
Abstract
Patients with the lysosomal storage disease mucopolysaccharidosis IIIA (MPSIIIA) lack the lysosomal enzyme N-sulfoglucosamine sulfohydrolase (SGSH), one of the many enzymes involved in degradation of heparan sulfate. Build-up of un-degraded heparan sulfate results in severe progressive neurodegeneration for which there is currently no treatment. Experimental gene therapies based on gene addition are currently being explored. Following preclinical evaluation in MPSIIIA mice, an adeno-associated virus vector of serotype rh10 designed to deliver SGSH and sulfatase modifying factor 1 (SAF301) was trialed in four MPSIIIA patients, showing good tolerance and absence of adverse events with some improvements in neurocognitive measures. This study aimed to improve SAF301 further by removing sulfatase modifying factor 1 (SUMF1) and assessing if expression of this gene is needed to increase the SGSH enzyme activity (SAF301b). Second, the murine phosphoglycerate kinase (PGK) promotor was exchanged with a chicken beta actin/CMV composite (CAG) promotor (SAF302) to see if SGSH expression levels could be boosted further. The three different vectors were administered to MPSIIIA mice via intracranial injection, and SGSH expression levels were compared 4 weeks post treatment. Removal of SUMF1 resulted in marginal reductions in enzyme activity. However, promotor exchange significantly increased the amount of SGSH expressed in the brain, leading to superior therapeutic correction with SAF302. Biodistribution of SAF302 was further assessed using green fluorescent protein (GFP), indicating that vector spread was limited to the area around the injection tract. Further modification of the injection strategy to a single depth with higher injection volume increased vector distribution, leading to more widespread GFP distribution and sustained expression, suggesting this approach should be adopted in future trials.
Introduction
Mucopolysaccharidosis type III (MPSIII) is a lysosomal storage disorder caused by a deficiency in enzymes that degrade heparan sulfate (HS). Loss of the lysosomal enzyme N-sulfoglucosamine sulfohydrolase (SGSH) 1 causes MPSIIIA, the most common MPSIII subtype. 2 SGSH deficiency leads to a cascade of pathological events, resulting in significant neurological manifestations, with a median life-span of 15 years. 2 Patients have mild visceral symptoms. However, the central nervous system (CNS) is severely affected, with neuropathology characterized by accumulation of HS, gangliosides, cholesterol, and neuroinflammation. 3 Therefore, the focus of new therapies is to treat the neurological manifestations associated with this disease. 4 Replacement enzyme approaches are used to treat somatic disease in other MPS subtypes. However, due to the inability of the enzyme to cross the blood–brain barrier, this approach is ineffective for the treatment of MPSIIIA. To overcome this, intra-cerebrospinal fluid (CSF) injection of recombinant human heparan N-sulfatase has been tested in both mice and dogs. 5,6 This treatment provides a reduction in HS levels, which results in improvements in neuropathology and associated symptoms. Nonetheless, repeat enzyme injections generated a humoral response. Recent clinical trials of intrathecal delivery of exogenous enzyme (NCT01155778 and NCT02350816), delivered via lumbar ports to direct enzyme toward the brain, were beset with device failure issues and were ultimately unsuccessful, despite CSF HS reductions. 7
Allogeneic hematopoietic stem-cell transplantation (HSCT) is currently used effectively to treat neurological disease in a handful of lysosomal disorders, 8 –10 including related MPSI, a disorder whose primary storage substrate is HS, similar to MPSIIIA. Although the success of HSCT stems from the migration of myeloid cells across the blood–brain barrier and cross-correction of neighboring cells, HSCT has been shown to be ineffectual in treating MPSIIIA. 11 –13 Mouse models have shown this is likely due to inadequate enzyme levels achieved in the brain of MPSIII mice. 14,15 Myeloid-specific hematopoietic stem-cell gene therapy, relying on gene addition in patients' own hematopoietic cells, has been shown to correct neurological manifestations in a murine model of MPSIIIA. 16 This study showed that elevating expression levels in the brain above a threshold of 8.5% of normal SGSH was sufficient to improve primary storage, neuropathology, and behaviour. 16,17
Another approach to treat lysosomal diseases is the use of adeno-associated virus (AAV) gene therapy vectors to deliver the therapeutic transgene. 18 A number of serotypes isolated from human and nonhuman primates have been shown to transduce the CNS efficiently, most prominently AAV9 and AAVrh10. 19,20 AAV9 can cross the blood–brain barrier and reach the CNS after intravenous administration. However, the quantity and manufacturing cost of vector required to reach the brain to achieve clinical efficacy in humans is extremely high and carries an elevated risk of inducing immune responses and systemic toxicity. 21 Therefore, a direct delivery approach into the brain parenchyma may be favorable.
Direct administration of AAV has been used to deliver therapeutic transgenes in a number of preclinical and clinical studies of neurological lysosomal disorders. 22 –26 In a recent study, 5-week-old MPSIIIA mice received a unilateral intracranial injection of AAVrh10, combining SGSH and SUMF1 in a single vector. 26 The inclusion of sulfatase modifying factor 1 (SUMF1) was based on the observation that co-expression of this factor enhances enzyme expression in a number of sulfatase deficiencies. 25,27,28 Dual gene delivery resulted in an ipsilateral restoration of SGSH and reduction in HS storage, and consequently a decrease in activated microglia. At later time points after treatment, a decline in GM3 ganglioside accumulation and ubiquitin-positive lesion formation was also observed. The same vector, SAF301, was later used in a Phase I/II clinical trial for MPSIIIA 23 in which patients received 7.2 × 1011 viral genomes simultaneously via six injection sites at two depths in 60 μL deposits bilaterally to the white matter anterior, medial and posterior to the basal ganglia. They reported no signs of toxicity. The treatment was well tolerated, and clinical assessment suggested possible improvements in neuropsychological and behavioral measures. Nonetheless, although these data are encouraging, they do suggest that this vector could be improved to increase therapeutic effect. This can potentially be achieved by codon optimizing the transgene, using a more robust promotor to drive gene expression, and/or improving the viral capsid.
This study compared the existing AAV vector used in the above clinical study, AAVrh10:PGK-SGSH-IRES-SUMF1 (SAF301), against two new variants: AAVrh10:PGK-SGSH (SAF301b) generated by the removal of SUMF1, and AAVrh10:CAG-SGSH (SAF302) generated by the exchange of the murine phosphoglycerate kinase (PGK) promoter with the chicken beta actin/CMV composite promoter in SAF301b. Expression from these vectors was assessed 4 weeks after intracranial injection into MPSIIIA mice. Vector distribution of the best vector, SAF302 carrying a green fluorescent protein (GFP) payload, was assessed via intrastriatal injections at two depths or a single-depth intrastriatal injection. It is reported that removal of SUMF1 from SAF301 slightly reduces enzyme activity while CAG-driven SGSH expression leads to a more effective treatment of MPSIIIA mice. The study showed that SAF302 was effectively distributed through the front and mid part of the brain, and was more effectively distributed by single injections, rather than injections at two depths, where vector spread was more limited to the tissue immediately surrounding the injection track. These data suggest that the SAF302 variant should be used in future clinical trials.
