
Abstract
Krabbe disease is an inherited neurodegenerative disease caused by mutations in the galactosylceramidase gene. In the infantile form, patients die before 3 years of age. Systemic adeno-associated virus serotype 9 (AAV9) gene therapy was recently shown to reverse the disease course in human patients in another lethal infantile neurodegenerative disease. To explore AAV9 therapy for Krabbe disease, we engineered a codon-optimized AAV9 galactosylceramidase vector. We further incorporated features to allow AAV9-derived galactosylceramidase to more efficiently cross the blood–brain barrier and be secreted from transduced cells. We tested the optimized vector by a single systemic injection in the twitcher mouse, an authentic Krabbe disease model. Untreated twitcher mice showed characteristic neuropathology and motion defects. They died prematurely with a median life span of 41 days. Intravenous injection in 2-day-old twitcher mice reduced central and peripheral neuropathology and significantly improved the gait pattern and body weight. Noticeably, the median life span was extended to 150 days. Intraperitoneal injection in 6- to 12-day-old twitcher mice also significantly improved the motor function, body weight, and median life span (to 104 days). Our results far exceed the ≤70 days median life span seen in all reported stand-alone systemic AAV therapies. Our study highlights the importance of vector engineering for Krabbe disease gene therapy. The engineered vector warrants further development.
Introduction
The infantile form of Krabbe disease (globoid cell leukodystrophy) is an autosomal recessive lysosomal storage disease caused by mutations in the galactosylceramidase gene. 1,2 This gene encodes for a lysosomal enzyme that digests galactosylceramide and psychosine. Galactosylceramide is a major constituent of myelin. Psychosine is a by-product of galactosylceramide synthesis and is normally kept at low concentrations. 3 Abnormal accumulation of psychosine leads to neural toxicity. 4 Patients present symptoms (such as irritability, cortical fisting, and poor head control) before 6 months of age, show rapid neurological deterioration, and die prematurely before 3 years of age. 5 Currently, hematopoietic stem cell transplantation is the only approved therapeutic option. 6 However, this therapy is associated with high procedure-related mortality, and importantly, it provides limited benefits and only works in presymptomatic patients. 7 There is a need for more effective therapies.
A variety of single and combined experimental therapies have been investigated in twitcher mice, the authentic mouse model for Krabbe disease. 8 –12 These include cell therapy, 13 gene therapy, 14 substrate reduction, 15 enzyme replacement, 16 anti-inflammation, 17 chaperones, 18 and lysosomal re-acidification. 19 Among these, gene therapy has attracted particular attention because it targets the genetic root of the disease. Specifically, a functional galactosylceramidase gene is delivered to affected subjects to replace the mutated gene. Early gene therapy attempts were based on the retroviral vector and adenoviral vector. 20,21 Unfortunately, these vectors are not suitable for clinical translation due to their low transduction efficiency in postmitotic cells and high immunotoxicity.
Recombinant adeno-associated virus (AAV) is currently the leading vector for treating monogenic inherited diseases because of its high in vivo transduction efficiency, low immunogenicity, and excellent preclinical and clinical safety profile. 22,23 In support, the number of AAV gene therapy clinical trials has doubled from 2012 to 2017. 24 Furthermore, three AAV gene therapy drugs have received regulatory approval for commercial use to treat Leber congenital amaurosis, hereditary lipoprotein lipase deficiency, and spinal muscular atrophy.
In 2005, two groups published AAV-mediated Krabbe disease gene therapy in the twitcher mouse model. 25,26 Since then, a number of different AAV serotypes including AAV1, AAV2, AAV5, AAV9, AAVrh10, and AAV-oligo001 have been tested. 25 –36 In these studies, the AAV vector was administered via either one route (intracranial, intracerebellar, intrathecal, intracerebroventricular, intravenous) or a combination of multiple routes. A modest life span extension was observed in twitcher mice when AAV was delivered via a single route. Specifically, the median life span merely increased from ∼40 days in untreated mice to ≤70 days in treated mice irrespective of the AAV serotype, age at injection, and delivery route. 25,26,28 –30,32,34 –36 In all these studies, the galactosylceramidase gene was expressed from a constitutive ubiquitous promoter (such as the cytomegalovirus promoter and the CAG promoter). 37 No attempt was made to optimize the transgene.
Recently, Sorrentino et al. reported that molecular engineering of the therapeutic gene significantly improved gene therapy for mucopolysaccharidosis type IIIA, a neurodegenerative lysosomal storage disease caused by mutations in the sulfamidase gene. 38 Specifically, the authors attached the coding sequence of the low-density lipoprotein receptor binding domain of apolipoprotein B (Apo-LDLR-BD) and the signal peptide of iduronate-2-sulfatase (IDS-SP) to the transgene. The chimeric enzyme expressed from the modified gene showed more efficient secretion from cells and enhanced ability to cross the blood–brain barrier (BBB).
