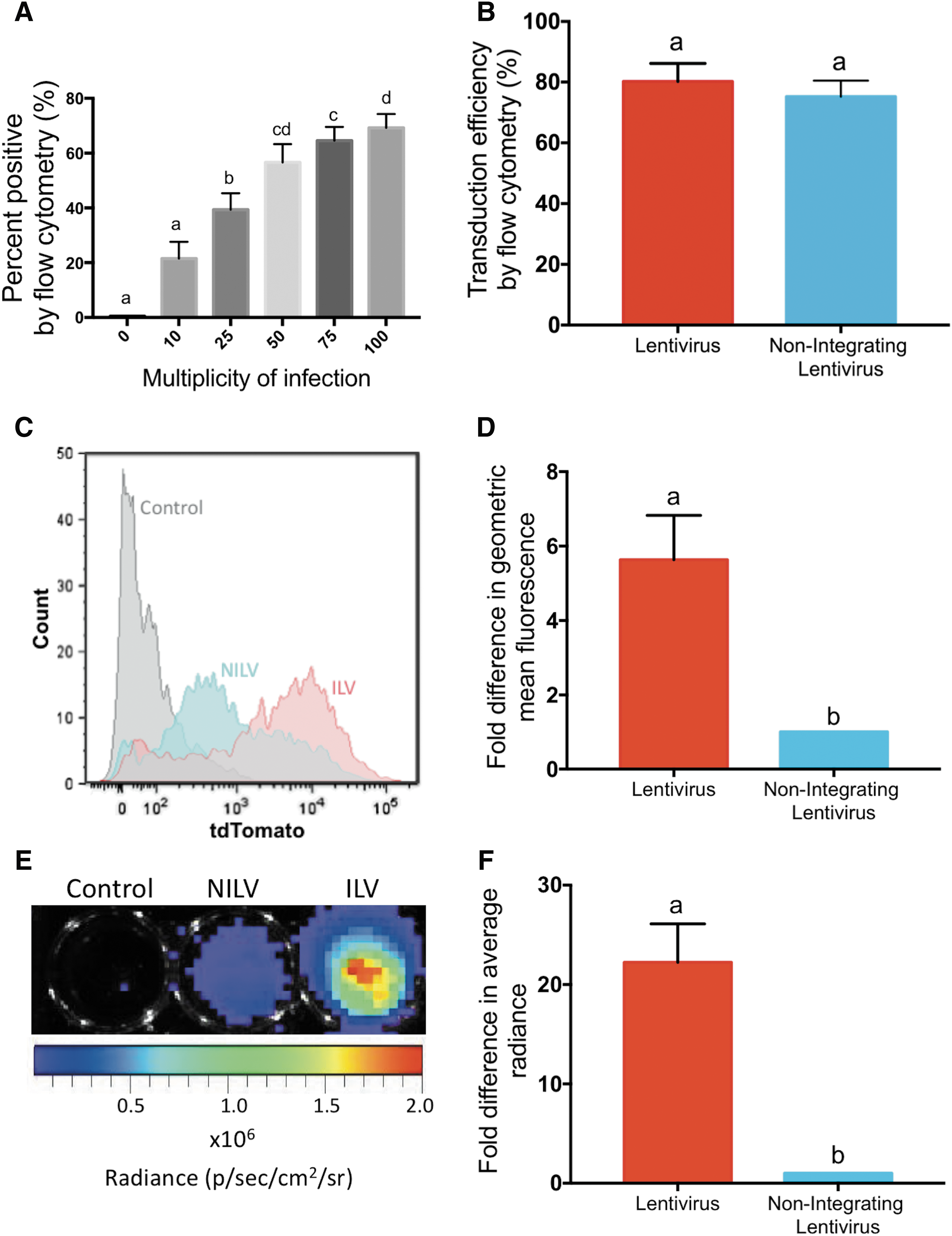
Evaluation of transduction efficiency and reporter gene expression in ILV- and NILV-transduced mesenchymal stem cells (MSCs) immediately after transduction.
