
Abstract
Recombinant adeno-associated viruses (rAAVs) have become favorable gene delivery vehicles for expressing therapeutic transgenes. Capsid engineering efforts to produce novel AAVs with improved transduction efficiencies, unique tissue specificities, and reduced host immunities are a direct response to the high demand for treatment needs that preexisting rAAVs cannot currently fulfill. New AAV capsids discovered by directed evolution methods, in silico design, or from natural proviral sequences ultimately require extensive characterization in relevant in vivo models. Consequently, quantitative screening of candidate capsid libraries now requires reliable high-throughput sequencing approaches. In this study, we have developed a vector/transgene tracking system that employs the indexing of a non-coding RNA. Specifically, a barcoded Tough Decoy (bcTuD) that express highly stable RNA transcripts that can be used as readouts for transduction efficiency. The pseudo-hairpin structure of the bcTuD contains a variable region that is amenable to barcode insertion, which can be detected by target amplicon sequencing. The described approach, named AAV-bcTuD screening, offers a new alternative for in vivo assessment of rAAV that can accurately quantify vector genomes and transcript abundances in tissues, as exampled by the demonstration in liver and brain infections. Proof-of-concept is provided to show that vector genome and transcript detection in tissues with this method is accurate and consistent for a vector dose range of upwards to four logs in a mixed vector injection, showing that this technique is robust, sensitive, and applicable for multiplexed screening of capsid performance in vivo.
Introduction
Adeno-associated virus (AAV)-based gene therapy platforms to treat an expanding range of human diseases have become increasingly promising. However, the toolkit of vectorized AAV serotypes currently used in clinical trials have limited cell and tissue tropism ranges, and this is therefore a significant bottleneck for the advancement of this technology. The discovery, engineering, and characterization of new AAV capsids as potential gene therapy vectors have become crucial for the continued growth of the field. Whether capsid discovery is accomplished by identification of naturally occurring isolates, 1 –5 directed evolution strategies, 6 –12 or in silico prediction approaches, 13,14 in vivo screening of capsid performance (i.e., transduction, biodistribution, cell specificity, sensitivity to neutralizing antibodies) is now performed by screening large libraries of candidate capsids in animal models. The pool of capsids tested at a single time can range from as few as tens of capsids to >1,000 capsids. 9,15 These studies are achievable through high-throughput screening methods. Unfortunately, the protein sequences of high performing capsids cannot be directly determined, particularly for screening capsid variation spanning the entire open reading frame. A practical solution has been to introduce DNA barcodes into the encapsidated vector genome that can be identified and quantified by next-generation sequencing (NGS) strategies. 16 Tissues successfully infected with a pool of these capsids can be harvested and subjected to DNA extraction and NGS analysis to quantify the abundance of barcoded reads. However, a potential caveat to this approach is that the abundance of vector genome copies detected in cells may not necessarily correlate with the level of transgene expression conferred by the capsid. For example, there is compelling evidence that the AAV capsid can affect the efficiency of second-strand synthesis and therefore may have direct roles in transgene expression. 17 Capsids that are efficient at all processes of vector transduction are ideally preferred. These processes, some of which still remain poorly understood, encompass tissue distribution, cellular uptake, intracellular trafficking, endosomal escape, nuclear import, uncoating, and transgene expression. In contrast, some capsids that can promote nuclease-free editing via homologous recombination 18 and not necessarily exhibit high transcriptional outputs are also desirable.
Many capsid screening methodologies are based on the detection of an index inserted into the 3′-UTR of a reporter gene 19,20 or expression of Cre recombinase. 21 Unfortunately, adaptive immunity to these foreign transgenes in large-animal models may mask the performance of candidate capsids, and can drastically limit vector persistence in transduced tissues or the ability to probe vector latency accurately. In addition, querying barcodes embedded in simple linear RNAs may be impacted by post-transcriptional regulatory mechanisms that can affect transcript abundances in unpredictable ways. These potential outcomes are best exemplified by multiple studies demonstrating that introduction of random sequences within untranslated regions can dramatically impact mRNA stability and processing. 22 –24 These constraints represent key challenges for studying cross-species translatability of engineered capsids. To address the current limitations in screening for in vivo capsid performance in small- and large-animal models, a vector transduction tracking system has been developed that employs the barcoding of Tough Decoy (TuD) RNAs, 25 stable non-coding RNAs with a pseudo-hairpin structure that contains a variable region amenable to barcode insertion. These barcoded TuD products (bcTuDs) are resistant to degradation and post-transcriptional processing, and are faithfully detected in different organs for an abundance range of upwards to four logs. The novel design of AAV-bcTuD screening provides a new alternative for AAV in vivo screening that can quantify relative amounts of vector genomes and transcripts in animal tissues. This report presents proof-of-concept demonstration of the strategy in murine animal models via two routes of vector administration: intravenous and intracranial injections.