We hypothesize that a similar engineering approach can improve AAV-mediated Krabbe disease gene therapy. We tested our hypothesis in neonatal twitcher mice. We found that the engineered vector significantly increased the median life span following a single intraperitoneal (by 150%) and intravenous (by 275%) injection, respectively. This is in sharp contrast to single systemic AAV delivery studies published previously. In these studies, the life span of treated twitcher mice only extended by ≤75%. 25,26,28 –30,32,34 –36 Our results have set the foundation to further develop systemic adeno-associated virus serotype 9 (AAV9) therapy for Krabbe disease with the engineered vector.
Materials and Methods
Experimental animals
All animal experiments were approved by the animal care and use committees of the University of Missouri and University of Kansas Medical Center and were in accordance with National Institutes of Health (NIH) guidelines. Homozygous twitcher mice were generated by mating heterozygous animals originally provided by Bruce Bunnell, PhD, Tulane University School of Medicine, New Orleans, LA. 39 Mice were PCR genotyped using a forward primer 5′-CACTTAATTTTCTCCAGTCAT and a reverse primer 5′-TAGATGGCCCACTGTCTTCAGGTGATA. The PCR product was digested with EcoRV. Wild-type mice yielded a 260 bp band. Homozygous twitcher mice yielded a 234 bp band. Heterozygous mice had both bands (Supplementary Fig. S1A). Both male and female mice were used in the study. Wild-type and heterozygous littermates were used as normal controls. All mice were maintained in a specific-pathogen-free animal care facility on a 12-h light (25 lux):12-h dark cycle with access to food and water ad libitum. In addition, mice were given 31M Nutrigel (ClearH2O, Portland, ME). All experimental animals were monitored regularly for gait, posture, and body weight. Animals were euthanized once they reach a moribund condition (severe wasting and/or loss of voluntary movement).
Generation of the modified galactosylceramidase construct
The engineered galactosylceramidase gene was synthesized by GenScript (Piscataway, NJ). The construct consists of (from the 5′-end) (1) the coding sequence of the human IDS-SP, (2) the codon-optimized mouse galactosylceramidase gene coding sequence, (3) the MycFlag tag, and (4) the coding sequence of the human ApoB-LDLR-BD. Transgene expression was controlled by the ubiquitous CAG promoter. The SV40 late polyadenylation signal was used for transcription termination. The whole-gene expression cassette was inserted between two AAV2 inverted terminal repeats. This cis-packaging plasmid was named pXP46 (Fig. 1A).

AAV9 galactosylceramidase vector engineering.
AAV preparation, purification, and titration
Recombinant AAV9 stock was generated using the conventional triple-plasmid transfection method in 293 cells according to our published protocol. 40 The three plasmids used in transfection were (1) the cis-plasmid pXP46, (2) the AAV9 helper plasmid pRep2/Cap9 (a gift from Dr. James Wilson at the University of Pennsylvania, Philadelphia, PA), and (3) the commercially available adenoviral helper plasmid (Agilent Technologies, Clara, CA). Crude AAV lysates were purified through three rounds of isopycnic CsCl ultracentrifugation followed by three changes of HEPES buffer at 4°C for 48 h. The endotoxin level (as determined by the limulus amebocyte lysate assay) was within the acceptable level recommended by the Food and Drug Administration. Viral titer was determined by quantitative PCR using the Fast SYBR Green Master Mix kit (Applied Biosystems, Foster City, CA) in an ABI 7900 HT qPCR machine. The forward and revise primers used for titration were 5′-CCAGACATGATAAGATACATTGATGAGTT and 5′-AGCAATAGCATCACAAATTTCACAA, respectively.
AAV delivery
Experiments were performed in two different locations. At the University of Missouri, 8 × 1011 viral genome (vg) particles of the vector was injected to 2-day-old twitcher mice via the facial vein (Supplementary Fig. S2). At the University of Kansas Medical Center, 6 × 1011 vg particles of the vector were injected intraperitoneally to 6- or 12-day-old twitcher mice. AAV injection was confirmed by PCR using a forward primer 5′-GCCACTACGTGGTGGCTAGC and a reverse primer 5′-GAGTGCTGGTGGGACATTGTC (Supplementary Fig. S1B).
Histochemical examination of galactosylceramidase expression
In situ histochemical staining of GALC was based on a published protocol. 41 Mice were fully anesthetized with isoflurane and perfused with 4% paraformaldehyde, tissue was removed, and immersed in 30% sucrose at 4°C until sunken. Tissue was frozen on powdered dry ice and sections of 50 μm thick were cut using a sliding freezing microtome. Sections were incubated in 0.027 M citrate/0.045 M sodium phosphate, pH 4.5, for 15 min at room temperature; 5 mg/mL taurodeoxycholate acid with 5 mg/mL oleic acid (stock in 100% ethanol) in citrate–phosphate buffer for 15 min at room temperature; and then incubated in the latter solution with 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, and 0.6 mg/mL X-gal (stock in dimethylformamide) for 6–6.5 h at 37°C protected from light. Sections were then rinsed twice in phosphate-buffered saline (PBS) and twice in H2O, mounted with 0.2% gelatin, air-dried overnight in dark, and coverslipped.
