
Abstract
CD20 is an effective immunotherapy target for CD20+ B-cell malignant cells. Monoclonal antibody, especially rituximab, has been a conventional strategy in the treatment of B-cell malignancies such as non-Hodgkin's lymphoma. However, treatment with monoclonal antibodies has not been enough to overcome the refractory/relapse problems. Chimeric antigen receptor engineered T (CAR-T) cells have exhibited excellent therapeutic effect on lymphocytic leukemia in recent years. In this study, a CD20-specific CAR was constructed and the cytotoxic efficacy of CD20 CAR-T cells on B-cell malignant cells was evaluated by CD107a degranulation, pro-inflammation cytokine production, and true lytic ability in vitro and in vivo. It was found that CD20 CAR-T cells possessed stronger cytotoxic ability against CD20 highly expressed cells. Furthermore, when histone deacetylase inhibitor was used to enhance the expression of CD20 antigen on the surface of B-cell malignant cells via inducing acetylation of H3K9 on CD20 promoter site, it revealed that the cytotoxicity of CD20 CAR-T cells against histone deacetylase inhibitor–treated B-cell malignant cells was significantly enhanced.
Introduction
CD20 is a nonglycosylated transmembrane phosphoprotein and takes a role in the development and differentiation of B cells as a calcium channel. 1 CD20 is not expressed on the surface of hematopoietic stem-progenitor cells but is highly expressed at the stages of late pre-B cells and mature B cells, while in the process of differentiation into plasma cells, the expression level of CD20 is downregulated. 2 Previous work showed that CD20 was not only expressed on normal B cells, but also highly expressed on the surface of malignant B cells. 3 CD20 is expressed by all cases of NHL and chronic lymphocytic leukemia (CLL) as well as about 40% of precursor B-cell acute lymphoblastic leukemia. 4 Therefore, CD20 is an attractive target for malignant B-cell treatment. Targeting the CD20+ B cells with therapeutic monoclonal antibodies, especially rituximab, has been verified as a valid strategy in the treatment of B-cell malignancies that displayed prominent antitumor activity and prolonged the overall survival of patients with multiple lymphoma subtypes in combination with chemotherapy or as long-term maintenance therapy. 5,6 However, despite CD20 monoclonal antibodies improve outcomes of B cell malignancies, more than 47,000 NHL patients die from their disease every year in China. Therefore, alternative therapeutic strategies are needed for the treatment of refractory/relapsed B cell malignancies. A hopeful approach is adoptive immunotherapy using chimeric antigen receptor engineered T (CAR-T) cells that specifically recognize and induce cytotoxic activities against malignant B cells. 7 –10
CAR is an artificial protein that contains extracellular, transmembrane, and intracellular signal transduction domains. The extracellular domain is composed of a signal peptide, antigen recognition domain, and hinge domain. 11 The antigen recognition domain usually consists of a single-chain variable fragment (scFv) 12 as well as the receptor 13 or ligand 14 of tumor-associated antigen. In the beginning, the intracellular signal transduction domain was only composed of the intracellular domain of CD3ζ, 15 which had little cytotoxic ability against tumor cells and didn't maintain persistent existence in patients' bodies. Following this, costimulatory domains such as 4-1BB and/or CD28 were introduced into the intracellular domain, inserted between the transmembrane domain and CD3ζ, which were capable of enhancing cytokine production, 16 T cell proliferation, 17 and in vivo ability of CAR-T cells. 18 Expecting that introducing costimulatory domains would be able to enhance the function of CAR-T, the affinity of scFv, 19,20 the length of hinge domain, 21 and the density of antigen on target cells 22 will also affect antitumor efficacy of CAR-T cells. It has been verified that increasing the expression level of antigen could augment the cytotoxic ability of CAR-T cells against target cells. 22
It has been reported that histone deacetylase inhibitors (HDACis) such as romidepsin and valproic acid could enhance CD20 expression at both protein and mRNA levels in Burkitt's lymphoma cell lines. 23 HDACs are a family of enzymes that catalyze the acetyl groups removing from central histones, which promote the compaction of chromatin and depress the transcription of relative genes. 24 In addition, HDACis are able to suppress the enzyme activity of histone deacetylase and to rescue genes expression by acetylation of H3, such as H3K27 and H3K9. 25
Therefore, in this study, we generated and characterized a specific anti-CD20 CAR construct containing the CD20 scFv that was previously established and validated in our institute. 26 Subsequently, we verified the cytotoxic activity of CD20 CAR-T cells against B-cell lymphoma cells in vitro and in vivo and demonstrated the relationship between their activity and expression level of CD20 on the surface of target cells. Furthermore, we investigated whether HDACi could augment the expression of CD20 antigen on surface of B malignant cells to enhance the cytotoxic activity of CD20 CAR-T cells.
Materials and Methods
Plasmid construction and lentivirus production
pCDH-CD20 CAR
The previously reported vector encoding heavy-chain and light-chain antibody targeting CD2026 was used to construct the CD20 CAR. The light chain was reamplified by polymerase chain reaction (PCR) using the following primers: 5′-GCTAGCGACATCGAGCTCACTCAGTCTCC-3′ (NheI) and 5′-AGAACCACCACCACCGGAGCCGCCGCCGCCAGAACCACCACCACCCCGTTTGATCTCCACCTTGG -3′ (linker). The heavy chain was PCR amplified using the following primers: 5′-GGTGGTGGTGGTTCTGGCGGCGGCGGCTCCGGTGGTGGTGGTTCTCAGGTGAAGCTGCAGCAGTC-3′ (linker) and 5′-CCGGAATTCTG AGGAGACGGTGACCGTGA-3′ (EcoRI). The CD20 scFv was connected in order by overlap PCR. The PCR product was digested and ligated into pCDH-EF1α-MCS-T2A-copGFP (pCDH) lentiviral vector containing 4-1BB-CD3ζ signaling domains which was previously described by An et al. 27
pCDH-CD20
CD20 coding region was amplified from cDNA obtained from Raji cells using the following primers: (XbaI) 5′- CTAGTCTAGAATGACAACACCCAG AAATTC-3′ and 5′- CGCGGATCCAGGAGAGCTGTCATTTTCTA-3′ (BamHI). The PCR product was digested and ligated into pCDH-EF1α-MCS-T2A-BFP lentiviral vector which replaced copGFP of pCDH by BFP.
pLV-CD20 promoter
The CD20 promoter region was amplified from genomic DNA obtained from Namalwa using the primers (ClaI) 5′-ATCGATTTTTATGT AGACTTTAAGATTGTCTTTTGG-3′ and 5′-GGATCCAGTGGGTGCAGTTTGTT TCTCA-3′ (BamHI) and cloned into pLV-firefly luciferase mPGK-Bsd-GFP lentiviral vector (Biosettia).