Methods
bcTuD vector construct
The 4.7 kb single-strand (ss)AAV-bcTuD construct was generated by cloning the scrambled TuD sequence
25
into an ssAAV-U6-TuD plasmid as the base construct described elsewhere.
26
A 3.8-kb stuffer sequence was inserted into the vector by conventional subcloning methods. Briefly, the 3.8-kb sequence was polymerase chain reaction (PCR) amplified from human genomic DNA H1 human embryonic stem cells (WA01, University of Wisconsin) using the following PCR primers containing restriction enzyme sites for cloning: forward 5′-
The 0.5 kb bcTuD scaffold cassette was generated via synthesis and propagated in a standard pUC57 cloning vector by GenScript. The scaffold contains two BsmBI restriction sites for directional cloning (Supplementary Fig. S1). The bcTuD scaffold was then subcloned with the ssAAV-U6-3.8 kb stuffer construct to generate the ssAAV-u6-TuD scaffold. To generate the bcTuDs, complementary oligos were annealed and ligated into the BsmBI-digested pAAV-bcTuD-scaffold construct (Supplementary Fig. S1). All oligos were purchased from Integrated DNA Technologies (IDT).
Recombinant AAV production
Recombinant AAV (rAAV) vectors were produced using the triple-transfection method in HEK293 cells and purified by CsCl gradient centrifugation. 27 Vectors were packaged with AAV2/2. Titers were determined by silver staining and Droplet Digital PCR (ddPCR) 28 with the following probe sets (Thermo Fisher Scientific): forward primer sequence 5′-AGCCCTAGGGATGAACCAGT-3′; reverse primer sequence 5′-AACCCAGGAGTCATTGCATC-3′; reporter sequence 5′-AATCTGAGCCACTGAGCCAT-3′.
Animals
Six- to eight-week-old male C57BL/6J mice (The Jackson Laboratory) were used as the in vivo model for viral biodistribution analysis. All animal procedures described in this study were approved by the UMass Medical School Animal Care and Use Committee.
rAAV injections
All surgical procedures were performed using aseptic techniques. For liver transduction analyses, mice were administered with a dose of 1 × 1011 vg of rAAV2 vector mix in a 200 μL volume through tail-vein injection. 29
For intra-hippocampal injections, mice were anesthetized intraperitoneally with a ketamine/xylazine cocktail. Mice were mounted to a stereotaxic surgery frame (Stoelting), and 2 μL rAAV2 vector mix was injected bilaterally into hippocampal sites using a monoinjector (World Precision Instruments) with a 33G needle attached to a 10 μL Hamilton syringe (Hamilton Company) and administered at a rate of 0.2 μL/min 30 with stereotactic coordinates AP = −2, ML = ±1.5, DV = 2.
Isolation of genomic DNA and total RNA
Four weeks after injection, the mice were euthanized, and liver or hippocampal tissues were immediately harvested and snap-frozen in liquid nitrogen. Liver samples were then homogenized in liquid nitrogen in an autoclaved mortar and pestle, and approximately 30 mg of powdered tissue was transferred into new tubes for DNA or RNA isolation. Brain hippocampal samples were directly homogenized for DNA and RNA extraction (left hippocampi for DNA, right hippocampi for RNA). The QIAamp DNA Mini Kit (Qiagen) was used for DNA extraction according to the manufacturer's instructions. The Direct-zol RNA MiniPrep kit (Zymo Research) was used for RNA extraction according to the manufacturer's instructions, with the exception of the on-column DNaseI treatment where double the volume of DNase I solution was used and incubation at 37°C was carried out for 1 h to ensure complete removal of vector DNA from RNA samples.
PCR and reverse transcription PCR amplification
Total DNA and RNA were quantified on a NanoDrop 1000 (Thermo Fisher Scientific). Total DNA (100 ng) was used for PCR to generate the target amplicon libraries with KOD Hot Start Master Mix (EMD Millipore). Due to the relatively small size of the pseudo-hairpin TuD transcript (118 nt), options for reverse transcription (RT)-PCR primers were limited. Therefore, a non-conventional PCR strategy was developed and validated whereby the RT-primer, forward primers, and reverse PCR primers are complementary to the Stem I structure and share sequence composition (Fig. 1).

Schematic of the ssAAV-U6-bcTuD vector genome.
Primer sequences
The gene-specific RT primer is: 5′-GACGGCGCTAGGATCATC-3′. Standard PCR procedures were used to amplify the bcTuD sequence for liver amplicons. PCR primers used for liver amplicons and RT-PCR were: forward primer 5′-GACGGCGCTAGGATCATCAAC-3′; reverse primer 5′-GACGGCGCTAGGATCATCTTG-3′. Total RNA (1 μg) from liver tissues and total RNA (100 ng) from hippocampal tissues were used for RT reactions with SuperScript III First-Strand Synthesis System (Invitrogen). RT product (1 μL) was then used for PCR with KOD Hot Start Master Mix. Minus RT control (–RT) reactions were run in parallel for all samples to confirm that samples were clear of any vector genomic DNA contaminants. The PCR products were run on 1% agarose gels and purified with the Zymoclean gel DNA recovery kit (Zymo Research). All PCR amplicons were verified by TOPO cloning (Thermo Fisher Scientific) and sequencing before submitting to the University of Massachusetts Medical School Deep Sequencing Core (UMASS DeepSeq Core) for high-throughput sequencing.