Light and electron microscopy
Enzymatic activity of galactosylceramidase was examined by histochemical staining according to a published protocol. 41 Periodic acid-Schiff (PAS) staining was performed as described previously. 42 Electron microscopy was performed according to our published protocol. 43 Briefly, the tissue sample was fixed in PBS containing 4% paraformaldehyde and 2% glutaraldehyde and then postfixed in 2% osmium tetroxide in 0.1 M cacodylate buffer. After dehydration in graded ethanol, the sample was incubated in propylene oxide for 20 min and then incubated overnight at 65°C in a half propylene oxide and half Embed 812 resin (Electron Microscopy Sciences, Inc., Fort Washington, PA). One-micrometer sections were stained with 1% toluidine blue for examination by light microscopy. Eighty-nanometer sections were stained with 3% uranyl acetate and Sato's lead citrate and viewed on a JEOL JEM-1400 transmission electron microscope at 100 KV.
AAV genome copy number quantification
Genomic DNA was extracted from the liver, spleen, pancreas, lung, kidney, tibialis anterior muscle, gastrocnemius muscle, heart, cerebrum, cerebellum, and spinal cord. The AAV genome copy number was determined by TaqMan qPCR using the TaqMan Universal PCR Master Mix (Thermo Fisher Scientific). The primers and probe were designed to target the SV40 polyA sequence. Specifically, the forward primer is 5′-ACAAACCACAACTAGAATGCAGTGA. The reverse primer is 5′-TGTTGTTGTTAACTTGTTTATTGCAGCTTA. The probe is 5′-ATAGCATCACAAATTTC.
Western blot
Whole tissue lysates were electrophoresed on an 8% sodium dodecyl sulfate–polyacrylamide gel. Western blot was carried out on an iBind Flex Western Device (Invitrogen). The flag-tagged galactosylceramidase was detected with a monoclonal anti-flag M2 antibody (1:200; Sigma). Alpha-tubulin was used as a loading control and was detected with a mouse monoclonal antibody (1:2,000; Sigma). Signal was detected using the ECL system (Bio-Rad) and visualized using the Li-COR Odyssey Imaging System (LI-COR Biotechnology).
Gait evaluation
The gait pattern was evaluated daily at the University of Missouri. The onset of the gangling walking and dragging walking was recorded. The gangling gait is defined as unsteady walk from side to side in a wobbly manner. The dragging gait is defined as hind limb dragging or paresis.
Tremor index quantification
The tremor phenotype of twitcher mice was studied at the University of Kansas Medical Center using a force plate actimeter according to the manufacturer's instruction (Basi, West Lafayette, IN). Specifically, the tremor index was calculated as the difference in the averaged power between the high-frequency range (13–20 Hz) and the low-frequency range (0–5 Hz). 44
Statistical analysis
Except for the survival curve, all other quantitative data are presented as mean ± standard error of the mean and are graphed as dot scatter plots. For the survival curve, data are graphed as a Kaplan–Meier plot. Statistical analysis was performed using GraphPad Prism V7 (GraphPad software, Inc., La Jolla, CA). The normality of data distribution was tested with the Shapiro–Wilk normality test. For mice treated at 2 days of age, statistical significance for the gait pattern, growth curve, and survival curve was determined using an unpaired Student's t-test, two-way ANOVA, and the log-rank (Mantel–Cox) test, respectively. For mice treated at 6–12 days of age, statistical significance for the life span, tremor index, and body weight was determined by one-way ANOVA. A p value of <0.05 was considered statistically significant.
Results
Construction and characterization of the engineered AAV9 galactosylceramidase vector
The construct was built on the codon-optimized mouse galactosylceramidase coding sequence. To enhance therapeutic benefits, we adopted a strategy described by the Fradli laboratory. 38 Specifically, we included the ApoB-LDLR-BD coding sequence (Fig. 1A). This domain has been shown to improve protein delivery to the central nervous system (CNS) by receptor-mediated transcytosis. 45 –47 We also included the signal peptide from IDS, a highly secreted sulfatase, to promote secretion of galactosylceramidase. 48 To facilitate the detection of the engineered chimeric galactosylceramidase, we further attached the MycFlag tag (Fig. 1A).
We next packaged the engineered construct in AAV9 and delivered intravenously to 2-day-old normal mice (n = 2). Three months after AAV administration, we examined galactosylceramidase expression by enzymatic staining (Fig. 1B). As have been reported, 30 nominal galactosylceramidase activity was detected in uninjected mice (Fig. 1B). In sharp contrast, nearly saturated galactosylceramidase expression was seen in the heart and skeletal muscle of AAV-injected mice. We also observed strong galactosylceramidase activity in the liver in AAV-injected mice. Galactosylceramidase levels were relatively lower in the brain compared with other organs in injected mice, but the expression was higher than that of uninjected mice (Fig. 1B).