Next, MSCs were transduced at an MOI of 75 with ILV or NILV, and tdT and luc2 expression were evaluated over time. Similar to what was observed with MDA-MB-231 cells, ILV-tagged MSCs exhibited stable tdT expression at 85.3 ± 5.3% (Fig. 3A and B) and consistent luc2 activity with an average radiance of 4.21 × 10 5 ± 4.15 × 10 4 photons/s/cm2/sr per 10 3 cells over time (Fig. 3C and D). In agreement with earlier findings (Fig. 2), successful gene transfer with NILV was evident immediately after transduction as 78.4 ± 17.0% of NILV-tagged MSCs displayed tdT expression. Differential tdT expression was evident by fluorescence microscopy (Fig. 3B), and luc2 activity was detected at 2.69 × 10 4 photons/s/cm2/sr per 10 3 cells immediately after NILV transduction (Fig. 3D).
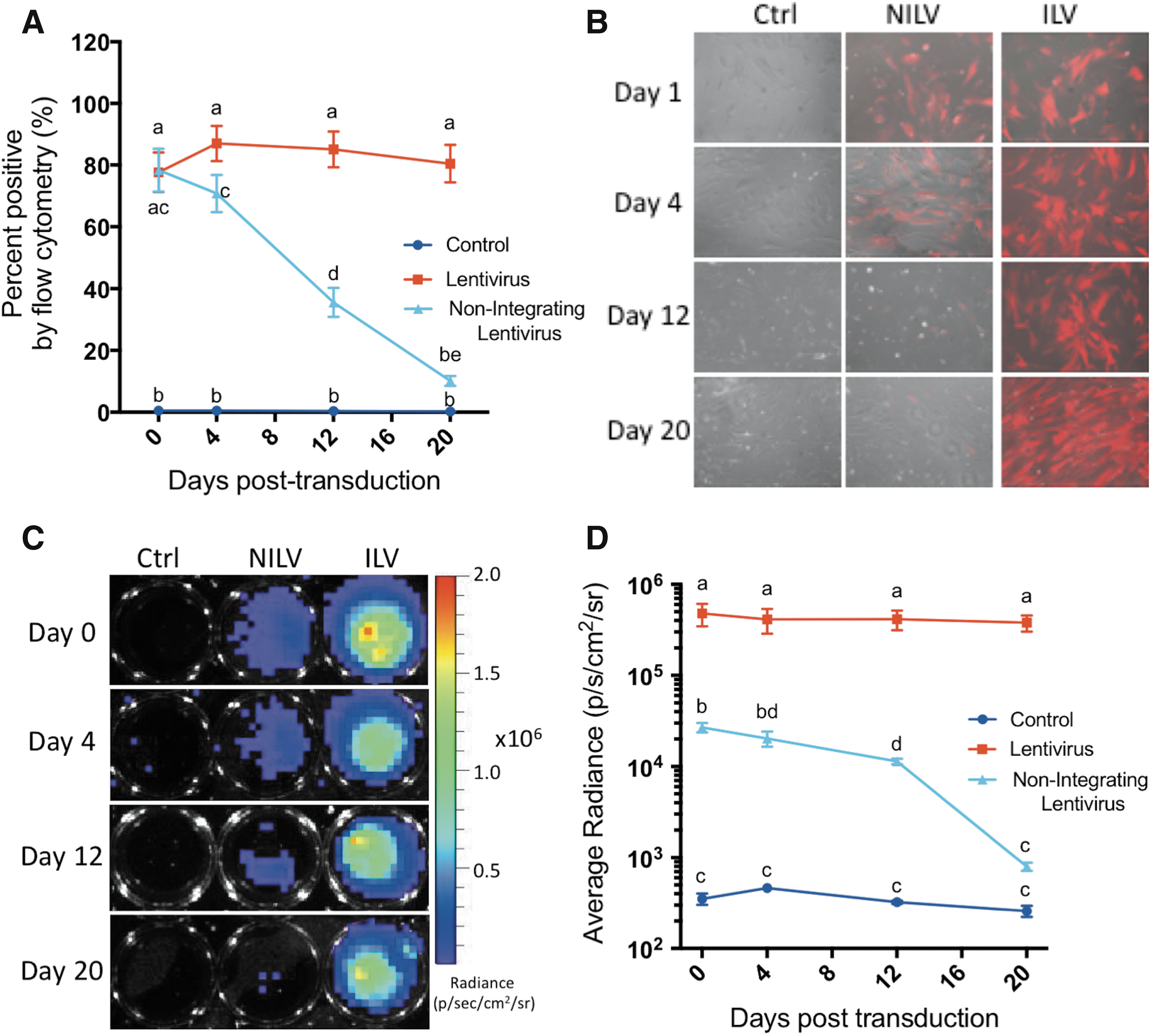
Longitudinal analysis of reporter gene expression in ILV- and NILV-tagged MSCs.