Lentiviral vectors were produced in 293T cells as previously described. 28
Cell lines
Daudi, Raji, Namalwa, and K562 hematological malignant cell lines were obtained from ATCC and maintained in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS). Lentiviral producer cell line (293T) was maintained in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) FBS and 2 mM
K562 cells was infected with lentivirus carrying pCDH-luciferase-RFP and pCDH-CD20 plasmids, followed by sorting of the highly expressed BFP+RFP+ cells by flow cytometry (BD FACSAria III) to generate polyclonal cells stably expressing CD20 (K562-CD20high) and unsorted cells as K562-CD20low. Namalwa cells were infected with lentivirus caring pLV-CD20 promoter plasmid (Biosettia), followed by clonal selection using 100 μg/mL blasticidin (Invitrogen) to generate stably polyclonal cells containing CD20 promoter derived firefly luciferase (Namalwa-CD20 Prom-Fluc).
T cells isolation and transduction
Peripheral blood of healthy donors was obtained from Tianjin Blood Center. All subjects signed an informed consent in accordance with the Declaration of Helsinki. CD3-positive T cells were isolated and enriched as previously described. 27 T cells maintained in X-VIVO15 (Lonza) with 5% FBS, 20 μL Dynabeads human T-activator CD3/CD28 (Stem Cell) and 200 U/mL rhIL-2 (R&D Systems) at a cell density of 1 × 106 per mL. After 24 hours, cells were infected with lentivirus carrying pCDH-CD20 CAR plasmid or empty vector (VEC) to generate CAR-T or VEC-T cells.
Analysis of CD20 expression
Namalwa, Raji, Daudi, and K562 cells were seeded into 12-well plates at a density of 2 × 105 cells/plate. Following either treatment or nontreatment with HDACis, cells were harvested and stained with allophycocyanin (APC)-conjugated anti-human CD20 antibody and APC mouse immunoglobulin 2b, κ isotype as control antibody (Biolegend) for 30 minutes on ice, and then washed and resuspended in phosphate-buffered saline for flow cytometry analysis (BD LSRFortessa). Fluorescence intensity of CD20 and isotype was calculated by FlowJo 7.6.1 software. Specific fluorescence intensity (SFI) of CD20 expression was calculated using the standard formula: fluorescence intensity of CD20/fluorescence intensity of isotype.
Analysis of direct cytotoxicity
CAR-T cells were incubated with target cells at effector to target cell ratios of 1:1, 1:2, 1:4, or 1:8 for 24 h or 48 h before flow cytometric analysis (BD LSRFortessa). VEC-T cells were used as control effector cells. Then cells were harvested and washed once, stained with APC-conjugated anti-human CD20 antibody and APC-Cy7-conjugated anti-human CD3 antibody (Biolegend) for 30 min on ice, and then washed and resuspended in phosphate-buffered saline with 1 μg/mL DAPI for flow cytometry analysis. The percentage of CD3−DAPI− cells represented the residual level of target cells.
Cytokine detection assays
CAR-T cells and VEC-T cells were co-cultured with target cells at a 1:1 ratio for 48 h, and the co-culture supernatant was harvested. Cytokines (interferon gamma [IFN-γ] and tumor necrosis factor alpha [TNF-α]) were detected using an ELISA kit (R&D Systems) according to the manufacturer's instructions. Three repeated wells were designed.
Degranulation assay
The degranulation of T cells was performed as previously described. 29,30 CAR-T cells (1 × 105) were co-cultured with 1 × 105 target cells in triplicate in 200 μL of medium with PE-conjugated anti-CD107a antibody (Biolegend) and 1000 U/mL rhIL2 for 1 h, then 100 μg/mL monensin (BD Biosciences) was added. After 4 h, cells were labeled with APC-Cy7-conjugated anti-human CD3 antibody and analyzed by flow cytometry (BD LSRFortessa). All living CD3+ CD107a+ cells were used for analysis.
Real-time PCR
Namalwa, Raji, or Daudi cells were seeded into 12-well plates at a density of 2 × 105 cells/plate and treated with indicated dosages of Romidepsin (Abcam) or suberanilohydroxamic acid (SAHA; MedChem Express) for 72 h. RNA extraction, cDNA synthesis, and real-time PCR were performed following a previously published protocol. 28,31 Signals were normalized to that of a housekeeping gene, GAPDH. Primers for CD20 amplification are 5′- CTTTGGGGGCTGTCCAGATT-3′, and 5′- ATGGCAGCAAAGAGGCTCAA-3′; and for GAPDH are 5′-TGCACCACC AACTGCTTAG-3′ and 5′- GGATGCAGGGATGATGTTC-3′. 32
Western blot
Namalwa, Raji, or Daudi cells were seeded into six-well plates at a density of 1 × 106 cells/plate and treated with indicated dosages of Romidepsin or SAHA for 72 h. Cell lysates were prepared in RIPA buffer described previously. 33 Cleared cell lysates were subjected to SDS-PAGE using 10% polyacrylamide gel and transferred to PVDF membranes. The primary antibodies were purchased from Cell Signaling (H3K9Ac, H3) and Sigma (β-actin).