Methods
Mice
Mice were housed in groups of two to five per cage and maintained at 21 ± 1°C with a constant humidity of 45–65% on a 12-h light/dark cycle (lights on at 08.00 and off at 20.00) with access to food and water ad libitum. These studies were approved by the Ethics Committee of the University of Manchester. They were conducted under license from the Home Office in accordance with British legislation and the European Communities Council Directive 86/609/EEC for the care and use of experimental animals. MPSIIIA mice 29 on a C57BL/6J background (B6.Cg-Sgshmps3a/6J) were maintained by heterozygote breeding and genotyped, as previously described. 3,15,30
AAV production and titration
The SAF301b transgene plasmid (ITR-PGK-SGSH-ITR) was constructed by digesting SAF301 (ITR-PGK-SGSH-IRES-SUMF-ITR) with SacI to remove the SUMF1 gene. This digestion gave rise to a deletion of 200 bp of the SGSH gene. This missing part was replaced by inserting a polymerase chain reaction (PCR) fragment generated with oligos F (5′-CCAGAGCTCTACGACCGGAGCCGGGA) and R (5′-CCAGAGCTCTCACAGCTCATTGTGGAGG) on SAF301. This fragment was purified and digested with SacI. Insert and vector were ligated overnight, microdialized, and electroporated into DH5α competent cells. Positive colonies were further analyzed by sequencing.
The SAF302 transgene plasmid (ITR-CAG-SGSH-ITR) was constructed by replacing the GFP coding sequence in the pTRUF-11 plasmid (ATCC, MBA-331) with SGSH cDNA into the NotI and SphI sites. The original pTRUF-11 plasmid was used to generate SAF302GFP. Recombinant AAVrh10 vectors were produced, purified, and titered, as previously described. 31
Intracranial injections
MPSIIIA mice (8–14 weeks old; n = 5–6 per group) received bilateral injections via two different strategies into the striatum. In the short-term arm of the study, injection volumes were 4 μL per animal, with a bilateral injection of 1 μL delivered at two depths using a Hamilton syringe. The co-ordinates used were 2 mm lateral to the bregma at two depths: 3 mm and 2 mm. All mice received a total of 4.1 × 109 genome particles in 4 μL for the delivery of SAF301, SAF301b, SAF302, or SAF302GFP, and they were sacrificed 4 weeks post injection. In the long-term arm of the study, injection volumes were 6 μL per animal, with a bilateral injection of 3 μL delivered at a single depth using a Hamilton syringe. All mice received a total of 6 × 109 genome particles in 6 μL of SAF302GFP, and they were sacrificed 4 months post injection. The coordinates used were 2 mm lateral to the bregma to a depth of 3 mm. For SAF302GFP distribution studies, five mice were injected per group.
Sample processing
For the determination of SGSH activity, the measurement of HS levels, and the colorimetric bead arrays, samples were homogenized and sonicated in homogenization buffer (0.5 mol/L NaCl, 0.02 mol/L Tris, pH 7–7.5) and centrifuged at 2,200 g for 15 min at 4°C, and the supernatant was collected. Protein concentration was determined using the Pierce BCA assay kit (Thermo Fisher Scientific, Loughborough, United Kingdom). For immunohistochemistry, brains were fixed in 4% paraformaldehyde for 24 h and then incubated in 30% sucrose 2 mmol/L MgCl2/phosphate-buffered saline for 48 h before freezing at −80°C.
SGSH assay
SGSH activity was measured in individual coronal fifths using the SGSH activity assay using 4-methylumbelliferyl-β-D-N-glucosaminide (4/MU), as previously described. 15,16
HS analysis
Glycosaminoglycan chains were purified, AMAC labeled, and analyzed by reversed-phase high-performance liquid chromatography (HPLC), as previously described. 14,32,33 Brain extract from each specific brain region representing regions 1–5 from two to three mice within each experimental group was pooled for analysis of total levels of HS. The two pools per group (one pool for SAF301 due to technical issues) were then analyzed independently for each region, running each pool twice over the HPLC. Results are presented as an average of these two biological pools. Only one pool was used for region 5 due to limiting sample.
Detection of anti-AAV antibodies
Total antibody responses against AAV capsid proteins were measured in brain homogenates with an enzyme-linked immunosorbent assay, as previously described. 31 Serum samples from naïve mice injected with an adjuvant plus the specific AAV vector used for each treatment group (SAF301, SAF301b, or SAF302) were used as a positive control. Antibody levels were measured across twofold serial dilutions of either the serum from positive control samples or brain homogenate collected from all experimental samples. All brain regions from each mouse within each group were pooled in appropriate volumes to generate a sample representing the whole brain for each mouse.
Evaluation of cytokine levels
The levels of interleukin (IL)-1α, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, RANTES, granulocyte-colony stimulating factor (CSF), granulocyte-macrophage CSF, and keratinocyte chemoattractant (KC) were measured using the BD cytometric bead array (CBA) Flex Set kits (BD Biosciences, Oxford, United Kingdom), as previously described. 3,34 Pooled samples from the right and left hemisphere of each specific brain region for each mouse were used. Five mice were analyzed per experimental group for each region of the brain (regions 1–5).
Vector copy number
Analysis of vector biodistribution was performed by quantitative PCR (qPCR). 31 Genomic DNA from tissue homogenate was extracted using a Qiagen DNeasy Blood and Tissue kit (Qiagen, Manchester, United Kingdom). A standard curve was generated based on specific amounts of a linearized SGSH plasmid containing native human SGSH cDNA and compared against GAPDH using naïve genomic murine DNA. The primer sequences used were SGSH F (5′-CCAGCATCAGAATGGGATGTA-3′), SGSH R (3′-GGCGACGTAGAGGAAGAAAG-5′), GAPDH F (5′-TGCACCACCAACTGCTTAGC-3′) and GAPDH R (3′-AGGAACATCATCCCTGCATCC-5′). SYBR Green amplifications were performed using the PowerUp SYBR Green master mix (Thermo Fisher Scientific) to a final volume of 25 μL, including 5 μL DNA. Plasmid amounts were calculated to give the numbers of double-stranded vector genomes per diploid genomic equivalent.