Double-barcode strategy was used to amplify the bcTuD sequence for brain amplicons. PCR primers used for brain amplicons and RT-PCR were:
The 8-nt barcodes used in this study are underlined.
Illumina-based high-throughput sequencing and bioinformatics data analysis
All gel-purified PCR fragments were quantified on a Qubit 3 fluorometer using Qubit HS DNA Assay Kit (Thermo Fisher Scientific). HiSeq libraries were prepared by adaptor ligation using barcoded adaptors for each sample by the UMASS DeepSeq Core using standard conditions. Equimolar amounts of each sample were mixed and spiked with 20% PhiX DNA to increase library complexity. Samples were run on a HiSeq 4000 Illumina apparatus with single-read 50 base sequencing. Since the full-length PCR amplicon is >100 bp in length, and the barcode is inserted into one side of the MBS region (Fig. 1), only half of the reads will cover the barcode. The read depth from HiSeq 4000 Illumina typically yields >300 million reads per lane, which is more than enough for profiling the relative abundances of vector genomes and transcripts, even after sacrificing half of the reads. The web-based Galaxy platform was used for data analysis. 31,32 Briefly, reads were demultiplexed by Barcode Splitter 33 using a 16 nt recognition sequence (8 nt of unique barcode +8 nt of trailing TuD MBS sequence), allowing 2 nt mismatches and 2 nt deletions. Since the bcTuD design for the brain study harbors two barcodes (first barcode for library identification and second barcode for capsid identification; Supplementary Fig. S3), reads were trimmed 29 nt following the initial demultiplexing step and demultiplexed again on the second set of barcodes.
ddPCR analysis
DNA or RNA samples from transduced liver tissues were used as template for ddPCR. Total DNA (1 μL; ∼50 ng) or gene-specific RT product (1 μL; see the PCR and RT-PCR amplification section above) was used for each ddPCR reaction. ddPCR™ Supermix for Probes (no dUTP; 10 μL; Bio-rad), 2 μL probe (2 μM), and 1.5 μL forward and reverse primers (10 μM) were mixed for ddPCR reactions. Each sample was tested in triplicate, and average values were reported. Negative controls (no template) for each probe were included to determine threshold baseline values. For increased specificity and sensitivity, ddPCR probes (IDT) were designed with locked nucleic acid (LNAs) and are indicated below, with a plus symbol (+) before each modified base:
Primers for ddPCR are the forward primer and reverse primer described above (see section PCR and RT-PCR amplification).
Statistical analysis
R 2 values for data fit to regression lines were calculated using Microsoft Excel. Statistical values by Pearson correlation were derived using GraphPad Prism 7 (GraphPad Software, Inc.).
Results
Design of the ssAAV-U6-TuD vector
Assessing transduction profiles for novel capsids in animal models should be obtained independently of any confounding immunological responses related to the transgene product. For instance, foreign transgenes such as standard reporter genes may present as antigens in nonhuman primates (NHPs). In addition, barcoding the 3′-UTR of Pol II transcripts may impact message stability via endogenous miRNAs or other host RNA modifying enzymes. Vectors that carry a U6-driven transgene would not be as strongly impacted by variability in message stability. 34 It was reasoned that the ubiquitously active U6-promoter would result in more reliable readouts for transgene expression from a mixed-pool of barcoded vectors. Importantly, ubiquitous viral Pol II promoters, such as cytomegalovirus, have been shown to be repressed in vivo 35 and therefore are not ideal for assessing the transduction profiles of capsids in a range of tissues.
In selecting the best U6 transcript, it was reasoned that short hairpin RNAs are not favorable, since modification of the stem-loop would require functional validation of each barcoded sequence. Furthermore, addition of a barcode outside of the stem-loop would not work, since the 5′ and 3′ ends of the molecule would be cleaved by Drosha and Dicer, respectively. 36 Likewise, single guide (sg)RNAs are not ideal, since sgRNAs may be turned over if not stabilized by the Cas9 nuclease. 37 Therefore, it was concluded that TuDs may be the ideal U6-driven non-coding RNA transgene for assessing the transduction efficacy of vectors. 25 The transcribed TuD resembles an imperfect hairpin, consisting of a stabilized stem-loop and two microRNA binding domains that are non-reverse complements to each other. Additionally, TuD RNAs have demonstrated high stability when expressed in vivo, 25,26 and rAAV-U6-TuD vectors have been used in animal studies with no long-term transgene-related toxicities. 26 It was hypothesized that the TuD miRNA binding site (MBS) variable region should be amenable to barcode insertion and will not be degraded or processed (Fig. 1).