Quantitative chromatin immunoprecipitation assay
Namalwa cells were seeded into T25 flasks at a density of 5 × 106 cells/flask and treated with 300 nM Romidepsin or 500 nM SAHA for 72 h. Quantitative chromatin immunoprecipitation (ChIP) assays were performed using Pierce™ Agarose ChIP Kit (Thermo Scientific) according to the manufacturer's protocol. Samples were immunoprecipitated with anti-H3K9Ac antibody (Cell Signaling)
31
or with anti-H3 antibody (Abcam) as the internal control. The DNA fragments were purified and subjected to quantitative PCR with different pairs of primers along the human CD20 promoters (Supplementary Table S1; Supplementary Data are available online at
Luciferase assay
Namalwa-CD20 Prom-Fluc cells were seeded into 12-well plates at a density of 2 × 105 cells/plate and treated with indicated dosages of Romidepsin or SAHA for 72 h. Cells were harvested and lysed with 250 μL 1 × passive lysis buffer (Promega)/sample. The samples were shaken at room temperature for 15 minutes, transferred 30 μL cell lysate into 50 μL LAR II reagent (Promega), and measured by luminometer (Berthold Technologies, Germany).
In vivo NOD/SCID murine studies
CD20 CAR-T efficiency assay
Six-week-old NOD/SCID female mice were purchased from Institute of Laboratory Animal Sciences (CAMS&PUMC, China). After adaptive feeding for 1 week, 14 mice were irradiated at 200 cGy and intravenously inoculated with 1 × 106 Raji cells. Two days after transplantation, mice were randomized into two treatment groups, CAR-T and VEC-T. Individuals were injected intravenously with 1 × 107 CAR-T cells or VEC-T cells, respectively, at days 3 and day 6 and 5 × 106 CAR-T cells or VEC-T cells, respectively, at day 15.
Combination therapy of CD20 CAR-T and SAHA
SAHA was first diluted to 200 mg/mL with DMSO and then diluted 1:100 with 0.1% carboxymethyl cellulose (CMC) to a 2 mg/mL suspension. Forty NOD/SCID mice were irradiated at 200 cGy and intravenously inoculated with 2 × 106 Namalwa cells on day 1. Four days after transplantation, mice were randomized into four treatment groups: (1) VEC-T + 0.1% CMC, (2) VEC-T + 30 mg/kg SAHA, (3) CAR-T + 0.1% CMC, and (4) CAR-T + 30 mg/kg SAHA. At day 5 and day 10, 1 × 107 T cells (CAR-T or VEC-T according to group type) were injected intravenously and at day 15, 5 × 106 CAR-T or VEC-T cells were injected. On days 4, 5, 6, 9, 10, 11, 14, 15, and 16, 300 μL 0.1% CMC or 2 mg/mL SAHA was administered to mice by gavage.
The overall survival was measured from the date of transplantation until death. Dead mice were dissected for pathological analysis to confirm the diagnosis of lymphoma. All animal experiments were approved by the Institutional Animal Care and Use Committee of Peking Union Medical College.
Immunohistochemistry staining
The lymph node tissues were incubated with antibody against CD20 (Abcam) at 1:100 dilutions. Images were recorded using a Nikon Eclipse-Ti fluorescent microscope (Nikon, Japan).
Isolation and culture of normal B cells
The detail of normal B cells isolation and culture are described in the Supplementary Data Materials and Methods.
Statistical analyses
Values were expressed as mean + standard error of the mean or mean ± standard error of the mean. Significance of survival data were assessed using a log-rank test, and others were determined by Student's t-test.
Results
CD20 CAR-T cells are reactive against human lymphoma cell lines in vitro
CD20 CAR was created with CD20-specific scFv fused with previously validated lentiviral constructs containing CD8α hinge and transmembrane domain, 4-1BB costimulatory domain, and CD3ζ signaling domain 12 (Supplementary Fig. S1A). CD20 CAR was transduced into primary donor T cells (CAR-T), and empty vector transduced T cells as control (VEC-T). The efficiency of CD20 CAR expressed on T cells was about 50%–60% (Supplementary Fig. S1B).
To validate the cytotoxic ability of CD20 CAR-T, three Burkitt's lymphoma cell lines with different CD20 expression level were used (Namalwa, Raji, and Daudi). Surface protein expression was assessed by flow cytometry (Fig. 1A). The SFI of CD20 in different cell lines was calculated, which was highest in Daudi and lowest in Namalwa (Fig. 1B). Degranulation assay was used to validate lytic function of T cells and quantified by increased cell surface CD107a expression. 34 After 5 h co-culture, significant degranulation of CD20 CAR-T cells were observed, but not in VEC-T cells (Fig. 1C and D). After co-culturing target cells with T cells for 48 h, pro-inflammatory cytokine release was measured using ELISA. The levels of IFN-γ and TNF-α productions were found to be significantly increased in CD20 CAR-T cells co-cultured with Raji and Daudi cells but not in those co-cultured with Namalwa cells (Fig. 1E). The true lytic capability of CD20 CAR-T cells were tested, and dose-dependent lysis data were observed (Fig. 1F and Supplementary Fig. S2). Surprisingly, it was found that in terms of degranulation, pro-inflammatory factor production, and true lytic capability, CAR-T cells showed more cytotoxic efficient on Daudi cells, which expressed the highest level of CD20 molecule.

CD20 chimeric antigen receptor engineered T (CAR-T) cells are reactive against human B-cell lymphoma cells in vitro and in vivo.
CD20 CAR-T cells are reactive against lymphoma in vivo
After confirming CD20 CAR-T cells' reactivity against human lymphoma cells in vitro, we investigated their antitumor activity in vivo. Immunocompromised NOD/SCID mice were systemically engrafted with 1 × 106 Raji cells by intravenous injection. Then the mice were administered 1 × 107 CD20 CAR-T cells or VEC-T cells on day 3 and day 6 and 5 × 106 CD20 CAR-T or VEC-T on day 15 intravenously (Fig. 1G). The median survival time in the CAR-T cell treatment group was extended significantly compared with that of the VEC-T group. Median survival times of the CAR-T and VEC-T groups were 29 days and 23 days (Fig. 1H), respectively. CAR-T treated mice showed a longer survival time compared with the VEC-T group (p < 0.05). All transplanted mice developed aggressive lymphoma with extensive infiltrations of Raji cells in bone marrow and lymph node, which was confirmed by pathological analysis (Fig. 1I).