A single intravenous injection of the engineered AAV9 galactosylceramidase vector significantly mitigated the clinical phenotype and increased the life span of twitcher mice treated at 2 days
In light of positive findings from normal mice (Fig. 1B), we injected the engineered vector to 2-day-old twitcher mice via the facial vein (Supplementary Fig. S2). Untreated twitcher mice showed characteristic clinical presentations, including ambulation difficulty, weight loss, hind limb stiffness and paralysis, and rapid progression to a moribund status (Supplementary Videos S1a, S1b, S1c, S1d). Therapy with the engineered AAV9 galactosylceramidase vector greatly ameliorated clinical signs and symptoms (Supplementary Videos S2a, S2b, S2c, S2d).
The gangling gait was detected in untreated mice at the age 31.8 ± 1.9 days (range: 26–34 days). AAV therapy delayed the onset of gangling walking to the age of 104.8 ± 5.7 days (range: 83–142 days) (p < 0.01) (Fig. 2A). In untreated mice, the onset of the dragging gait was at the age 45.0 ± 0.6 days (range: 44–46 days). In treated mice, it was delayed to the age of 121.5 ± 8.0 days (range: 93–167 days) (p < 0.01) (Fig. 2A). Twitcher mice were able to maintain a normal body weight in the first 3 weeks of life but showed significant weight loss thereafter (Fig. 2B, C). AAV gene therapy significantly prevented weight loss. The body weight did not show statistically significant difference between AAV-treated twitcher mice and normal mice between 4 and 11 weeks of age, although the body weight of normal mice appeared higher (Fig. 2B). All untreated twitcher mice died by 7 weeks of age (median life span: 41 days; range: 32–49 days) (Fig. 2D). The life span of AAV-treated mice was significantly increased with the minimum, median, and maximum age reaching 16 weeks (112 days), 21.4 weeks (150 days), and 25.7 weeks (180 days), respectively (Fig. 2D) (p < 0.0001).
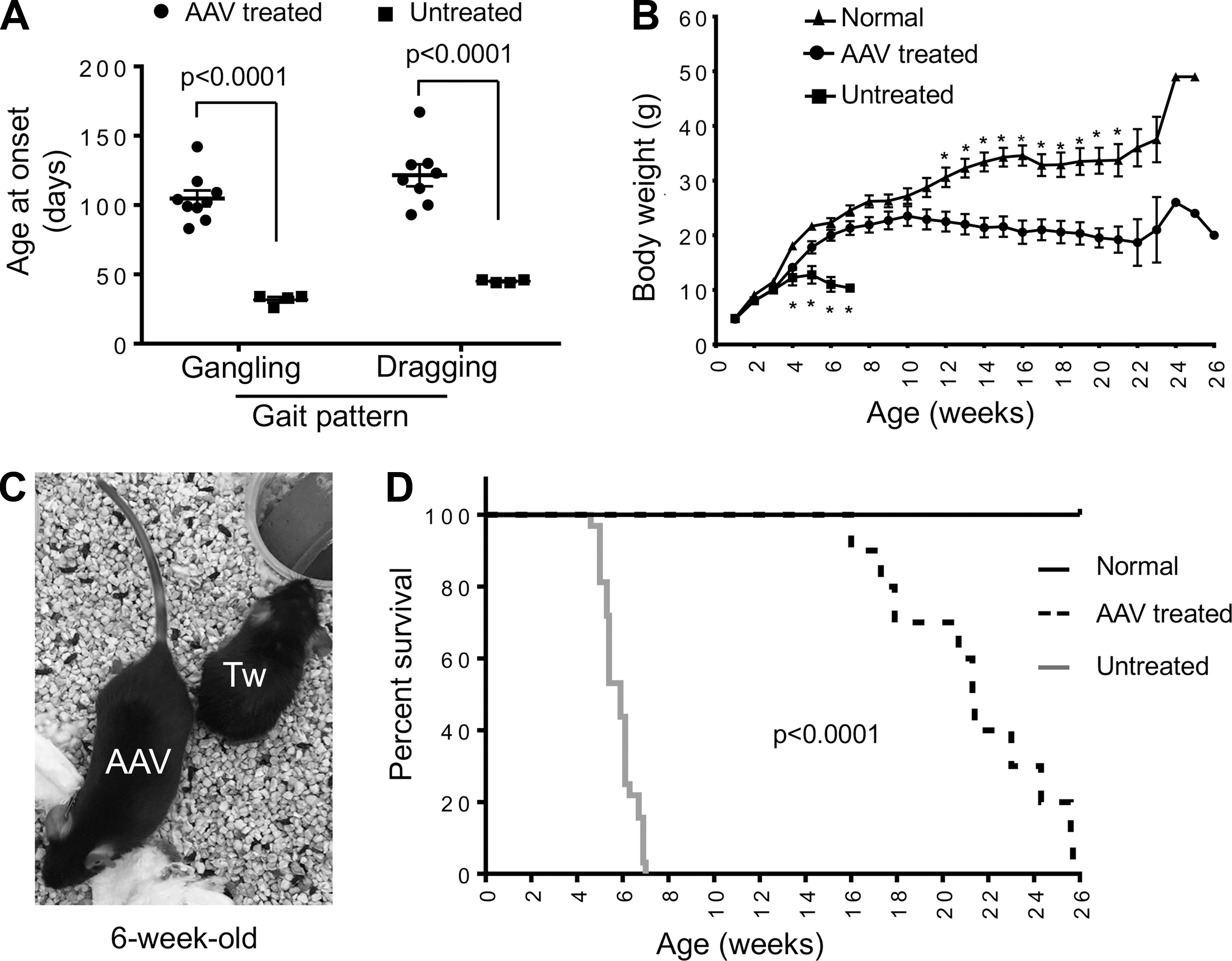
Intravenous delivery of the engineered AAV9 galactosylceramidase vector to 2-day-old twitcher mice resulted in significant amelioration of clinical manifestations.