The rAAV-U6-TuD construct is based on a previously designed ssAAV-TuD vector. 26 The TuD scaffold used to house the barcode is based on the scrambled TuD cassette, which lacks any binding specificity to any mouse or human miRNAs. 25,38 A BsmBI restriction site was inserted into the 5′ MBS sequence (Supplementary Fig. S1) for simple cloning of annealed oligos with compatible overhangs. To ensure proper packaging, a 3.8-kb stuffer sequence from an intergenic region within human chromosome 18 was inserted downstream of the TuD cassette (Fig. 1A). The stuffer sequence was selected based on its lack of overlap with any annotated gene sequence, regulatory elements, or any repetitive sequences (Supplementary Fig. S2).
In vivo testing: systemic delivery and liver transduction
To demonstrate the capacity for a bcTuD RNA to track transduction efficiency of AAV capsids properly when delivered into tissues, a workflow for detecting the vector DNA and the expressed bcTuD in rAAV-treated tissues was designed (Fig. 2). (1) rAAV-bcTuDs with distinct barcodes are generated individually by large-scale preparation. In this study, the conventional triple transfection method was used to generate rAAV2 vectors (i.e., rAAV2/2 with AAV2 ITRs and capsid as the test ITR/capsid platform). (2) Injection of mixed viruses into mice. For this first proof-of-concept demonstration, six bcTuD vectors mixed at a 10-fold dosage series (9E10, 9E9, 9E8, 9E7, 9E6, and 9E5) were systemically delivered by tail-vein administration, so that each mouse received a total of approximately 1E11 vg/mouse (Fig. 3A). (3) Tissue collection after vector administration at desired timepoints. The tissue chosen to probe first was the liver. Based on previous studies, peak TuD expression in the liver was between 3 and 4 weeks post injection. 26 Therefore, tissues were collected on week 4, and total DNA and RNA were extracted. (4) Library construction by PCR amplification of the bcTuD from DNA samples to detect vector genome abundance and transgene-specific RT-PCR of RNA samples to detect bcTuD transcript abundance. (5) Target amplicon sequencing and analysis. Note that for this study bcTuDs were only packaged into AAV2 capsids in order to remove any confounding factors related to serotype differences, such as variance in the ratios of empty versus full capsids, differences in capsid stability in the cell, the ability for different serotypes to traffic efficiently, the ability of capsids to escape from late endosomes or lysosomes, the varied efficiencies for capsids to uncoat before reaching the nucleus, and differences in second-strand synthesis once unpacked in the nucleus. 39
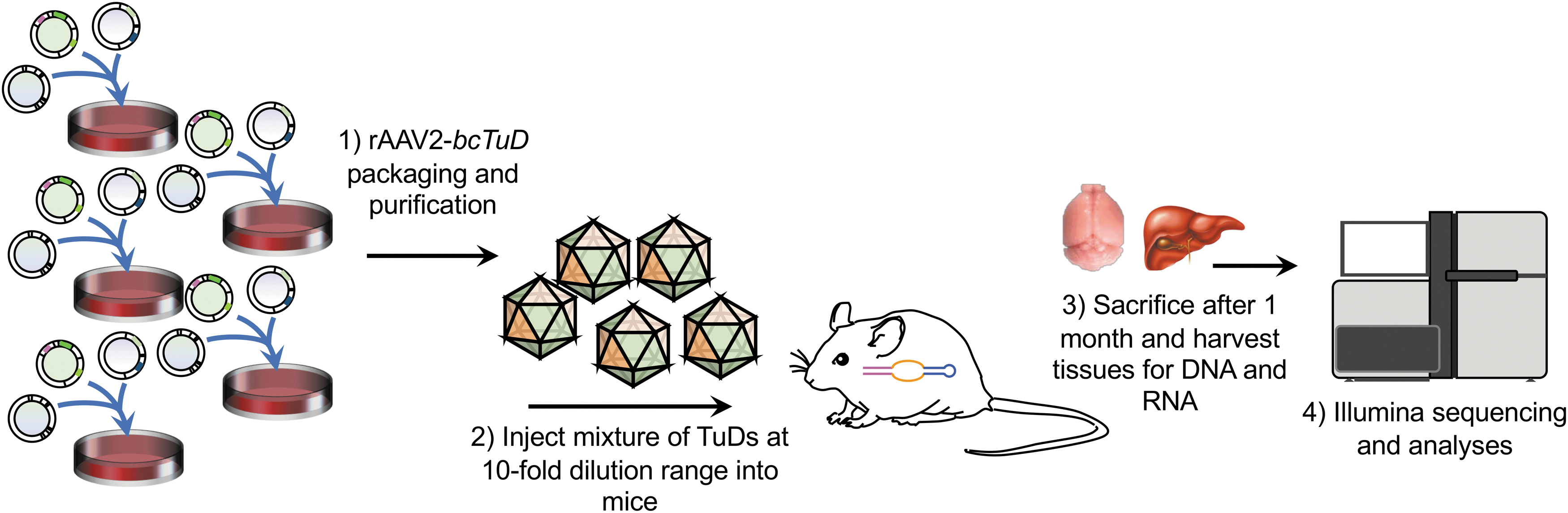
Summary workflow for the in vivo detection of bcTuDs delivered by AAV2.