CD20 CAR-T cells exhibit high cytotoxic activity toward CD20-high expressed target cells
As shown above, CAR-T cells revealed higher cytotoxic activity toward Daudi cells that expressed a higher level of CD20 than that of Raji and Namalwa. To verify this finding, CD20-low expressed K562 cells (K562-CD20low) and CD20-high expressed K562 cells (K562-CD20high) were established, and the expression level of CD20 was detected by using flow cytometry analysis (Fig. 2A). The SFI of CD20 expression was calculated (Fig. 2B). Degranulation assay was performed and revealed that higher CD107a expression was observed in CD20 CAR-T cells co-cultured with K562-CD20high cells (Fig. 2C and D). After co-culturing for 48 h, CD20 CAR-T cells secreted more IFN-γ and TNF-α cytokines in co-cultured supernatant with K562-CD20high cells than that with K562-CD20low cells (Fig. 2E); the true lytic capability of CD20 CAR-T cells showed similar results (Fig. 2F and Supplementary Fig. S3). In brief, CD20 CAR-T exhibited higher cytotoxic activity toward CD20 higher expressed cells, whether CD20 expression was endogenous or exogenous.
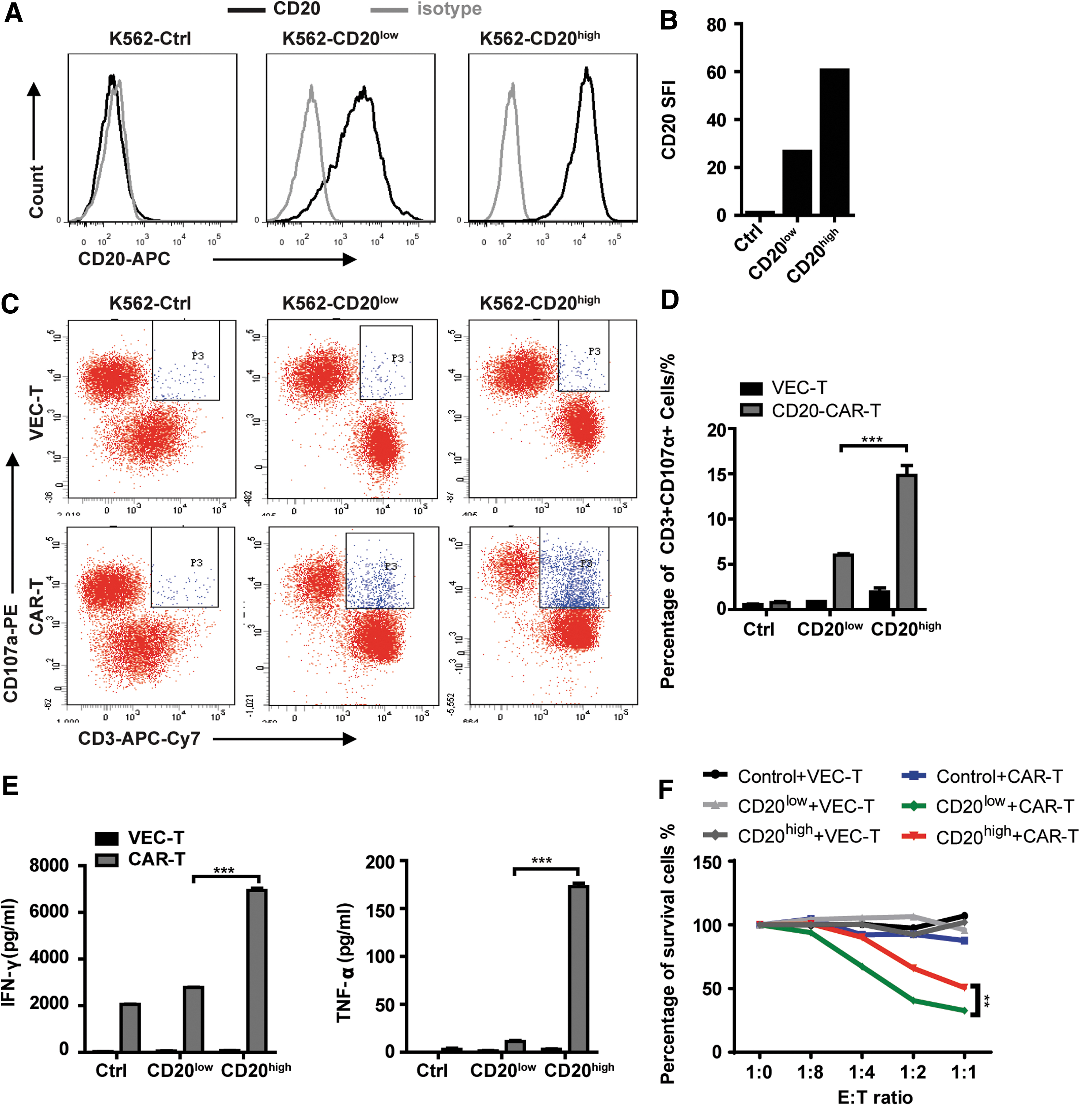
CD20 CAR-T cells exhibit high cytotoxic activity towards CD20 high expressed target cells.
HDACis enhance the expression of CD20 antigen on B-cell lymphoma cells
To improve the cytotoxicity toward CD20+ target cells, enhancing CD20 expression on lymphoma cells may be an effective strategy. A recent research has found that HDACis, such as romidepsin and valproic acid, could upregulate CD20 expression of lymphoma cells. 23 Therefore, lymphoma cells were treated with romidepsin and SAHA in various concentrations in this study, and accordant results were obtained that the CD20 SFI of lymphoma cells was enhanced (Fig. 3A and B), especially the proportion of CD20+ Namalwa cells was significantly increased (Fig. 3C), and the mRNA expression level of CD20 was upregulated in all three cell lines (Fig. 3D) by romidepsin and SAHA treatment. It is worth mentioning that the concentrations of romidepsin and SAHA we used were much lower than the IC50 of both lymphoma cells and T cells, and the two HDACis at these low concentrations hardly affect the growth (Supplementary Fig. S4A) and apoptosis (Supplementary Fig. S4B and C) of lymphoma cells.