The in vitro proliferation rate of MSCs has been documented to be slower than that of MDA-MB-231 cells, with doubling times of approximately 80 h. 36 With this known difference in growth rate we anticipated that reporter gene expression would be retained for a greater period of time in the slowly proliferating MSCs. Four days after NILV transduction 71.5 ± 15.1% of MSCs successfully retained tdT expression (Fig. 3A) and luc2 expression was reduced by only 24% to an average radiance of 2.03 × 10 4 ± photons/s/cm2/sr per 10 3 cells (Fig. 3D). Flow cytometric analysis further revealed that after 12 days of proliferation 36.6 ± 10.1% of NILV-tagged MSCs were still tdT positive and expression only reduced to approximately 10% after 20 days of growth (Fig. 3A). Similarly, 42% of original luc2 activity was still evident by BLI analysis on day 12 after NILV transduction, and NILV-tagged MSCs were still detectable with an average radiance of 7.95 × 10 2 ± 1.65 × 10 2 photons/s/cm2/sr per 10 3 cells 20 days posttransduction. These results collectively demonstrated that NILV could be used to successfully and safely deliver reporter genes to MSCs and permit longitudinal tracking at detectable levels for multiple weeks posttransduction under slowly proliferating cell conditions in vitro.
Longitudinal bioluminescence imaging of ILV- and NILV-tagged MSCs in mice
Longitudinal in vivo BLI of MSC viability was performed with a stem cell model of transplant rejection. In this model, MSCs are injected into the hind limb of nude mice, whereby cells are lost over time due to cell death or rejection. Our previous work in this model involved injection of 10 6 perfluorocarbon-labeled MSCs, and fluorine-19-based MRI showed that the fluorine signal slowly declined over time and was detectable for approximately 2 weeks. 37 Because of the greater sensitivity of BLI compared with MRI, for our current study we injected 4 × 10 4 ILV- or NILV-transduced MSCs into the hind limb and BLI was performed on days 0, 3, 6, 10, 14, and 17 postimplantation. Cell implants were easily discernable in both ILV- and NILV-tagged MSC-injected mice (Fig. 4A and B, respectively). In agreement with our in vitro assessments, approximately 38-fold greater signal was detected from ILV-tagged compared with NILV-tagged cells (p < 0.05); however, the average radiance was observed to similarly diminish over time for both ILV- and NILV-tagged MSC implantations (Fig. 4C and D). Despite the minimal number of cells injected, both cell populations were reliably detectable by BLI for up to 2 weeks in vivo. Seventeen days after cell implantation mice were sacrificed and hind limb muscle was isolated and assessed for the retention of tdTomato-positive cells. Fluorescence microscopy of DAPI-stained sections identified nucleated red fluorescent cells in select tissue sections of ILV-transduced MSC-injected mice (Fig. 5A) and NILV-transduced MSC-injected mice (Fig. 5B). In both cases, the red fluorescence-positive cells were found in connective tissue neighboring the isolated muscle.

Longitudinal viability tracking of ILV- or NILV-tagged MSCs in vivo in mice.