Immunohistochemistry
Brain sections (30 μm) were examined, and appropriate slices were identified. Free floating immunofluorescent staining was performed on coronal sections taken at the bregma +1.7, +0.26, −1.18, and −2.62mm according to the mouse brain atlas. Sections were blocked in 5% goat serum, 1% Triton-X-100 in Tris-buffered saline (TBS) for 1 h at room temperature. Sections were then incubated overnight in primary antibodies made up in blocking solution at 4°C on a rocker. Sections were washed four times in TBS and then incubated with secondary antibody diluted in blocking solution for 1 h. Following one wash in TBS, sections were incubated in DAPI (300 nM) for 15 min. After four washes, sections were mounted and cover-slipped. The primary antibodies used for the co-labeling experiments in this study were as follows: chicken anti-GFP (Abcam, Cambridge, United Kingdom), rabbit anti-NeuN (Abcam), and rabbit anti-glial fibrillary acidic protein (GFAP; DakoCytomation, Ely, United Kingdom). Secondary antibodies used were AlexaFluor 488 goat anti-chicken (Life Technologies, Warrington, United Kingdom) and AlexaFluor 594 goat anti-rabbit (Life Technologies). Images were acquired on a 3D-Histech Pannoramic-250 microscope slide-scanner using a 20 × objective (Zeiss, Oberkochen, Germany) and the DAPI, TRITC, and FITC filter sets. Snapshots of the slide-scans were taken using the Case Viewer software (3D-Histech). Quantification of GFP (n = 5 mice per group) was performed using ImageJ, thresholding the total GFP-positive area versus total area of the brain determined from NeuN staining.
Statistical analysis
Statistical analysis was performed using GraphPad Prism Version 7 (GraphPad Software, Inc., La Jolla, CA). All data were analyzed by analysis of variance and Tukey's post hoc test. Where standard deviations were unequal, data were log transformed to achieve normal distributions. Significance was assumed at p < 0.05.
Results
In order to determine the brain distribution, promoter strength, and therapeutic activity of the new variants of the SAF301 vectors, MPSIIIA mice received intrastriatal injections of three vector variants expressing SGSH (SAF301, SAF301b, and SAF302) and one variant where the transgene was exchanged with GFP (SAF302GFP) via bilateral injections at two depths (Fig. 1A). SAF301 is the original vector used in a previous MPSIIIA clinical trial 23 composed of an mPGK promoter controlling the expression of SGSH and SUMF1, which are separated by an IRES sequence, leading to the production of separate proteins from the same transcript (Fig. 1B). The rationale for addition of SUMF1 was to determine if increased expression of SUMF1, an enzyme responsible for activating the sulfatase activity of SGSH by altering a cysteine residue in the active site to formylglycine, would aid enzyme activity. 25 SAF301b is a variant of the SAF301 vector in which the SUMF1 gene has been removed; SAF302 is altered to include expression of the SGSH transgene via the CAG promoter. Additionally, a variant of SAF302 expressing GFP was injected in order to assess vector distribution and promoter strength in the brain. All vectors were pseudotyped with the AAVrh10 capsid.
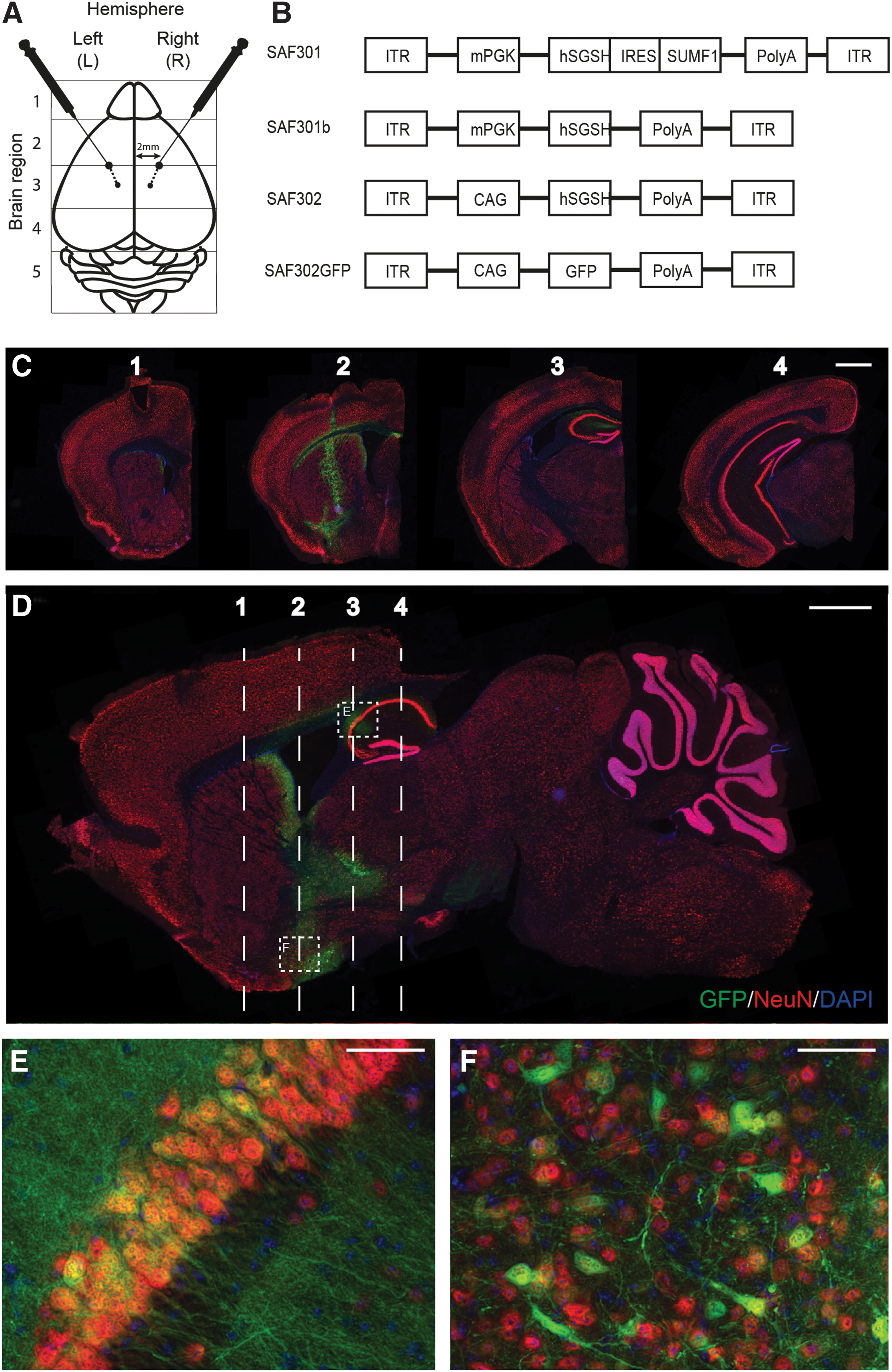
Intrastriatal injection of adeno-associated virus (AAV) vectors in mucopolysaccharidosis IIIA (MPSIIIA) mice.