On toluidine blue staining (Fig. 3, top panels), the peripheral nerve from the normal mouse had intact myelin and normal distribution of axons (Fig. 3). Untreated twitcher mouse sciatic nerves displayed characteristic features of Krabbe disease, such as demyelination and axon density reduction (Fig. 3). AAV therapy greatly improved histology with no obvious myelin loss and no increase in the spacing between myelinated axons (Fig. 3). The cerebellum was examined by PAS staining (Fig. 3, bottom panels). Abundant infiltration of foamy macrophages was detected in untreated twitcher mice, but none was found in normal mice (Fig. 3). Foamy macrophage infiltration was greatly reduced in AAV-treated mice (Fig. 3).

Systemic treatment of 2-day-old twitcher mice using the engineered AAV9 galactosylceramidase vector attenuated neuropathology. Top panels: representative photomicrographs of sciatic nerve toluidine blue staining from a normal mouse, an untreated twitcher mouse, and an AAV-treated twitcher mouse. Bottom panels: representative PAS staining photomicrographs of the cerebellum from a normal mouse (170 days old), an untreated twitcher mouse (48 days old), and an AAV-treated twitcher mouse (170 days old). Arrowhead, foamy macrophage. PAS, periodic acid-Schiff.
To study the expression of the engineered galactosylceramidase gene, we performed Western blot with an antibody that recognizes the flag tag (Fig. 4A). A positive band with the expected molecular weight (∼81 kDa) was consistently detected in the heart of AAV-treated twitcher mice (Fig. 4A). This band was also found in some skeletal muscle samples in treated mice. Surprisingly, we failed to see the flag positive band in other tissues in treated mice. To determine whether this was due to poor AAV transduction, we quantified the AAV genome copy number by TaqMan PCR (Fig. 4B). The highest amount of the AAV genome was detected in the liver (>40 copies per diploid genome), followed by the lung (>10 copies per diploid genome), heart and spinal cord (>3 copies per diploid genome), cerebrum and pancreas (>1 copy per diploid genome), and cerebellum and tibialis anterior muscle (>0.5 copy per diploid genome). The gastrocnemius muscle, spleen, and kidney had very low but detectable levels of the AAV genome (Fig. 4B).

Bodywide evaluation of the expression of the engineered galactosylceramidase and the biodistribution of the AAV9 vector.
Intraperitoneal AAV9 therapy in 6- to 12-day-old twitcher mice prevented weight loss, reduced tremor index, and extended the life span
To evaluate therapeutic effects of our engineered vector in older mice, we delivered AAV intraperitoneally to three 6-day-old and three 12-day-old twitcher mice (Fig. 5). We measured the body weight and the tremor index at 5 weeks of age (35 days) and recorded the life span. We did not see a significant difference in all the three parameters examined between mice treated at the age of 6 and 12 days (Supplementary Fig. S3). For this reason, all the data from AAV-treated mice were combined for subsequent analysis. Weight loss in untreated twitcher mice started at 3 weeks of age (Fig. 2B). At 5 weeks of age, the body weight of twitcher mice dropped to ∼50% of that of normal mice (p < 0.0001) (Fig. 5A). This weight drop was completely prevented by AAV treatment (Fig. 5A). The twitching phenotype was quantified by the tremor index at 5 weeks of age (Fig. 5B). Untreated twitcher mice had a significantly higher tremor index than the other two groups (p < 0.0001). Similar to what we saw with the body weight (Fig. 5A), AAV treatment prevented the twitching phenotype, and their tremor index was identical to that of normal mice (Fig. 5B). Compared with that of untreated twitcher mice (median: 41 days; range: 32–49 days), the life span of AAV-treated twitcher mice was significantly increased (median: 104 days; range: 78–112 days) (p < 0.0005) (Fig. 5C).

Intraperitoneal injection of the engineered AAV9 galactosylceramidase vector to 6- to 12-day-old twitcher mice significantly reduced clinical presentation.