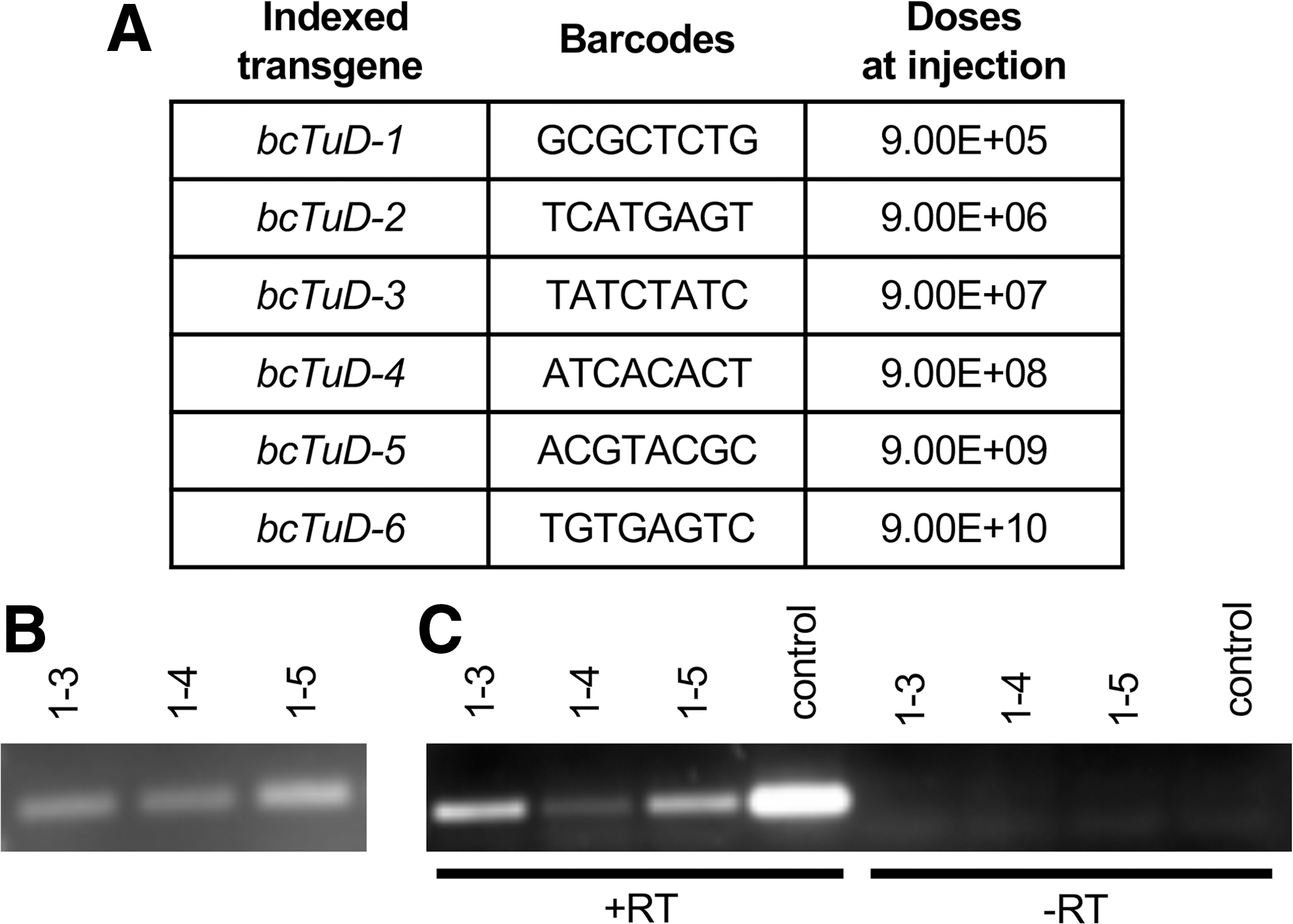
Detection of bcTuDs in transduced mouse livers.