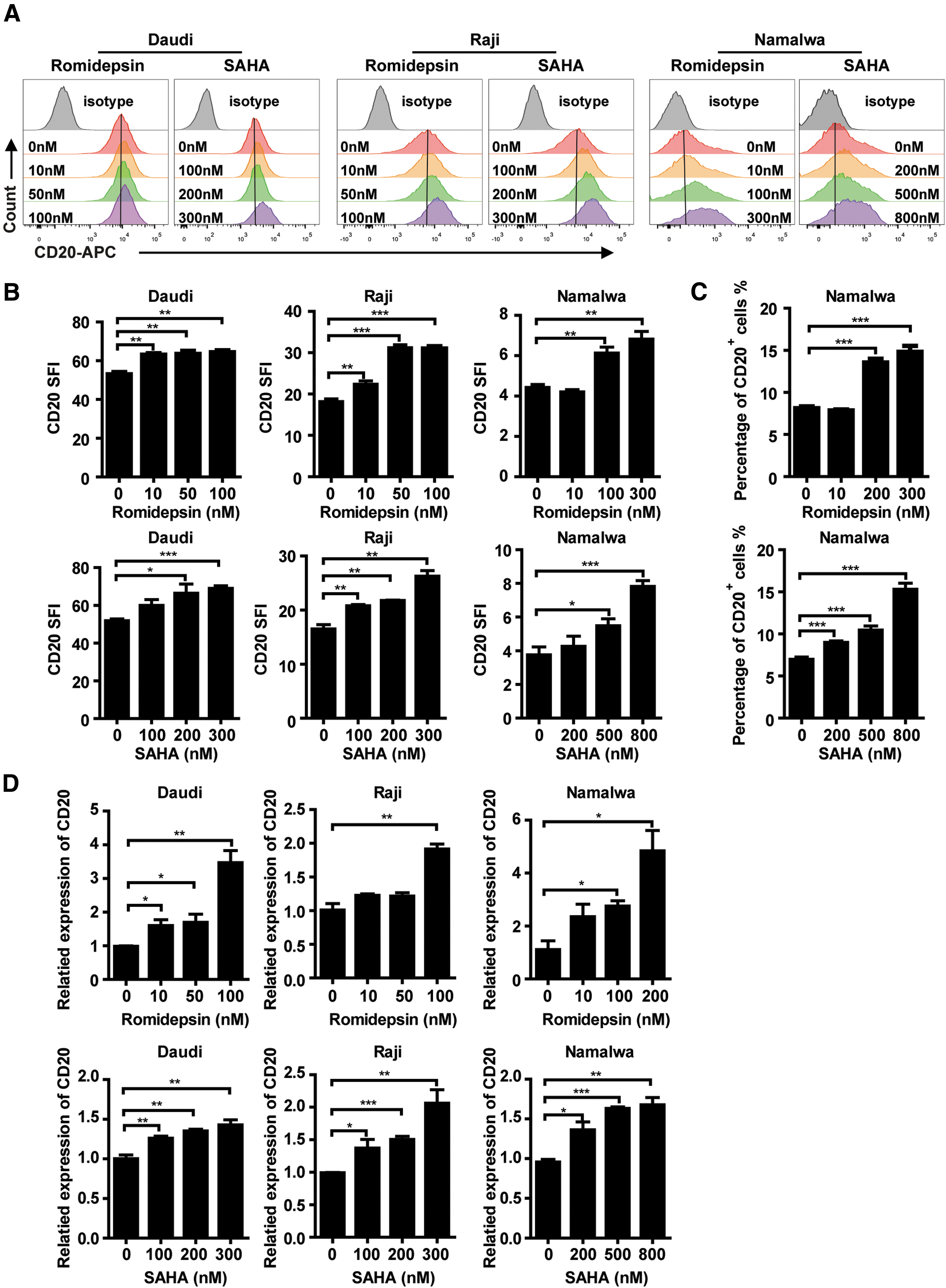
Histone deacetylase inhibitors (HDACis) enhance the expression of CD20 antigen on B-cell lymphoma cells.
In addition, we isolated normal B cells from three healthy donors and verified that HDACis could increase the expression of CD20 on normal B-cell surface (Supplementary Fig. S5A and B), similar to lymphoma cells, the low dose of HDACis had no effect on the apoptosis of normal B cells either (Supplementary Fig. S5C and D).
HDACi upregulate the CD20 expression through H3K9 acetylation
To investigate why HDACi could upregulate the CD20 expression, Western blotting was applied to detect the acetylation level of H3K27 and H3K9, which were frequently acetylated sites in active transcribed genes. 35 Results showed that the expression level of H3K9Ac increased in all three lymphoma cells in a dose-dependent manner when treated with romidepsin or SAHA (Fig. 4A), but no changes of H3K27Ac was detected (data not shown). To further examine whether these HDACis contributed to the acetylation of H3K9 on CD20 promoter site, ChIP assay was performed to test the acetylation level of H3K9 occupancy on the CD20 promoter following HDACis treatment. Chromatin DNA associated with H3K9Ac was immunoprecipitated from control cells or cells treated with romidepsin or SAHA, and quantified by real-time PCR using pairs of primers amplifying the binding regions in CD20 promoter (Fig. 4B). Romidepsin (Fig. 4C, upper panel) and SAHA (Fig. 4C, lower panel) did increase the acetylation level of H3K9 on the promoter region of CD20. Then a lentivirus-based plasmid containing CD20 Prom-Fluc was constructed and stably expressed in Namalwa cells (Namalwa-CD20 Prom-Luc). Luciferase assay was further performed, and results showed that the expression of firefly luciferase was enhanced in Namalwa cells by HDACis treatment (Fig. 4D), thus unveiling the regulatory mechanism of HDACi on the expression of CD20.