On toluidine blue staining, myelin of the sciatic nerve from AAV-treated twitcher mice was largely preserved, revealing a greatly improved histology compared with that of untreated twitcher mice (Fig. 6A). However, the myelination of the axon was not completely normalized in AAV-treated twitcher mice. In particular, some myelin-disrupted axons were found in treated mice under an electron microscope (Fig. 6B). Nevertheless, most axons showed a normal pattern of myelination in the peripheral nerve of AAV-treated mice (Fig. 6B).

Systemic treatment of 6- to 12-day-old twitcher mice using the engineered AAV9 galactosylceramidase vector reduced characteristic neuropathology in the sciatic nerve.
Discussion
In this study, we tested single systemic delivery of an engineered galactosylceramidase AAV9 vector as a stand-alone therapy for infantile form Krabbe disease in neonatal twitcher mice. One-time AAV9 administration greatly ameliorated neuropathology and clinical presentation. Specifically, treatment reduced demyelination, axon loss, and foamy microphage infiltration (Figs. 3 and 6). AAV-treated mice also showed significantly improved body weight and motor function (Figs. 2 and 5). Untreated twitcher mice had a median life span of 41 days and they all died at or before 49 days (Figs. 2D and 5C). Notably, AAV9 therapy significantly increased the life span by a large margin (Figs. 2 and 5). Intraperitoneal injection in 6- to 12-day-old twitcher mice extended the median life span to 104 days, with the longest survival to 112 days (Fig. 5C). Intravenous injection in 2-day-old twitcher mice resulted in a median life span of 150 days and the longest-lived mouse survived to 180 days (Fig. 2D).
Pathological accumulation of psychosine in the nervous system leads to progressive demyelination and neurological damage in Krabbe disease. Restoration of galactosylceramidase expression in the nervous system with AAV gene therapy should remove toxic psychosine and attenuate neurological deterioration. To correct defects in the CNS, various AAV vectors, such as AAV2, AAV5, AAV9, AAVrh10, and AAV-oligo001, have been delivered directly to the CNS of neonatal twitcher mice by intracranial, intracerebellar, intrathecal, and intracerebroventricular injection. 25,27,31,32,34 –36 Although these treatments significantly prolong the life span, the increase is modest and the median life span fails to exceed 70 days in treated twitcher mice. The limited effect on the life span suggests that treating CNS alone is insufficient.
To overcome this issue, investigators have combined CNS-targeted AAV therapy with traditional therapies, such as bone marrow transplantation (BMT) and substrate reduction therapy. Karumuthil-Melethil et al. reported a median life span increase from <56 days with AAV9 intrathecal injection alone to 79 days following a combined therapy of intrathecal AAV9 delivery and BMT in 10- to 11-day-old twitcher mice. 32 Lin et al. showed a median life span of 111 days when they supplemented intracranial AAV5 injection with BMT in 3-day-old twitcher mice. 35 Hawkins-Salsbury et al. demonstrated a very impressive median life span of 299 days in 2- to 3-day-old twitcher mice when they combined AAV5 CNS delivery with both BMT and substrate reduction. 31 Collectively, these studies suggest that an effective therapy for Krabbe disease may require treating CNS, peripheral nervous system, and possibly other tissues.
Systemic AAV injection can deliver a therapeutic gene throughout the body. 49 Hence, this approach may better meet the needs of Krabbe disease gene therapy than CNS-targeted delivery. The first systemic AAV gene therapy for Krabbe disease was conducted by Rafi et al. in 2005 using AAV1. 26 Interestingly, intravenous injection to 1-day-old twitcher mice merely extended the median life span to ∼55 days. This is perhaps not surprising considering the fact that AAV1 cannot efficiently cross the BBB. 50,51 Recent studies suggest that AAV9 and AAVrh10 are highly potent in reaching the CNS from the peripheral vein. 52 –56 Stand-alone intravenous AAV9 injection has not been tested in twitcher mice before our study. However, systemic AAVrh.10 delivery has been reported in several studies. 28 –30 Unexpectedly, treatment in 2- and 10-day-old twitcher mice only extended the median life span to ∼55 and ∼70 days, respectively. 28 –30 This is similar to what have been reported with CNS-targeted AAVrh.10 therapy. 32 These findings suggest that some unknown factors may have hindered systemic Krabbe disease gene therapy with AAVrh.10 in the context of the twitcher mouse model.
Several laboratories have shown that intravenous AAV9 injection can effectively ameliorate lysosomal storage diseases, such as mucopolysaccharidosis IIIA, mucopolysaccharidosis IIIB, gangliosidosis-1, and metachromatic leukodystrophy. 57 –60 Systemic AAV9 gene therapy has also resulted in impressive bodywide correction in mouse models of neuromuscular diseases, such spinal muscular atrophy and Duchenne muscular dystrophy. 61 –63 Most recently, Mendell et al. reported a marvelous reversal of the catastrophic disease course in type I spinal muscular atrophy patients with systemic AAV9 therapy. 64 Several clinical trials have been initiated in last couple of years to treat mucopolysaccharidosis IIIB and Duchenne muscular dystrophy using intravenous AAV9 delivery. 65 –67 In light of the rapid preclinical and clinical progress of systemic AAV9 gene therapy, we evaluated this AAV serotype in twitcher mice by intravenous injection.