Distribution of SAF302GFP after intrastriatal injection at two depths
Injected MPSIIIA animals receiving SAF302GFP via bilateral injections at two depths were sacrificed 4 weeks after injection, and brains were harvested. Coronal and sagittal sections were stained for GFP alongside neuronal marker NeuN and nuclear marker DAPI (Fig. 1C–E) to allow determination of GFP-expressing cells. Vector spread is illustrated in coronal sections 1–4 (Fig. 1C), with the positioning of coronal images 1–4 shown by the annotations on the sagittal image (Fig. 1D). The injection site is clearly visible at the bregma on coronal section 2 (Fig. 1C), with GFP-positive staining from the cortex through the striatum depicting the needle track. Little GFP-positive staining was evident within the cortex, except immediately surrounding the needle insertion site. However, clear transduction of cells within the hippocampus and striatum was evident (Fig. 1D), spreading both anterior and posterior from the injection site. High-magnification images of these regions stained with NeuN clearly demonstrate predominant neuronal transduction with this vector (Fig. 1E and F), with GFP detected in the nucleus and perinuclear cytoplasm of neurons and along neuronal processes, projecting from NeuN-positive neuronal cell bodies. Co-staining of GFP and NeuN in sagittal sections across the hemisphere also allowed vector distribution to be ascertained across the sagittal plane (Supplementary Fig. S1A–F). Clear vector spread in both directions away from the injection site (2 mm lateral to the bregma) both toward and away from the midline was apparent. Wild-type (WT) animals were also injected with SAF302GFP at equivalent sites. No differences in vector distribution were seen between WT and MPSIIIA animals (data not shown).
Increases in SGSH enzyme activity are dependent upon vector design and position relative to the injection site
The efficacy of the three therapeutic AAV-SGSH vectors (SAF301, SAF301b, and SAF302) were compared 4 weeks after injection by assessing SGSH enzyme activity across the brain following division of the brain into five distinct regions from front to back, as depicted in Fig. 1A. The five brain regions from the left and right hemisphere were processed independently (n = 5 animals per treatment, 10 independently injected hemispheres per group; Fig. 2).
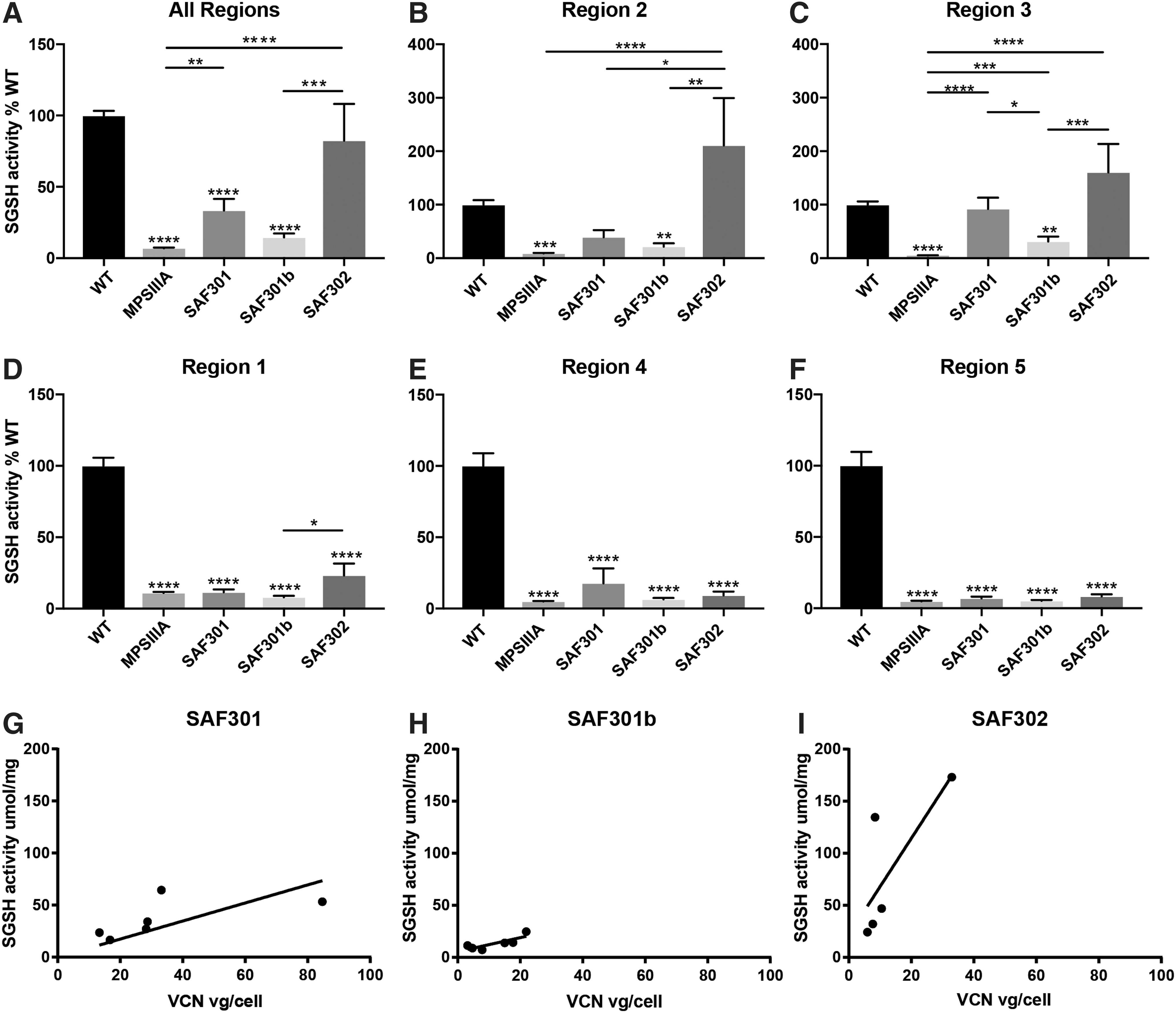
N-sulfoglucosamine sulfohydrolase (SGSH) activity levels in AAV-treated MPSIIIA mice vary with vector type and location relative to the injection site, with the highest level of enzyme activity observed with SAF302.
The injection sites lay on the border of regions 2 and 3. Injection with all three vectors resulted in increases in enzyme levels in region 2 of the brain compared to untreated animals. However, only treatment with SAF302 resulted in significant increases in SGSH levels, reaching approximately twofold the normal level measured in WT animals (p < 0.0001; Fig. 2B). Furthermore, treatment with SAF302 was significantly better than with SAF301 (p < 0.01) and SAF301b (p < 0.001). Likewise, in region 3, significant improvements in enzyme activity were observed following treatment with both SAF301 and SAF302 (p < 0.0001; Fig. 2C), with SGSH levels equal to or greater than WT enzyme. Results suggested SAF301 was superior to SAF301b, with little improvement in SGSH activity noted following SAF301b injection.