Using optimized procedures established by in vitro trials (data not shown), it was possible to yield target-amplicon bands of both vector genomes (DNA, Fig. 3B) and bcTuD transcripts (RNA, Fig. 3C) successfully by PCR to amplify the bcTuD sequence. Amplicons were then subjected to high-throughput sequencing library construction by adaptor ligation using barcoded adaptors for each sample. Libraries to query vector genomes and bcTuD transcripts for two animals (four libraries total) were multiplexed on a single flow cell. A total of ∼34 million high-quality reads were initially obtained (Supplementary Table S1). Importantly, the overall quality spanning the 8-nt barcodes required for vector identification have average Phred scores ≥36 by FastQC analysis (Supplementary Fig. S3A). In this study, unmatched reads account for approximately 80% of the total counts. About half of the reads were anticipated to be unmatched, since the barcode was only inserted into the first MBS region. The low-complexity nature of the library may also lead to lower base-calling scores. Absolute barcode counts show that mice receiving a total dose of 1E11 GC, with a six-point, 10-fold dilution range spanning 9E10–9E5, results in the detection of all barcoded transgenes for both vector genomes and bcTuD transcripts (Fig. 4). However, read counts related to vector doses <1E7 appear to be beyond the linear range of representation. Removal of data points related to 9E5 and 9E6 doses resulted in R 2 > 0.95 for both vector genome and transcript data. Importantly, the slopes for vector genomes and transcripts within samples are quite similar (Fig. 4C), suggesting that detecting bcTuDs by DNA and RNA strongly correlate with each other. Pearson correlation coefficients showed positive correlation between the two animals when detecting vector genomes (r = 1, p = 3.9e-6) and bcTuD transcripts (r = 1, p = 2.1e-3), and when comparing vector genomes and transcripts within the transduced tissues (r = 1, p = 7.5e-4 for mouse 1-5 and r = 1, p = 5.1e-3 for mouse 1-3).
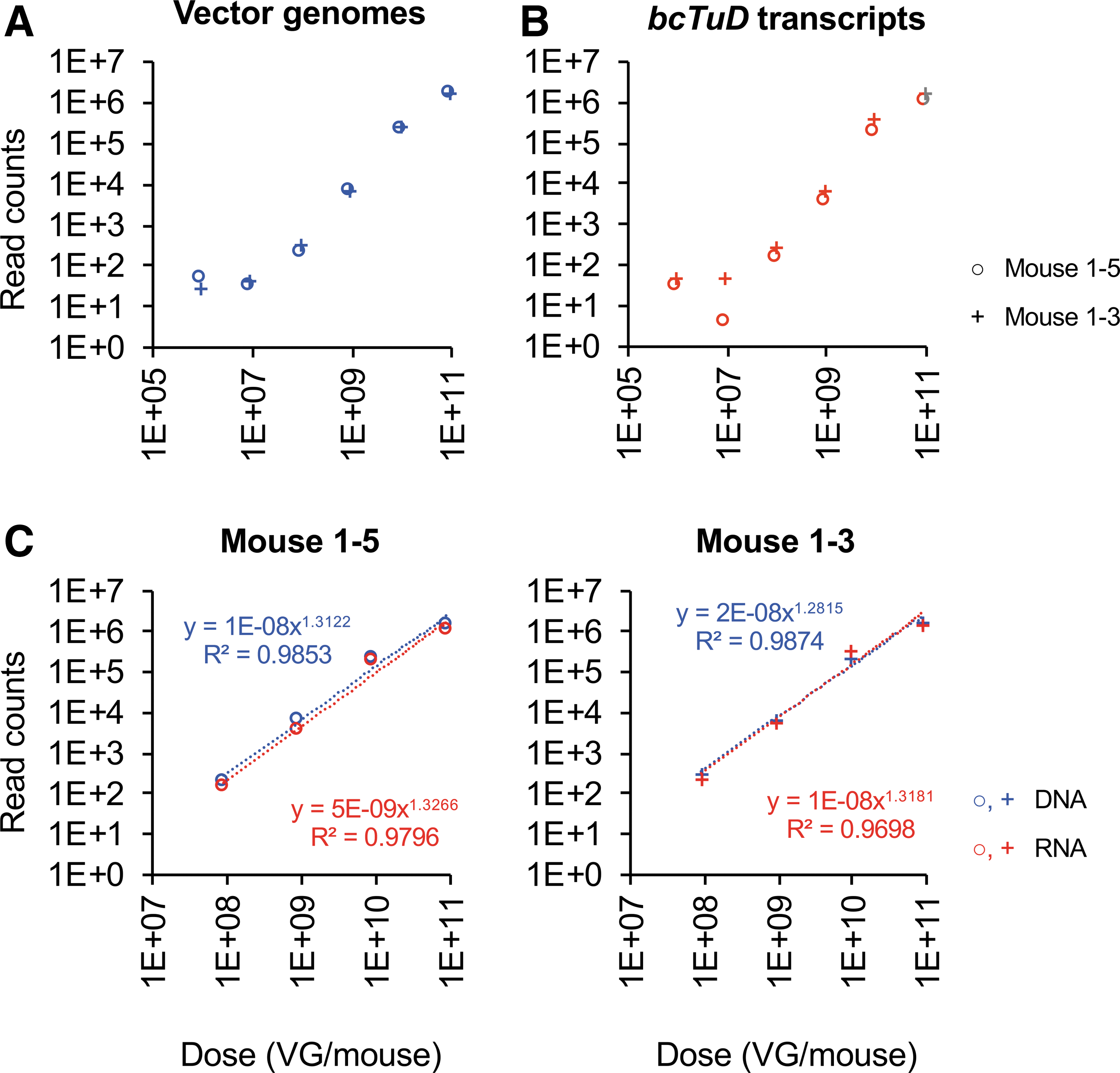
Analysis of target amplicon sequencing of barcoded rAAV-bcTuD vectors from livers. Two mouse liver samples were selected (1-5 and 1-3) for Illumina sequencing.