We first engineered an AAV9 vector (Fig. 1A). Several considerations were taken in vector construction. First, we codon-optimized the endogenous mouse galactosylceramidase gene coding sequence. This strategy has been widely used to enhance protein expression. 68,69 Second, we optimized galactosylceramidase secretion and CNS delivery. For lysosomal storage disease therapy, it is well established that therapeutic efficacy can be greatly improved by boosting secretion of the therapeutic enzyme and/or enhancing the ability of the enzyme to cross the BBB. 38,47,70 –75 For our vector, we utilized a design published by Sorrentino et al. 38 Specifically, the IDS-SP was attached to the N-terminal end of galactosylceramidase. This signal peptide facilitates secretion. 48,76 The MycFlag-tag-conjugated ApoB-LDLR-BD was added to the carboxyl terminus of galactosylceramidase. The MycFlag tag allows detection of the chimeric protein. ApoB-LDLR-BD enhances penetration through the BBB (Fig. 1A).
Galactosylceramidase expression from the engineered AAV9 vector was characterized in vivo by systemic injection in newborn normal mice. Physiological galactosylceramidase expression in normal mice was very low (undetectable in the heart and skeletal muscle and faint in the liver and brain by enzymatic staining) (Fig. 1B). 30 Consistent with our previous publications, 77,78 systemic AAV9 injection resulted in bodywide expression of the engineered galactosylceramidase (Fig. 1B). Robust galactosylceramidase activity was seen in the heart, skeletal muscle, and a subset of liver cells in AAV-injected mice (Fig. 1B). The parental construct that we used to build our vector contains the IDS-SP for improved secretion and the ApoB-LDLR-BD for enhanced BBB crossing. The parental construct has been shown to result in a sustained increase of the activity and level of the therapeutic enzyme in both the serum and brain in different types of lysosomal storage disease. 38 In our study, we did not directly test whether our engineered construct had indeed increased secretion and BBB crossing. Based on the findings from the parental construct, we speculate that systemic AAV9 injection might have turned transduced tissues into protein factories to produce and secrete galactosylceramidase to the circulation and then to enter the CNS to digest excessive psychosine. Besides peripheral contribution, therapeutic galactosylceramidase can also be produced in the CNS in situ following systemic AAV9 injection because AAV9 can cross the BBB. 53,56 Indeed, we observed increased galactosylceramidase activity in the brain of AAV-injected mice by enzymatic staining (Fig. 1B).
To determine the therapeutic outcome of a single AAV9 systemic delivery, we treated 2-day-old twitcher mice. We observed significant improvements in the gait, body weight, and life span (Fig. 2). Histological amelioration in the nervous system supported clinical findings (Fig. 3). Unexpectedly, in the flag tag Western blot, we only found the chimeric galactosylceramidase band in the heart and skeletal muscle (Fig. 4A). The enzymatic staining results in Fig. 1B as well as our previously published studies suggest that intravenous AAV9 injection in newborn mice consistently leads to the highest transgene expression in the heart, followed by the skeletal muscle. 77,78 The liver and other internal organs show much lower transgene expression. 77,78 We speculate that the absence of the galactosylceramidase band in nonmuscle tissues might be due to the limited detection sensitivity of the flag tag Western blot, rather than a lack of AAV gene transfer. Indeed, the biodistribution study revealed high amounts of the AAV genome in the liver, lung, heart, and nervous system (Fig. 4B).
In a clinical scenario, it is very likely that many patients will not have immediate access to gene therapy right after birth. To determine whether gene therapy started at a later age can slow disease progression, we treated 6- and 12-day-old twitcher mice (Figs. 5 and 6). Consistent with what we saw in 2-day-old twitcher mice, demyelination was greatly reduced, although not completely prevented (Fig. 6). Furthermore, we observed significant improvements in the tremor index, bodyweight, and life span in mice treated at the older age (Fig. 5).