In areas more distal to the injection site (brain regions 1, 4, and 5), little difference in SGSH enzyme activity levels was apparent between untreated MPSIIIA mice and all AAV treated MPSIIIA mice, and they were all significantly different from WT (p < 0.0001; Fig. 2D–F, respectively). Thus, the improvement in SGSH enzyme activity in treated MPSIIIA mice varied significantly across brain regions (p < 0.0001), with regions closest to the injection site showing the highest activity. When the enzyme activity across all regions of the brain as a whole was studied (Fig. 2A), both SAF301 and SAF302 significantly increased SGSH levels compared to untreated MPSIIIA animals (p < 0.01 and p < 0.0001, respectively). However, only SAF302 recovered enzyme activity to near WT levels.
Vector copy number (VCN) was determined by qPCR on pooled regions across the hemisphere of injected animals (Fig. 2G–I and Supplementary Fig. S2). VCN was not significantly different in brain tissue treated with any of the AAV-SGSH vectors (Supplementary Fig. S2), although it is noted that one animal within the SAF301 group had a recorded VCN of 85—more than three times higher than other mice within this group. VCN was also plotted against enzyme activity for each mouse in SAF301, SAF301b, and SAF302 treatment groups (Fig. 2G–I, respectively), with comparisons suggesting an improved functional outcome per VCN following intrastriatal injection of SAF302.
Reductions in HS accumulation in the brains of MPSIIIA mice after SAF302 treatment
The primary storage material, HS, was measured in brain extracts from regions 1–5 from five untreated MPSIIIA, WT, and AAV-injected animals (SAF301, SAF301b, and SAF302). Due to sample size limits, individual regions were processed separately, with each coronal fifth of the brain from two or three animals pooled together in two independent biological samples per region prior to analysis. The amount of HS (μg) per milligram of brain protein was then determined by reversed-phase HPLC (Fig. 3). Total HS for the whole brain was calculated by averaging the values obtained across brain regions for each mouse and presenting it as fold increase over WT (Fig. 3A). A significant 9.2-fold increase was seen in untreated MPSIIIA mice compared to WT mice (p < 0.0001) due to the accumulation of un-degraded HS as a direct consequence of the absence of SGSH. Little change in total HS was achieved compared to untreated MPSIIIA animals following treatment with SAF301. Overall, HS levels in SAF301b and SAF302 were reduced to 6.8- and 4.7-fold above WT, respectively, indicating a clear trend toward decreased HS following treatment with these vectors versus MPSIIIA. However, elevation of HS compared to WT remained, presumably reflecting the incomplete transduction of all cells within these regions.
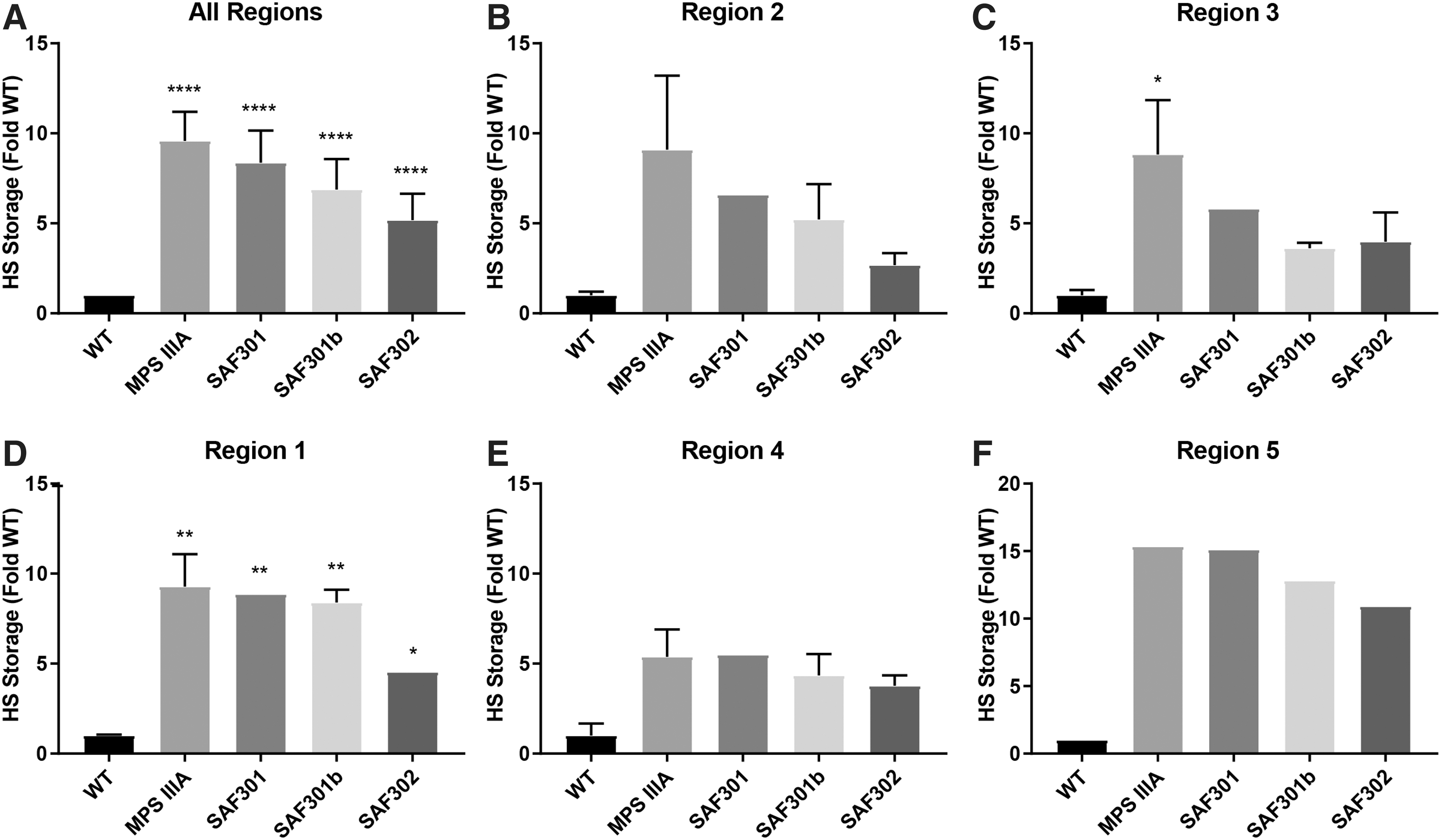
Total amount of heparan sulfate (HS) determined by reversed-phase high-performance liquid chromatography analysis of AMAC-labeled disaccharides.
When individual brain regions were studied, total HS was increased in untreated MPSIIIA mice compared to WT mice in all regions, as expected (Fig. 3B–F). In regions 2 and 3, close to the injection sites, clear trends toward a reduction in HS levels were observed with all AAV treatments. However, in region 2, SAF302 appeared to provide superior reductions in HS accumulation compared to the other two vectors, reducing HS by 3.4-fold over MPSIIIA. In region 3, treatments with both SAF301b and SAF302 were equivalent and provided enhanced reductions in HS compared to SAF301. There were also reductions in total HS levels in regions 1, 4, and 5 with SAF302, whereas SAF301 and SAF301b had little effect. Notably, limitations from pooling of the brain sections limited the ability to demonstrate significant differences in individual regions.