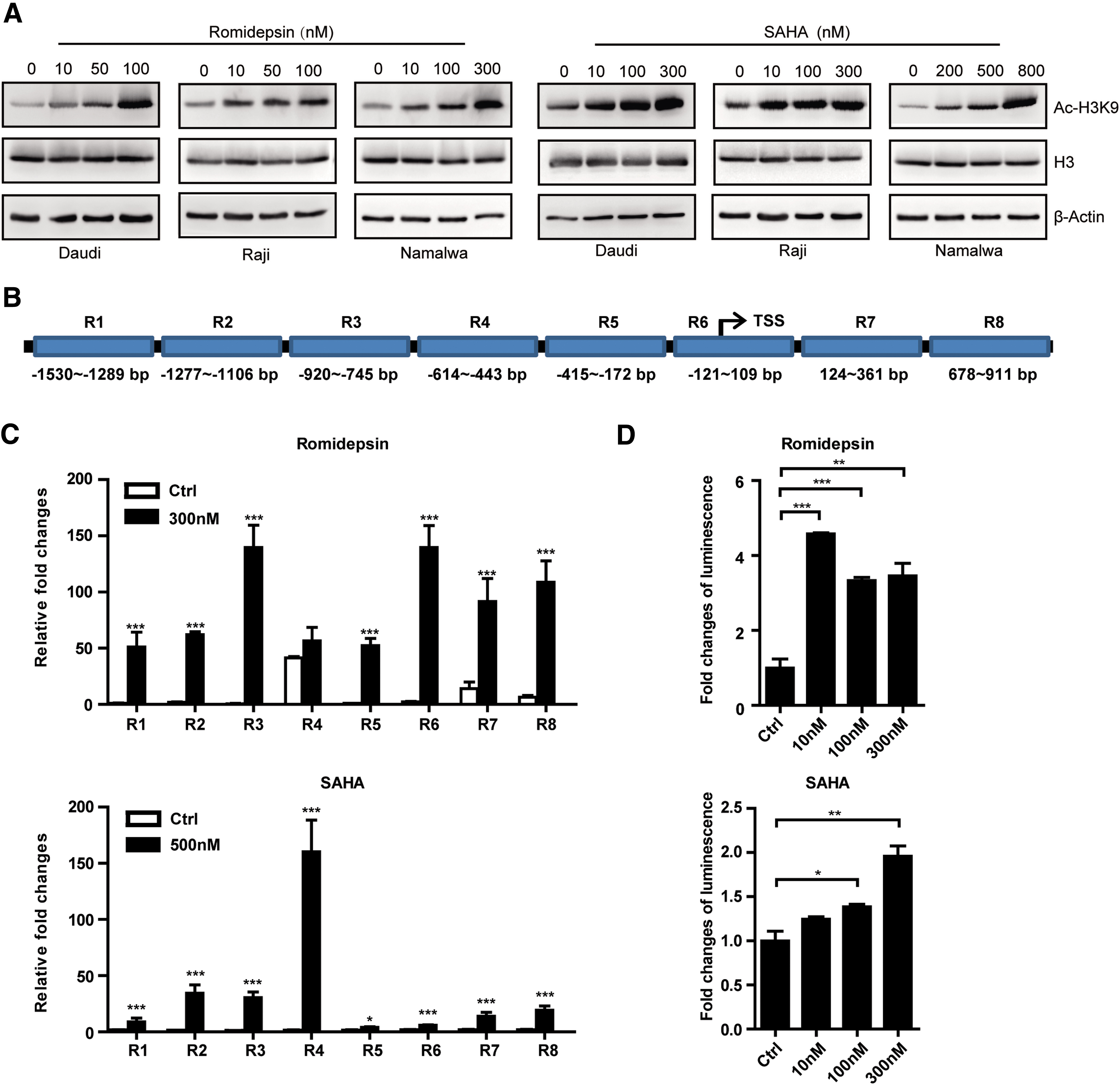
HDACi promote the CD20 expression through H3K9 acetylation.
HDACi enhance the cytotoxic effect of CD20 CAR-T cells in vitro
To test whether HDACi-induced upregulation of CD20 expression on target cells could sensitize their response to CD20 CAR-T cells' attack, lymphoma cells that had been preincubated with romidepsin or SAHA for 48 h were washed, seeded in fresh media, and co-cultured with CD20 CAR-T cells or VEC-T cells. The proportion of CD107a positive cells was significantly increased in CD20 CAR-T cells co-cultured with HDACis pretreated target cells for 5 h, especially with Namalwa cells (Fig. 5A and B). CD20 CAR-T cells secreted significantly more IFN-γ and TNF-α in the co-cultured supernatant of HDACi pretreated target cells for 48 h (Fig. 5C). In addition, the true lytic capability of CD20 CAR-T cells were also tested, and similar results were obtained, showing that CD20 CAR-T imparted more cytotoxic ability to HDACis-pretreated lymphoma cells (Fig. 5D and Supplementary Figs. S6, S7, and S8).