More than 12 AAV gene therapy studies have been conducted in twitcher mice. 25 –36 While various endpoints have been used in different studies, all have reported the life span. Using the life span as a bench marker, our results far exceed what have been reported in all previous single-route AAV therapy studies irrespective of the AAV serotypes (AAV1, AAV2, AAV5, AAV9, AAVrh10, and AAV-oligo001), age at injection (1- to 10-day-old), and delivery route (intravenous or direct injection to the CNS). In most of these studies, the median life span rarely surpassed 60 days. 25,26,28 –30,34,35 However, we observed a median life span of 150 and 104 days in mice treated at 2 and 6–12 days of age, respectively (Fig. 2D). Vector engineering is the primary difference between our study and previous studies. Our construct has the built-in features of improved protein production, secretion, and CNS penetration, but the vectors used in previous studies do not. The superior efficacy seen in our study reemphasizes the importance of vector engineering for lysosomal storage disease gene therapy. 38,47,70 –75
While our results are highly encouraging, the treated mice in our study did still die prematurely. The cause of death is not clear, but undoubtedly, it suggests that the therapy described here is not sufficient to completely stop disease progression. Our current study has focused on the clinical improvement in treated twitcher mice. Future studies are needed to comprehensively quantify galactosylceramidase expression (at both mRNA level and protein level), galactosylceramidase activity in the serum, every region of the brain and spinal cord, as well as all peripheral organs and tissues. Future studies should also include quantitative biochemical and histological evaluation of neuroinflammation and neuropathology, and detailed analysis of correction in every cell type (e.g., neurons and oligodendrocytes) in the CNS. These studies will validate our presumed mechanism of action (via improved secretion, uptake, and crossing the BBB) and reveal whether the current dosage has resulted in efficient restoration of the galactosylceramidase activity throughout the brain and in all peripheral organs.
In light of above discussion, we believe that there is still room for improvements. For example, it may be worthwhile to systemically compare the ApoB-LDLR-BD with other BBB-penetrating peptides, such as the anti-human transferrin receptor antibody, 74 the fragment from insulin-like growth factor II, 79 and the 16-lysine residue-linked low-density lipoprotein receptor-related protein-binding amino acid segment of apolipoprotein E. 70 Such studies may allow us to identify a more potent CNS-crossing peptide to be incorporated in the AAV vector. Besides the expression cassette, there is also a good chance to further enhance AAV therapy using alternative AAV capsids. Recent studies have identified a series of novel AAV variants that are more effective than AAV9 in transducing the CNS via systemic injection. These include AAV9.HR, AAV-B1, AAV-PHP series, AAV-AS, and AAV-Anc80L65. 80 –85 Combination therapy is another direction to be explored in the future. This approach has yielded by far the best outcome. In one study, Hawkins-Salsbury et al. combined intracranial/intrathecal AAV5 injection with BMT and substrate reduction. This resulted in a median life span of 299 days. 31 Most recently, Marshall et al. combined intravenous, intracranial, and intrathecal AAV9 injection and achieved a median life span of 263 days. 33 The addition of BMT further extended the median life span to 285 days. 33 It is very likely that a similar combination therapy using our engineered AAV9 vector may yield a median life span exceeding these reported by Hawkins-Salsbury et al. and Marshall et al. 31,33
Clearly, more studies are needed to completely characterize pathological, biochemical (e.g., psychosine levels), enzymatic (e.g., galactosylceramidase serum levels and activities), and neuroinflammatory changes in treated twitcher mice. Despite the limitation of the study, our results have provided the important proof-of-principle for an improved approach for Krabbe disease gene therapy through vector optimization and engineering.
In summary, we have developed an engineered AAV9 vector for Krabbe disease gene therapy. Systemic delivery with our engineered AAV9 vector resulted in unprecedented improvement in twitcher mice. Large animal study is an important step before clinical trials are initiated. Krabbe disease has been described in dog, sheep, and nonhuman primate. 86 –89 Gene and cell therapies have been conducted in the canine and primate models. 90 –92 Future tests of our engineered vector in these models will lay the foundation for translating systemic AAV9 therapy to Krabbe disease patients.
Footnotes
Acknowledgments
This work was funded by the Kansas City Area Life Sciences Institute—Patton Trust Research Grant (KCALSI-15-6). Core support was provided by the Kansas Intellectual and Developmental Disabilities Research Center, NICHD (P30 HD 02528) and the Electron Microscopy Research Laboratory at the University of Kansas Medical Center, supported in part by the NIH COBRE grant 9P20GM104936 (the JEOL JEM-1400 was purchased with funds from the NIH grant S10RR027564). We thank Dr. Alessandro Fraldi (Telethon Institute of Genetics and Medicine, Italy) for providing a plasmid containing the human IDS-SP and human ApoB-LDLR-BD. We thank Dr. James Wilson (University of Pennsylvania) for providing the AAV9 packaging plasmid. We thank Dr. Bruce Bunnell (Tulane University School of Medicine, New Orleans, LA) for providing the breeder mice that were used for the eventual production of twitcher and control mice used in this study. We thank Barbara Fegley for preparing sections for microscopic analysis. We thank Dr. Lixing W. Renecker (Department of Ophthalmology, School of Medicine, the University of Missouri) for kindly providing the paraffin-embedding device.
Author Disclosure
D.D. is a member of the scientific advisory board for Solid Biosciences and an equity holder of Solid Biosciences. D.D. is an inventor on a patent (unrelated to the current study) licensed to Solid Biosciences. The Duan laboratory has received research supports (unrelated to the current study) from Solid Biosciences. S.M.L. has received funding from ApoPharma, Inc.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Video S1a
Supplementary Video S1b
Supplementary Video S1c
Supplementary Video S1d
Supplementary Video S2a
Supplementary Video S2b
Supplementary Video S2c
Supplementary Video S2d
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.