To validate that the detection of vector genomes and bcTuD transcripts by high-throughput sequencing truly reflects transduction of liver tissues, the bcTuDs were detected by ddPCR. Since the sequencing results revealed that the detection of the bcTuDs related with vector doses spanning 9E10–9E7 are within the linear range of representation, ddPCR probes specific to barcodes associated with these four vector dose points were designed (Fig. 3A). Regression analysis of resulting ddPCR for both detected vector genome and transcripts in liver tissues showed strong linearity (R 2 > 0.9) within the analyzed dose range (Supplementary Fig. S4A and B). Importantly, with the exception of vector genome detection for mouse 1-5, which appears to be skewed by the 9E7 dose point, it was observed that linear regression and slopes were comparable with high-throughput sequencing data (compare to Fig. 4C). Pearson correlation coefficients showed positive correlation between the two animals when detecting vector genomes (r = 1, p = 4.0e-6) and bcTuD transcripts (r = 1, p = 1.2e-4), and when comparing vector genomes and transcripts within transduced tissues (r = 1, p = 2.4e-5 for mouse 2-1 and r = 1, p = 2.3e-5 for mouse 2-3).
In vivo testing: intracranial injection and brain transduction
The central nervous system, specifically the brain, is a highly desirable target organ for rAAV-based therapies. In turn, accurate quantification of vector transduction to target tissues of the brain is also needed. To assess the capacity of bcTuDs to track transduction efficiency in the brain, a three-point, 10-fold dilution range spanning 2E7–2E9 was chosen for intra-hippocampal injections. This range was chosen, since liver tissue data suggested read counts related to vector doses <1E7 were beyond the linear range of representation (Fig. 4). This study also employed a double-barcoding strategy. The double-barcoded TuD library therefore contained a primary barcode that permits multiplexing several libraries into one sequencing lane, and a second barcode that functions as a readout for capsid transduction, which is similar to the index used in the liver study. In addition, the inclusion of the first barcode acts to increase complexity of the target amplicon library and, in turn, increases the amount of quality reads. A vector mix with 2.22E9 dose per injection (2 μL in volume) was administered bilaterally into hippocampal sites (single site). Target-amplicon bands of both vector genomes (DNA, Fig. 5B) and bcTuD transcripts (RNA, Fig. 5C) were yielded successfully.

Detection of bcTuDs in transduced mouse brains.
As before, libraries include amplicons from vector genomes and TuD transcripts for two animals (four libraries total) multiplexed on a single flow cell. A total of ∼297 million reads were obtained for this study (Supplementary Tables S2 and S3). Read positions spanning both first and second barcodes were of high sequencing quality, with Phred scores ≥32 for the first barcode and ≥40 at for the second barcode (Supplementary Fig. S3B). These data demonstrate that the dual-barcode strategy can yield quality sequencing results without relying on indexed Illumina adaptors. Unmatched reads account for about 60% of the total reads, nearer to the expected 50% outcome of non-barcoded read events than observed in the liver study. Analysis of reads show that mice receiving a total dose of 2.22E9 GC, with a 10-fold dilution range spanning 2E7–2E9, resulted in the detection of all barcoded transgenes for both vector genomes and bcTuD transcripts (Fig. 6). Regression for both vector genome and transcript barcodes detected in brain tissues show strong linearity (R 2 = 0.92) and similar slopes within the analyzed dose range (Fig. 6C). Pearson correlation coefficients showed positive correlation between the two animals when detecting vector genomes (r = 1, p = 0.011) and bcTuD transcripts (r = 1, p = 0.006), and when comparing vector genomes and transcripts within the transduced tissues (r = 1, p = 0.031 for mouse 2-1 and r = 1, p = 0.036 for mouse 2-3), further suggesting that detecting both bcTuD vector genomes and transcripts is highly accurate and strongly correlate with each other.

Analysis of target amplicon sequencing of barcoded rAAV-bcTuD vectors from brains. Two mouse brain samples were selected (2-1 and 2-3) for Illumina sequencing.