Reductions in neuroinflammation were most significant in MPSIIIA mice treated with SAF302
Neuroinflammation was assessed using CBA analysis to quantify a set of inflammatory cytokines in brain extracts from regions 1–5 of AAV-treated MPSIIIA mice (SAF301, SAF301b, and SAF302), and these were compared to equivalent brain regions of age-matched WT and MPSIIIA untreated mice. CBA values were averaged across brain regions for each mouse to give an overall reading for each analyte. Neuroinflammation was more evident in MPSIIIA mice compared to WT mice, as shown by significant increases in MIP-1α/CCL3 (p < 0.0001), MCP-1/CCL2 (p < 0.0001), and IL-1α (p < 0.05; Fig. 4A–C, respectively). Total levels of RANTES/CCL5 and KC/CXCL1 were, however, similar between WT and MPSIIIA mice at this early disease stage (Fig. 4D–E, respectively). An overall reduction in MIP-1α was only seen following treatment with SAF302 (p < 0.01). MCP-1 was significantly reduced following treatment with SAF301 (p < 0.01), SAF301b (p < 0.05), and SAF302 (p < 0.01) compared to untreated MPSIIIA mice. Finally, IL-1α was reduced following treatment with all three vectors. However, this was only significant with SAF302 (p < 0.01). Data for each of the analytes in individual brain regions are shown in Supplementary Fig. S3, demonstrating the varying response to vector treatment dependent upon position relative to the injection site, with the most significant effects noted close to the injection site in regions 2 and 3.

Assessment of neuroinflammation and AAV antibody responses in AAV-treated MPSIIIA mice.
Absence of brain-specific antibody response to AAV-SGSH vectors in injected MPSIIIA mice
Generation of antibodies against AAV capsid proteins can significantly reduce the effectiveness of AAV vector therapies. 35 Therefore, total antibody responses to AAV were analyzed in the brain of untreated mice and of mice treated with SAF301, SAF301b, and SAF302. Positive control samples were generated by subcutaneously injecting naïve mice with each AAV type mixed with an adjuvant known to induce antibody responses. Serum collected from these mice was used as a positive control sample. Anti-AAV immunoglobulin G antibodies were undetectable in untreated groups. However, high antibody responses were generated in positive control mice treated with adjuvant plus each AAV type. No anti-AAV antibodies were detected in mice treated with SAF301, SAF301b, or SAF302 (Fig. 4F–H, respectively), suggesting there is no antibody response to AAV within the brain at least.
Sustained, neuron-specific gene transduction following intrastriatal injection of SAF302GFP at a single depth
To investigate whether distribution could be enhanced using a larger injection volume at a single depth as opposed to delivery over two depths (as in Fig. 1) and to evaluate long-term expression of the vector, a second GFP distribution study was conducted to assess mice 4 months after injection. MPSIIIA mice received a stereotactic injection of AAV-SAF302GFP at 8–14 weeks of age via bilateral injections at a single intrastriatal depth (Fig. 5A; n = 5 animals). Animals were then sacrificed at approximately 6 months of age, which was around 4 months post injection. Coronal and sagittal sections were co-labeled with NeuN, GFP, and DAPI to give a comprehensive overview of vector distribution throughout the brain (Fig. 5B and C). Imaging of the brain sections demonstrated enhanced vector distribution in the animals that received injections at single depth compared to the animals in the short-term study receiving injections at two depths (compare Fig. 5B to Fig. 1C). Maximal GFP expression was present in the locality of the injection site, reducing in scope and intensity with increasing distance away from this region. Coronal section 2 shows GFP-positive cells throughout the striatum radiating out from the injection site. GFP-positive cells were also visible along the striatum and cortex touching the white matter tract of the external capsule and within the corpus callosum. Expression was considerably greater in coronal section 3 compared to the short-term study, with more extensive spread of GFP-positive cells within the hippocampus, indicating increased spread of the vector. Internal capsule staining was also apparent, with vector spread apparent in the cerebral peduncle arising from the internal capsule in coronal section 4 (Fig. 5B). High-magnification images of the hippocampus and the striatum denoted by the dashed boxes in the sagittal section shown in Fig. 5C confirm the neuronal specificity of the SAF302GFP vector (Fig. 5D and E, respectively) with strong co-localization of GFP with neuronal nuclear marker NeuN, with GFP extending along neuronal processes. Co-staining of GFP with astrocyte marker GFAP in both short-term animals injected at two depths and long-term animals indicated a proportion of astrocytes were also transduced alongside neurons (Fig. 6). However, the proportion of astrocytes targeted was reduced compared to neurons. No cells within the cerebellum were transduced following intrastriatal injection of SAF302GFP in the long-term study, similar to results achieved in the short-term study arm.
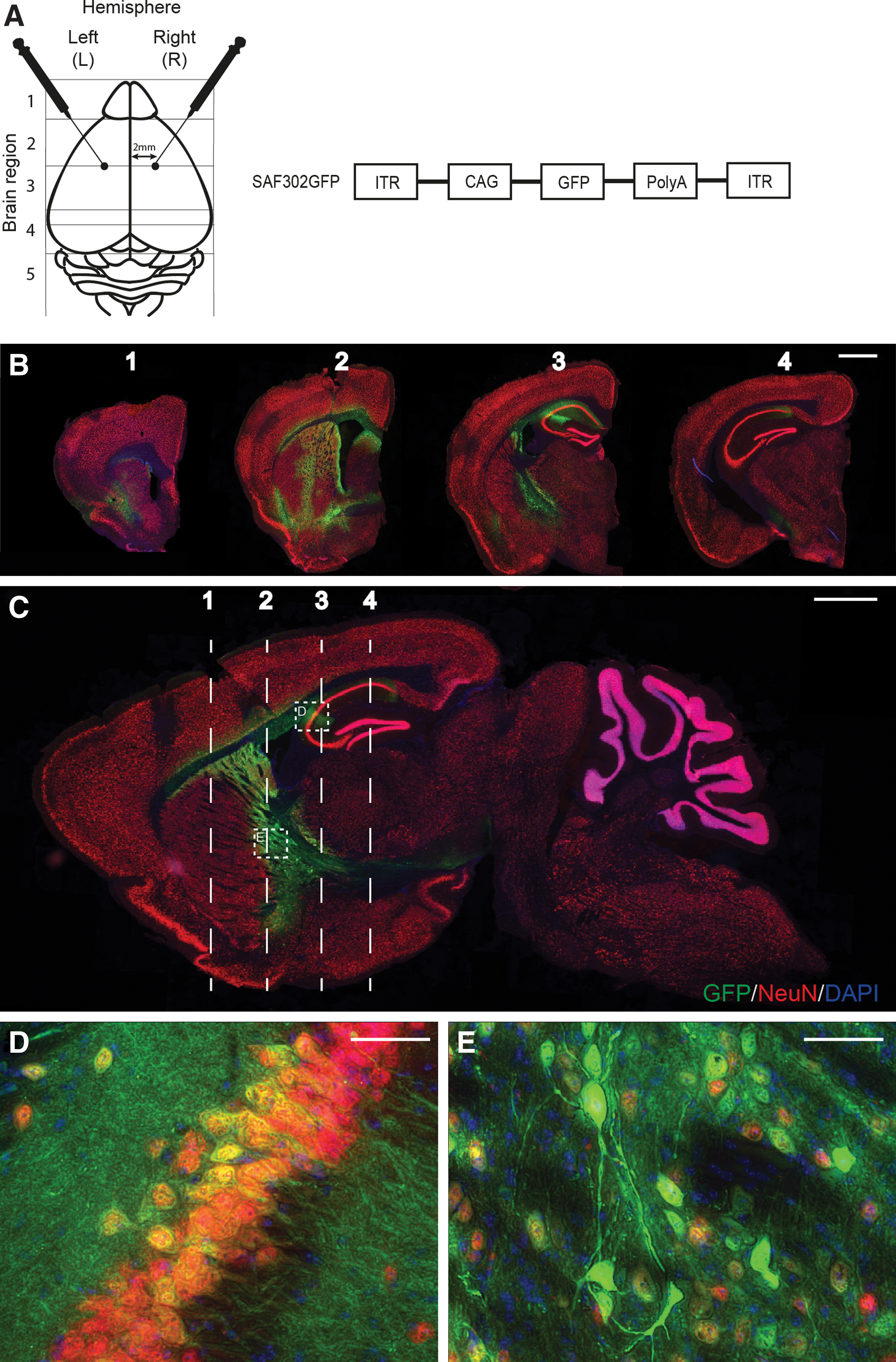
SAF302GFP expression is sustained at 4 months post injection in the neurons of MPSIIIA mice.