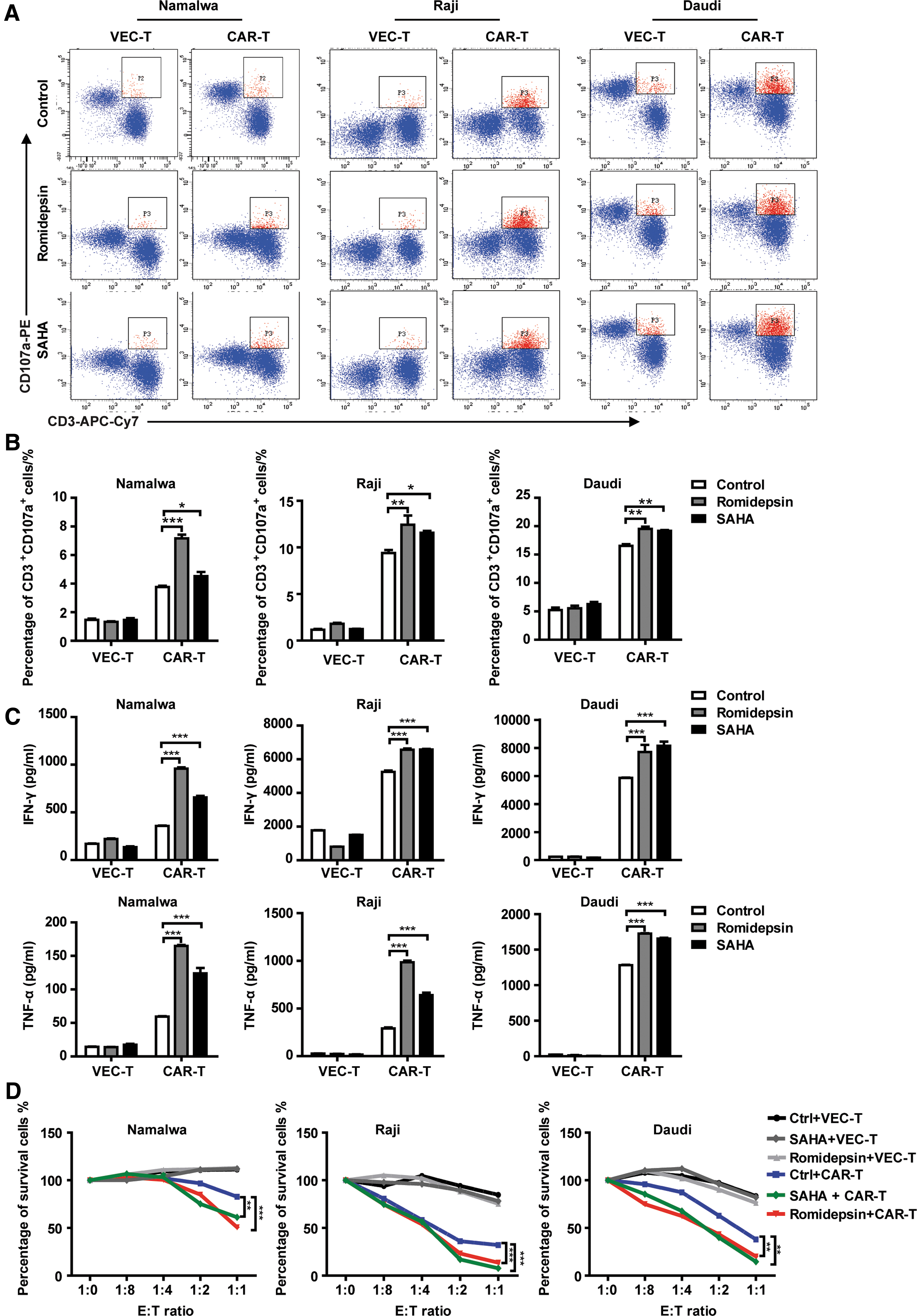
HDACi enhance the cytotoxic effect of CD20 CAR-T cells in vitro.
HDACi enhance the cytotoxic effect of CD20 CAR-T cells in vivo
To further demonstrate that HDACi augmented cytotoxic activity of CAR-T cells, SAHA was used in combination with CAR-T cells to treat Namalwa inoculated mice. The treatment regimen was shown in Fig. 6A. In the two CAR-T groups, the survival of mice treated in combination of 30 mg/kg SAHA was significantly prolonged compared with that in combination with 0.1% CMC (p = 0.0437). However, there was no difference in the survival time between the two VEC-T groups either in combination with SAHA or 0.1% CMC. Due to the low expression of CD20 on the surface of Namalwa, CAR-T cells in combination with 0.1% CMC failed to prolong the survival of the mice compared with that of VEC-T in combination with 0.1% CMC. However, when combined with SAHA, CAR-T cells exhibit powerful antitumor effect to significantly prolong the survival of mice compared with that of VEC-T in combination with 0.1% CMC (p = 0.006) (Fig. 6B). All transplanted mice developed aggressive lymphoma with extensive infiltrations of Namalwa cells in bone marrow and lymph node, which was confirmed by pathological analysis (Fig. 6C). Regardless of whether the mice were treated with VEC-T or CAR-T, the expression level of CD20 in the lymph nodes of SAHA-treated mice was higher than that of the control group (Fig. 6D).

HDACi enhance the cytotoxic effect of CD20 CAR-T cells in vivo.
Taken together, these findings demonstrated that CD20 CAR-T cells we generated could specifically target CD20+ lymphoma cells and their cytotoxic capability could be enhanced by HDACi via the induction of CD20 antigen expression.
Discussion
Here, we described a CAR specific for human CD20 for the targeting of B-cell malignant cells. Our initial data using endogenous CD20 antigen expressed in Burkitt's lymphoma cell lines Namalwa, Raji, and Daudi confirmed the viability against cell surface antigen CD20 with CAR-T cells as demonstrated by in vitro co-culture assays resulting in T cell degranulation, cytokine secretion, target cell lysis, and in vivo lymphoma mice model. From these results, we found that our CD20 CAR-T cells possessed not only effectively cytotoxic activity against CD20+ Burkitt's lymphoma cells, but also more cytotoxic activity against CD20 highly expressed cells as demonstrated by increased percentage of CD3+CD107a+ degranulation, augmentation of IFN-γ and TNF-α output from T cells, and enhanced true lysis ultimately. In addition, it was clear that CD20 CAR-T cells specifically eliminated Daudi target cells with the highest CD20 expression in low E:T ratio (1:8). Therefore, CD20-low and CD20-high expressed K562 cells were established, on the one hand to confirm the feasibility of targeting CD20+ cells with CAR-T cells, and on the other hand to provide evidence for CAR-T cells, and revealed high cytotoxic ability toward high antigen expressed target cells in a true CD20− acute myeloid leukemia (AML) cell line. Despite only moderate ability against Raji in vitro, CD20 CAR-T cells did significantly prolonged the survival of mice bearing lymphoma in vivo, implying that systemic delivery of CD20 CAR-T cells to tumor-bearing mice resulted in efficient lysis of lymphoma cells. The median survival time of Daudi-bearing mice was about 30 days without any treatment; if the mice were treated with 1 × 107 CD20 CAR-T cells 7 days after Daudi cell engraftment, the CAR-T cells were unable to significantly prolong the survival of mice (Supplementary Fig. S9). This result suggested that CD20 CAR-T may be effective at conquering only minimal tumor cells burden. However, many NHL patients achieve remission through chemotherapy or radiotherapy but ultimately relapse because of minimal residual disease, which is usually undetectable by conventional methods. 36,37 Therefore, CD20 CAR-T cells could be used as an effective strategy for patients with conventional treatment induced remission or minimal residual disease conditions.