Discussion
AAV vectors are among the most promising gene therapy tools due to their safety and ability to mediate long-term transgene expression. 40,41 Biodistribution analysis and transduction profiling is a key step in the evaluation of novel AAV capsids and vector platforms. 20 Classical approaches for assessing tissue tropism in vivo, such as visualization of luciferase transgene activity by bioluminescence live imaging, quantification of vector genomes by quantitative PCR of tissue lysates, or detection of fluorescent transgene products by microscopy of fixed tissue sections, are all reliable when testing individual capsids. However, when screening larger numbers of candidate vectors, NGS approaches are more informative for assessing tissue transduction efficiencies and are far more cost-effective and humane. Barcode-tagged AAV libraries allow a spectrum of AAV capsid phenotypes to be addressed. 16,20 Previous versions of barcoded AAV screening strategies that quantify vector genomes in tissues may not necessarily reflect the capacity that the vector can confer transgene expression. For example, a novel AAV capsid may exhibit high levels of cellular entry in target tissues but may not traffic properly within the cell, may fail to escape from endosomes, may become rapidly degraded through proteasome activity, or may display stalled second-strand synthesis. The most important property for a rAAV vector is its capacity to confer therapeutic levels of transgene expression. Recent AAV barcode-transgene methods can quantify both AAV vector genomes and can indirectly assess transcriptionally active vectors in cells. 21 However, long-term overexpression of foreign transgenes such as Cre, eGFP, or other foreign proteins can cause immunological response and inflammation. 42,43 This is especially problematic with large-animal models, such as NHPs, where exogenous products can present as antigens. 44,45 In addition, barcoding the 3′-UTR of Pol II transcripts may impact message stability via endogenous miRNAs. 46
TuD RNAs were originally developed to act as decoy targets for microRNAs, and are on their own promising biotherapies. 25 For example, scAAV-delivered TuDs designed to inhibit miR-122 function can simultaneously lower high- and low-density lipoprotein levels, 26 and a rAAV9-miR-25-TuD vector was able to enhance cardiac function in a pressure-overload murine HF model. 47 The stand-out feature of TuD RNAs as potent biotherapies is their in vivo stabilities. Here, the scrambled TuD was utilized as a scaffold to harbor a barcode for transgene identification. Importantly, the variable region selected for barcode insertion will not be degraded or processed, since the unique bulge of the TuD protects it from endonucleolytic targeting. 48 This study did not find any noticeable shortcomings from using TuD RNAs as the scaffold to house an index. Thus far, AAV-TuDs have yet to demonstrate any toxicity effects, suggesting that although they are considerably stable, they are not known to accumulate in the cell to cause toxicity. The study also showed that following a 4-week period, where AAV-U6-TuDs have reached peak levels of expression in the liver following tail-vein administration, the detection of the TuDs were still within a linear range of detection. This observation suggests that they do not accumulate in tissues and have not reached a point of saturation to skew the quantification of barcoded reads. In this study, in vivo detection of the bcTuD cassette and bcTuD transcripts strongly correlated with each other within a 10-fold dose range spanning four logs by systemic injection (large volume) and three logs in intracranial injection (small volume), indicating that rAAV-bcTuD vectors are a reliable tool for assessing capsid variant characteristics and can potentially be used to reveal the role of AAV capsids in viral trafficking, uncoating, and transcription. It should be noted that a pre-injection “input” library was not included in this study, since a decision was made to test the platform with a single capsid serotype. However, when testing a multitude of capsid candidates, an input library should also be sequenced in parallel to ensure that bcTuD abundances truly reflect capsid performances in vivo and are not skewed by the variability inherent to vector titer quantification among different serotypes. Importantly, it was possible to validate the detection of the bcTuDs by ddPCR, and no biases related to the library design and sequencing strategy in the data were revealed. It should be noted that because the method relies on target amplicon sequencing, low library complexity will be a challenge to base-call quality. The use of a PhiX spike-in is highly recommended. Additionally, the inclusion of phased adaptors may be a means of improving sequencing quality. Finally, it should be noted that this study only represents a proof-of-concept demonstration that bcTuDs can be used to quantify transgene abundance accurately and have many theoretical advantages over barcoding linear RNAs. However, before the true value relative to linear RNA barcoding is established, a high diversity library and a side-by-side comparison should be established. Ultimately, this approach is potentially a valuable tool for profiling new AAV capsid designs, particularly in large-animal models where transgene immunity has become a major factor that influences transduction data.
Footnotes
Acknowledgments
We would like to thank Nicholas Kaiser and Xavi Anguela for insightful scientific discussions and guidance. We also thank members of the Gao Lab, The UMMS Vector Core, Maria Zapp, and Ellie Kittler of the UMMS Deep Seq Core. G.G. holds support from grants under the National Institutes of Health (P01AI100263, R01NS076991, P01HD080642, and R01AI121135). M.X. is supported by an Inter-Institutional grant, Horae Postdoctoral Fellowship.
Author Disclosure
G.G. is a scientific co-founder of Voyager Therapeutics and Aspa Therapeutics, Inc., and holds equity in these companies. G.G. is an inventor on patents with potential royalties licensed to Voyager Therapeutics, Aspa Therapeutics, Inc., and other biopharmaceutical companies.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.