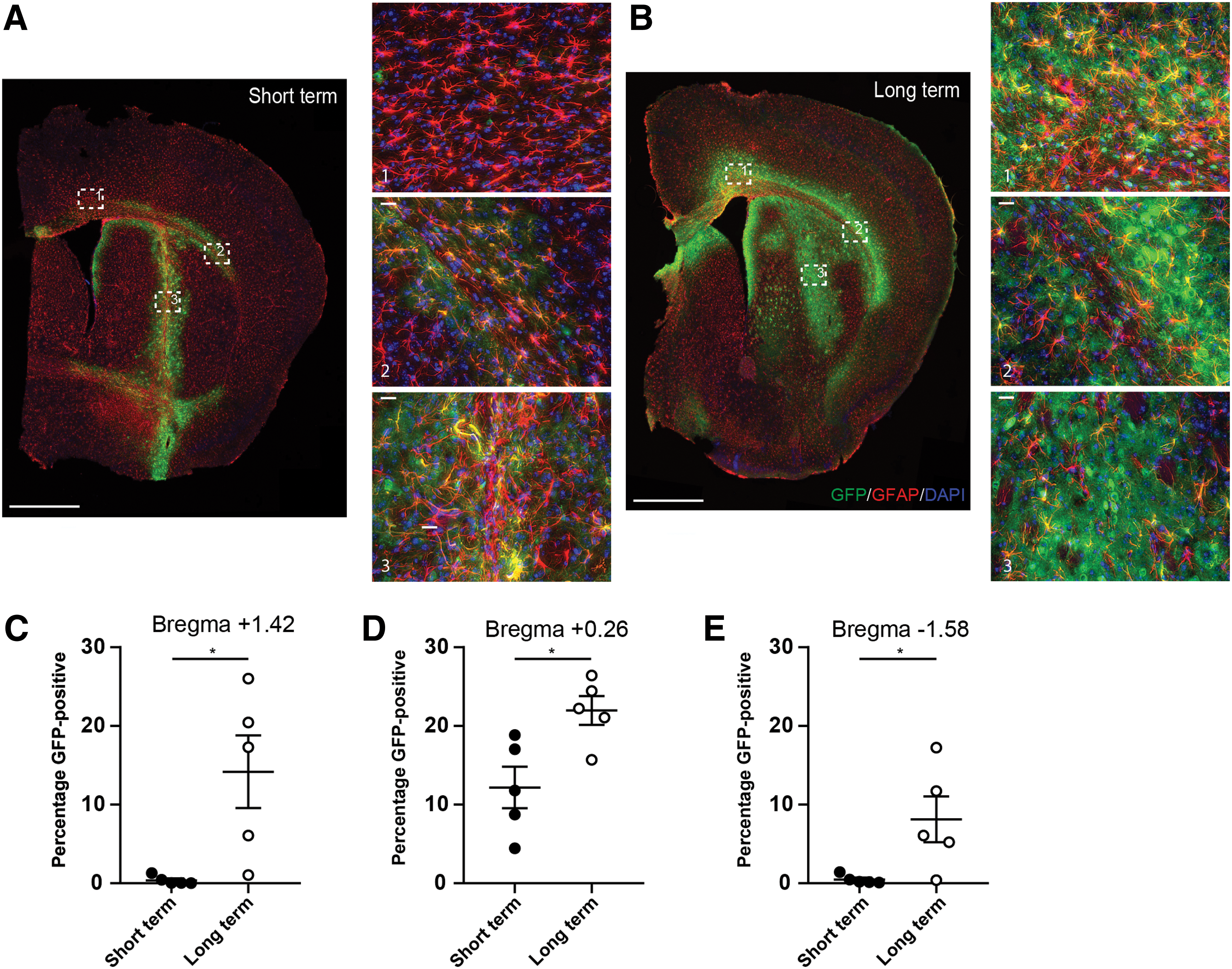
Clear transduction of GFAP-positive astrocytes is seen near the injection site following treatment with SAF302GFP.
Expression of GFP across the bregma following use of the two injection strategies was quantified as the percentage of GFP-positive cells relative to total brain area (Fig. 6C–E; n = 5 animals per group). In the vicinity of the injection site (bregma +0.26), the single injection strategy (long-term) led to increased global GFP compared to the two-depth strategy (short-term; Fig. 6D). Increased transduction of cells distal to the injection site was also apparent with the single bolus injection, whereas little transduction from the double-height injection at distal sites was seen (Fig. 6C and E).