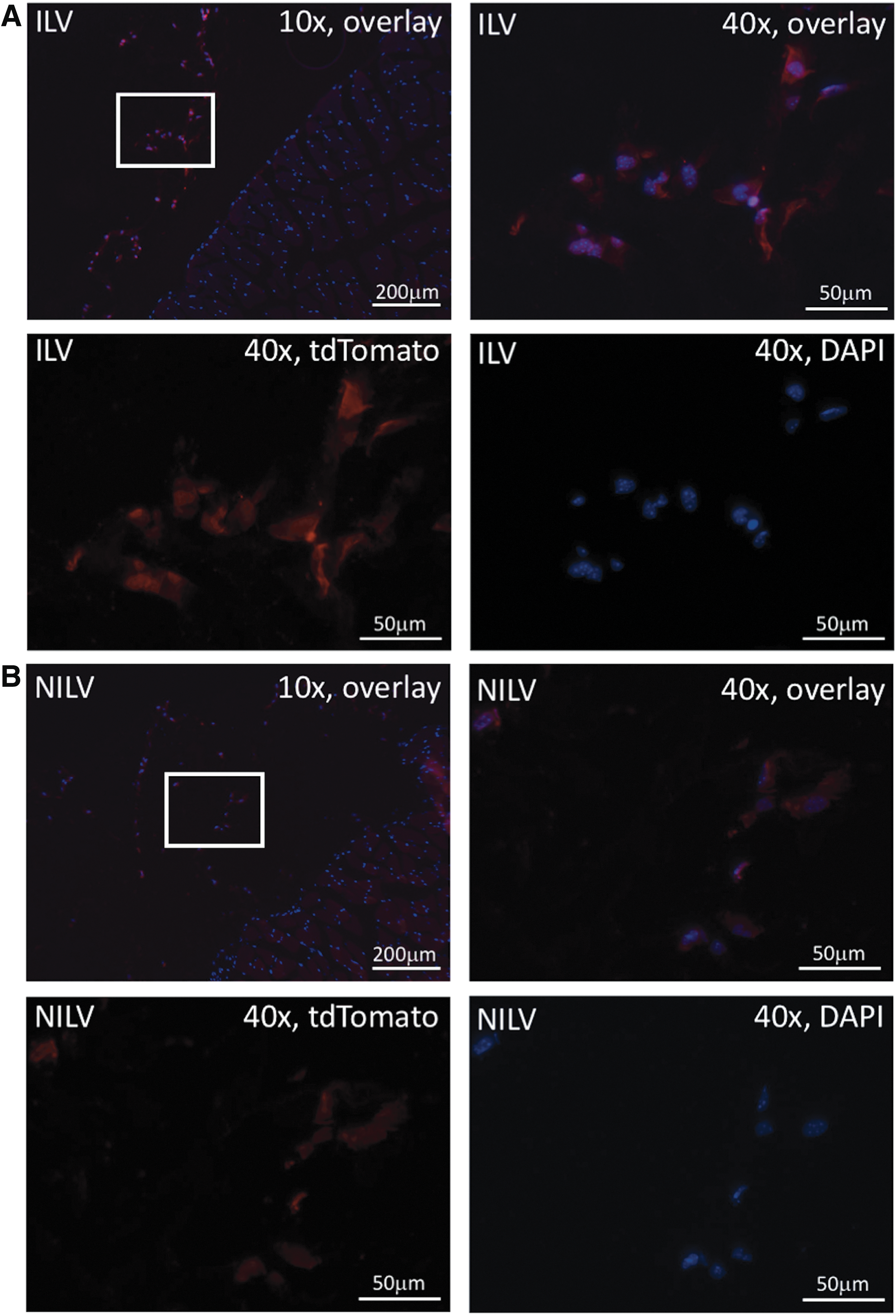
Fluorescence microscopy of endpoint mouse hind muscle after intramuscular injection with ILV- or NILV-transduced MSCs.
Discussion
In this study we present NILVs as an efficient means of episomal reporter gene expression in MSCs for noninvasive assessment of cell viability by molecular imaging. Previous work using NILVs in MSCs or progenitor cells has focused mainly on efforts to carry out site-specific gene modifications, using zinc finger nucleases 38 –40 or nuclease-assisted homologous recombination. 41 NILVs have also been used in human stem cells to safely identify differentiated cells by employing a fluorescent reporter driven by a differentiation-specific promoter and enabling fluorescence-activated cell sorting of differentiated cells from the pluripotent parent population. 42 Work by Verghese and colleagues has investigated the addition of a scaffold/matrix attachment region (S/MAR) to prolong transgene expression from NILV episomes in hematopoietic progenitor cells 43 and evaluated their use as an allogeneic stem cell transplant treatment for Fanconi anemia. 44 In our study, we successfully employed NILVs to transfer reporter genes and safely track the location and viability of transplanted MSCs in vivo over time. Despite reduced signal intensity and the minimal number of cells injected, live NILV-transduced MSCs were reliably visualized for up to 2 weeks postimplantation.