Studies have reported that some NHL patients with lower levels of CD20 expression had a poor prognosis. 38,39 CD20-low lymphoma clones remain a potential obstacle for CD20 CAR-T immunotherapy because CD20 CAR-T cells demonstrated attenuated lytic activity against target cells with lower CD20 expression. Nevertheless, we and others have confirmed that HDACi definitely upregulates CD20 expression in Burkitt's lymphoma cells. 23 Romidepsin is a depsipeptide obtained from bacteria, which was approved by the U.S. Food and Drug Administration for cutaneous T-cell lymphoma in 2009 and other peripheral T-cell lymphomas in 2011. 40 SAHA, also known as vorinostat, was one of the first histone deacetylase inhibitor 41 approved by Food and Drug Administration for the treatment of cutaneous T-cell lymphoma in 2006. It is possible to enhance target cells with surface antigen expression under the critical value for recognition or lysis of CD20 CAR to levels high enough to stimulate T cells. Indeed, degranulation, cytokine production, and lytic ability from CD20 CAR-T cells was augmented after co-culturing with romidepsin or SAHA pretreated Burkitt's lymphoma cells. In addition, we have indicated that the functions of CAR-T were enhanced by the treatment with the addition of HDACi in vivo. Therefore, in patients with low CD20 expression levels, HDACi preconditioning before CD20 CAR-T treatment may enhance the cytotoxic activity of CD20 CAR-T on tumor cells in order to prolong survival of patients.
In addition to increasing the density of antigen on target cells, the moderate ability of CD20 CAR-T cells against CD20-low expressed target cells may also be conquered by optimizing the structure of CAR. It is reported that the activity of CAR-T cells can be enhanced by modifying the length of hinge domain, which could change the interspace between target cells and CAR-T cells, thus affecting the formation of synaptic junction and influencing the proliferation and antitumor activity of CAR-T cells. 21,42 Beyond that, alter the affinity of scFv may be an effective approach to improve the cytotoxic ability of CAR-T cells. Chmielewski et al. demonstrated that if the affinity of scFv was low (Kd >10−8 M), CAR-T cells had only cytotoxic activity against antigen-high expressed target cells, and if the affinity of scFv was higher (Kd <10−8 M), their cytotoxic activity was not relevant to the density of antigen. 19 Turatti et al. revealed that if the expression level of antigen was under suboptimal expression levels, when the affinity of scFv was Kd ≅10−6 , the cytotoxic activity of CAR-T cells was better than high scFv affinity (Kd <10−8 ) in some cases. 20 Although interaction between scFv and antigen is necessary to construct the immunological synapse and to stimulate the cytotoxic ability of T cells, if the combination was too strong to separate CAR-T cells from target cells, this would decrease the overall antitarget effect of CAR-T cells. 43 Therefore, we anticipate modifying the CD20 CAR-T cells by mutating the scFv to an appropriate affinity that may improve the antitumor efficacy.
In addition to NHL, CD20 expression is also reported on normal B cells. Healthy B lineage tissues remain a potential off-target by CD20 CAR-T cells. In recent innovations, several studies attempted to attenuate the risks of off-tumor toxicity. It is reported that mRNA of CD20 CAR was electroporated into T cells, resulting in transient expression of CD20 CAR and restricting the persistence of CAR-T cells. 44 In addition, an inducible suicide gene in combination with CD20 CAR was constructed into T cells at the same time, which was used for initiating apoptosis of CAR-T cells by the small molecule inducing dimerized drug AP1903. 45 But our study confirmed that the CD20 CAR-T we used had no killing effect on normal B cells (Supplementary Fig. S10). This may be due to the low expression level of CD20 on the surface of normal B cells (SFI ≅20) which escaped from the CAR-T cell killing.
A successful CD20 CAR-T cell platform has the potency for therapeutic benefit in various diseases beyond NHL. CD20 has also been described in CLL. 4 When using CD20-specific monoclonal antibodies, such as rituximab, the treatment outcome was moderate for CLL that response rates were low and many patients relapsed and/or became refractory to treatment. 46 In addition, CD20-specific antibody had successfully depleted CD20+ B cells in patients in several autoimmune diseases, such as rheumatoid arthritis, 47 thrombotic thrombocytopenic purpura, 48 and so on. In a word, CD20 CAR-T cells have the potential to exceed direct toxicity of antibody and may unfold an encouraging new approach to enlarge the use of CAR-T cells in cancer, as well as in other inflammatory related diseases.
Footnotes
Acknowledgments
Thanks Wei Zhou (School of Medicine, Nankai University, China) for helping to perform the immunohistochemistry staining of lymph nodes. This work was supported by grants from the National Key Research and Development Program for Precision Medicine (2017YFC0909800), National Natural Science Foundation of China (81430004, 81700163), Foundation for Innovative Research Groups of the National Natural Science Foundation of China (81421002), CAMS Initiative Fund for Medical Sciences (2016-I2M-1-007, 2017-I2M-1-015), PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (2017310024), and a State Key Laboratory of Experimental Hematology Research Grant (Z18-06).
Author Disclosure
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.