Discussion
Mucopolysaccharide diseases, especially subtypes with substantial neurological involvement, including MPSIIIA, are ideal candidates for gene therapy. In MPSIIIA patients the brain is most affected. Therefore, direct delivery to the brain using AAV may prove beneficial. Vector-mediated delivery and cross-correction are two potential approaches to restore enzyme levels. If a number of cells can express the deficient SGSH enzyme, it can be taken up by surrounding cells. As a result, for clinical efficacy, a therapeutic vector that produces sufficient levels of enzyme is critical to enable cross-correction. This study shows superior SGSH expression following intracranial injection of SAF302, providing higher levels of enzyme to the brain compared to SAF301 and SAF301b. SAF301 has previously been used in a preclinical study 26 and clinical trial. 23 Winner et al. 26 demonstrated that in mice receiving SAF301 via a unilateral injection, high levels of SGSH could be detected close to the injection site, with substantially less expression detected as the distance from the injection region increased. Vector-mediated delivery of SGSH improved HS catabolism, reduced microglial activation, and led to reduced GM3 ganglioside accumulation and ubiquitin-positive lesion formation with improvements noted over time. 26 In patients, intracerebral administration of SAF301 at six sites per hemisphere in the white matter anterior, medial and posterior to the basal ganglia, suggested a cognitive benefit in the youngest patient. However, the therapeutic effect was less clear in older patients with presumably higher disease burden. Increasing vector potency by altering vector design may increase the clinical efficacy of this vector. These studies were designed (1) to determine the role of SUMF1 in the SAF301 vector and (2) to determine whether improving vector design by introducing a strong ubiquitous promotor would increase SGSH expression in a murine model of MPSIIIA.
For all vectors analyzed, the therapeutic efficacy was clear in the brain areas proximal to the injection site. Similar findings were reported by Winner et al. 26 who utilized a unilateral intrastriatal injection protocol and reported reduced primary and secondary pathology only in areas within or directly connected to the striatum. All variants increased SGSH enzyme levels in MPSIIIA mice, but given that no differences in VCN were seen, despite equivalent titers delivered, SAF302 was clearly superior. The removal of SUMF1 from SAF301 to generate SAF301b resulted in a 50% reduction in SGSH expression, consistent with the findings by Fraldi 25, 27 whereby SUMF1 activity is required for SGSH processing. Modification of the promoter via exchange of the relatively weak PGK promoter with the stronger CAG promotor containing an intron acceptor site led to production of a superior vector. SGSH activity levels were similar to WT levels in mice injected with SAF302 yet only reached 30% of WT levels in mice treated with SAF301. Additionally, in brain regions adjacent to the injection site, injections with SAF302 increased SGSH activity considerably above WT, whereas levels obtained with SAF301 were much below WT levels. Thus, the SAF302 variant was most effective overall. 36 A vector containing the CAG promotor and SGSH-IRES-SUMF1 might be the ideal scenario, although co-expression of SGSH with SUMF1 in a lentiviral context has not proven beneficial in increasing SGSH activity (data not shown).
Deficiency of the SGSH enzyme causes a progressive accumulation of un-degraded HS. From previous studies using a different therapeutic strategy, lentiviral-mediated HSCT in the MPSIIIA mouse model led to global increases across the brain in SGSH to ∼7% of WT, which was necessary to reduce HS levels and to correct neuroinflammation, 16 with similar results obtained in MPSIIIB 14 and MPSII 37 mouse models. In MPSIIIC animals, injection of AAV-HGSNAT resulted in a 46% reduction in HS, leading to associated decreases in neuroinflammation. 38 All studies led to improvements in disease-associated changes in behavior. For this treatment to be effective in the clinic, SGSH levels need to be sufficiently increased to lower HS storage and associated neuroinflammation significantly. A trend toward reduced HS storage and decreases in neuroinflammatory markers MIP-1α, MCP-1, and IL-1α was observed in each brain hemisphere injected with SAF302. Importantly, when regions 2 and 3, which lie at the border of the injection site, were studied in isolation, significant decreases in HS storage were observed. Thus, it can be concluded that SAF302 produces sufficient enzyme levels close to the injection site to prevent HS accumulation successfully.
Injection of animals with the SAF302GFP vector allowed vector distribution in the CNS of MPSIIIA mice to be evaluated using two different delivery methods, although vector amounts and time of sacrifice are not directly comparable (3 × 109 viral genomes per injection site in the long-term study harvesting animals 4 months post injection and 1 × 109 per site at two depths [2 × 109 total] in the short-term study harvesting animals at 4 weeks post injection). In the short-term study with injections at two depths and in the long-term study with a single-depth injection, a distinct vector distribution pattern was observed with most intense GFP-positive staining and thus cell transduction visible within regions 2 and 3, near the injection site. Co-staining experiments highlighted neurons as the primary cell type transduced. However, a proportion of astrocytes were also targeted. There was no evidence of GFP staining within the cerebellum (region 5), thereby providing an explanation for the drop-off seen in enzyme and HS levels, as well as neuroinflammation reduction in regions 4 and 5. Although cells types transduced in the short-term GFP study were similar to those seen in the long-term study, despite the altered injection schedule and increased vector spread, it was clear that injections at two depths were less efficient than a single intrastriatal injection, despite the reduced volume. The vector appeared to distribute more along the injection track in the injections at two depths, rather than spreading laterally from the end of the injection site as per the intrastriatal injections used in the long-term SAF302GFP study. This probably reflects reflux of the vector as the needle is withdrawn, rather than creating a larger lateral pressure that delivering a single bolus of virus achieves.
Twelve injection sites per patient (6 trajectories, 2 depths) were used in the SAF301 clinical trial, targeting white matter regions of the brain, leading to reported disease stabilization. 23 Six similar trajectories were used together with two additional trajectories in the cerebellum in a recent clinical trial in MPSIIIB (16 injection sites per patient), leading to small improvements or a decrease in projected neurocognitive decline, suggesting that if treatment is administered early in the disease course, the treatment may have some benefit for patients. 24 The results indicate that one injection site per hemisphere leads to modest vector spread and suggest that the delivery technique will need to be optimized for full clinical efficacy.
In summary, the present studies show that the SGSH AAV vector variant SAF302 produced a greater in vivo benefit relative to SAF301, and highlight the value of promoter choice in vector design. GFP expression was substantial, regardless of whether the mice were sacrificed at 4 weeks or 4 months post injection, confirming stability of the CAG promoter over time. The SAF302 vector could be a significant improvement over SAF301 for a future clinical trial in Sanfilippo disease.
Footnotes
Acknowledgments
The work was funded by grants from Lysogene SAS. The Bioimaging Facility microscopes used in this study were purchased with grants from BBSRC, Wellcome, and the University of Manchester Strategic Fund. Special thanks go to Roger Meadows for his help with microscopy.
Author Disclosure
This study was funded by a commercial grant from Lysogene SAS, who were also involved in the study design. B.B. and C.O'L. are shareholders in Phoenix Nest, Inc., and have licensed a related AAV program for MPSIIIC. B.B. is a scientific advisory board member and shareholder in Orchard Therapeutics Ltd. and has licensed unrelated stem-cell gene therapy programs for MPSIIIA and MPSIIIB. E.H. is funded by a Rare Disease Consortium Award from Pfizer, Inc., to develop AAV vector technology. A patent #WO2015121501 on “Adeno-associated virus vector” has been deposited by R.M.L. and E.H. R.M.L. is a consultant to various gene therapy companies. M.L. is a scientific advisory board member of Spark Therapeutics.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.