Safety is a major regulatory concern for future clinical applications of reporter gene imaging. Both viral and nonviral approaches have been utilized to engineer stem cells. Adeno-associated virus (AAV), adenovirus (AdV), herpes simplex virus (HSV), and ILV have all been studied for use in stem cell gene editing and other gene-based gene therapies. 13,45 AAV is a small virus that can safely deliver a predominantly episomal DNA vector with diminished pathogenicity and immunogenicity in humans. 46 One considerable disadvantage of this viral vector, however, is its small transgene capacity, which restricts its delivery of multiple reporter or therapeutic genes. 46,47 AdV vectors has an effective large gene transfer capacity and shows safe transient episomal expression of transgenes 1 ; however, MSCs lack the AdV receptor, making them largely resistant to AdV transduction. 48 In addition, the prevalence of AdV and HSV among the healthy population has resulted in a broad preexisting immunity against these viruses, thereby necessitating the use of rare serotypes 47 and complex production strategies. 13 Although preexisting immunity is a reason for alarm for viral gene therapy approaches, it is less likely to hinder gene delivery with aims of enabling imaging-based cell tracking. Of greater concern for both vectors, however, is whether an HSV or AdV vector coinfects a cell with an underlying latent wild-type infection. The prevalence of these viruses in the general population and the opportunity for genetic recombination represent a considerable safety concern, particularly for patient-derived therapeutic cells. 13,49
Nonviral episomal vectors have also been used previously to introduce exogenous reporter genes into MSCs. MSCs are known to be difficult to transfect. 50 Many nonviral systems using cationic liposomes or nanoparticles have been developed and applied to MSCs but usually suffered from low transfection efficiency (less than 35%) and short duration of reporter gene expression (less than 7 days). More recently, a minicircle microporation-based nonviral gene delivery system has shown more promising results with up to 66% transfection efficiency and fluorescent reporter gene expression persistence for more than 7 days. 50 Although these impressive results successfully widened the practical application of nonviral episomal vectors they still fail to compare with the gene transfer efficiency and duration of reporter gene expression we report here using NILVs.
Site-specific genome editing is another approach that has been used in the effort to introduce exogenous genes into stem cells and other progenitor cell types without incurring the risk of insertional mutagenesis. 51,52 Multiple methods, including the use of φC31 site-specific recombinase 52 or zinc finger nucleases, 51 have been successfully used to introduce multiple stable molecular imaging genes into progenitor cells while maintaining pluripotency and self-renewal. These methods, however, require substantial cell expansion, and are usually single-cell derived due to low editing efficiency, making them less attractive to the clinical applications where large cell numbers (>10 6 ) and low passage number (<7) are desired.
ILVs exhibit several advantageous traits that have made them the vector of choice for MSC transduction including high transduction efficiency, robust transgene capacity, and long-term reporter expression. 1,15 Despite no evidence of ill effects due to ILV transduction in MSCs, 16,17 previous instances of insertional mutagenesis observed with γ-retroviral transduction of therapeutic cells in a clinical trial have maintained genetic integration as a safety concern for cellular therapeutics aimed at clinical use, 53 particularly for non-life-threatening conditions where MSCs have been proposed as a potential therapy (e.g., osteoarthritis, macular degeneration, etc.). To address this concern, NILVs represent an ideal alternative as they can effectively deliver genetic material while minimizing nonspecific integration events, thereby maximizing safety and, it is hoped, broadening the potential applications.
Although NILVs drastically reduce integration, there still remains a low rate of residual integration, which may preclude NILV use for certain applications. Extensive previous work from other groups has investigated the rates of residual integration when using different variants of NILV. Catalytic triad mutations (D64, D116, and E152; two of which were used in our double mutant) have been found to show relative integration frequencies about four orders of magnitude lower than wild-type virus. 28,33,34 These class I mutations have also been shown to cause no reduction in DNA synthesis and reduced integration frequencies compared with other forms of NILVs such as those possessing mutations in the U3 or U5 att site. 54 It has also previously been shown that a combination of catalytic domain mutation (D64V) and att site mutation does not result in a further reduction in integration efficiency, and thus offers no addition safeguard from insertional mutagenesis. 33 Integration site analysis has also suggested that the minimal integration observed with catalytic domain mutants is not mediated by integrase but by other background recombination events. 33 In fact, estimates of integration frequency from NILVs in cultured cells are within the range described for plasmid transfection. 34,55 Our choice to use multiple integrase mutations confers an additional safeguard as it minimizes the potential of reversion back to an integrating phenotype. 34 To our knowledge our work represents the first time that this approach has been applied to enable viability imaging of an MSC population.
The potential safety benefits gained in the use of NILVs come at a cost of transgene expression. We and others have observed that despite efficient transient transduction, NILVs confer lower transgene expression levels than that seen with their integrating counterparts. 26,34,56,57 Several approaches have been utilized in an attempt to combat this known consequence of episomal expression including the use of strong promoters and enhancing elements, as well as the removal or reduction of inhibitors to episomal transgene expression. 57 In our NILV formulation we applied both of these tactics by using a transfer plasmid with a strong mammalian constitutive promoter (pEF1α) and a self-inactivating 3′ long terminal repeat. This resulted in sufficient transgene expression to enable the visualization of a small cell number transplant in vivo, using BLI. Several groups have shown that DNA episomes can become organized into chromatin structures and silenced through the action of chromatin-remodeling histone deacetylases. 58,59 Quite interestingly, Pelascini and colleagues were successful in rescuing NILV transgene expression in a variety of cell types in vitro including MSCs, using histone deacetylase inhibitors. 58 Our future efforts will aim to further improve detection by employing strategies to combat transgene silencing as well as the use of new BLI reporters with greater sensitivity. 59
The dual-modality imaging approach used in our current study is a valuable tool that can be utilized to track the in vivo fate of MSCs and enable the monitoring of cell distribution, viability, and migration in real time. ILVs have a history of safe use and offer higher transgene expression; thus, our ILV formulation may be ideal and sufficient for preclinical evaluations. However, as regulators continue to look for ways to minimize potential risks in cell-based therapy, NILV represents a safer approach to gene-based imaging for clinical studies. The optical imaging techniques used as proof of principle in this paper are ideal for imaging small animals but are not practically feasible for large-animal or human studies. 10 For clinical translation, NILVs can be engineered to express human reporter genes and visualized by sensitive clinical imaging modalities such as PET and MRI. Reporter gene imaging of therapeutic cells in humans has been demonstrated using PET imaging. In these first-in-human studies a specific PET tracer was used to image HSV thymidine kinase (HSV-tk) in engineered cytotoxic T lymphocytes to monitor chimeric antigen receptor therapy for the treatment of high-grade gliomas. 60,61 To avoid immunogenicity several human reporter genes have been developed and tested preclinically, including thymidine kinase 2 for PET imaging 62,63 and most recently organic anion-transporting polypeptide IB3 for MRI. 64
In summary, NILVs are an efficient gene delivery system that we have shown can safely engineer MSCs to express imaging reporter genes, allowing viable cell in vivo tracking over time. To proper validate MSC-based therapeutic strategies the fate of administered cells should be further studied and image-based techniques, specifically reporter gene-based imaging, can serve as a vital tool to monitor the viability, biodistribution, and trafficking of MSCs in vivo. Lentiviral vectors are the most effective means to confer genes in MSCs and, by employing a nonintegrating approach, the incidence of insertional mutagenesis is minimized and safety is maximized. We believe NILVs may have broad clinical applications for expression of imaging reporters and other gene products in numerous cell-based therapies. Our future work will aim to improve reporter expression, explore clinically relevant reporters, and apply this approach to clinically relevant models of disease.
Footnotes
Acknowledgments
The authors thank Dr. Armand Keating (Krembil Research Institute, Cancer Clinical Research Unit, Princess Margaret Cancer Centre) for supplying human mesenchymal stem cells. This work was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (J.A.R.).
Author Disclosure
The authors have no conflicts of interest to disclose.