
Abstract
Endothelial cells (EC) are targets in gene therapy and regenerative medicine, but they are inefficiently transduced with adeno-associated virus (AAV) vectors of various serotypes. To identify barriers hampering efficient transduction and to develop an optimized AAV variant for EC transduction, we screened an AAV serotype 2-based peptide display library on primary human macrovascular EC. Using a new high-throughput selection and monitoring protocol, we identified a capsid variant, AAV-VEC, which outperformed the parental serotype as well as first-generation targeting vectors in EC transduction. AAV vector uptake was improved, resulting in significantly higher transgene expression levels from single-stranded vector genomes detectable within a few hours post-transduction. Notably, AAV-VEC transduced not only proliferating EC but also quiescent EC, although higher particle-per-cell ratios had to be applied. Also, induced pluripotent stem cell-derived endothelial progenitor cells, a novel tool in regenerative medicine and gene therapy, were highly susceptible toward AAV-VEC transduction. Thus, overcoming barriers by capsid engineering significantly expands the AAV tool kit for a wide range of applications targeting EC.
Introduction
Adeno-associated virus (AAV) vectors are one of the most frequently applied gene delivery systems in preclinical research and in human clinical trials. 1 Three AAV vector-based gene therapies, Glybera, Luxturna, and Zolgensma, have already received marketing authorization in the Western World. 2 Further are expected to follow in the foreseeable future, because clinical benefit is reported for a wide range of monogenetic diseases. 3 First-generation AAV vectors are based on naturally evolved serotypes or variants of AAV. Adeno-associated viruses are replication-deficient and nonpathogenic members of the Parvoviridae, with AAV serotype 2 (AAV2) as the best characterized representative and vector prototype. 4
AAV vectors deliver a single-stranded DNA genome into proliferating or post-mitotic cells. Cell transduction is mediated by the AAV capsid that is assembled from 60 monomers, which share the so-called common VP3 region. 5 This region is identical in amino acid sequence to the major capsid protein, VP3 (60 kDa), and contains the receptor binding motifs. In contrast, the two minor capsid proteins, VP1 (90 kDa) and VP2 (72 kDa), contribute distinct domains such as those required for endosomal escape and nuclear delivery located within their N-terminal extensions. 5 Efficiency of in vivo gene transfer was greatly improved by using capsids of serotypes or variants others than AAV2 for packaging of recombinant AAV genomes. 4 Nevertheless, relatively high vector doses are required to obtain therapeutic effects, 6,7 and distinct clinically relevant cell types such as keratinocytes or dendritic cells are refractory toward transduction. 8,9
Nonsusceptibility due to lack of receptors is overcome by cell surface targeting. 5,10 The same strategy is used to re-direct AAV's tropism toward a predefined cell type or a more precisely predefined cell receptor. 5,10 In particular, the variable loops at the peaks of the AAV capsid and the N-terminus of the VP2 proteins accept tropism-modifying alterations without impairing capsid assembly. 5,10 Lack of knowledge on peptides directing the particle toward a receptor that initiates a productive infection without interfering with capsid assembly fostered the development of viral libraries composed of capsid-engineered AAV variants that are subjected to high-throughput selection screens. Although libraries were initially based on AAV2 and displayed 7mer peptides of random sequence at capsid position 58711 or 588, 12 the portfolio has been expanded to other serotypes, to libraries displaying larger peptides or being composed of shuffled capsids. 13 Through this powerful technology, a whole tool kit of AAV mutants with significantly improved gene delivery properties for in vitro and in vivo usage have become available. These efforts also increased our knowledge of AAV's infection biology and on cell surface molecules suited for cell entry targeting. 5,8,14
Here, we focused on endothelial cells (EC), a cell type highly relevant in translational and basic research, but poorly transduced by AAV vectors with naturally occurring serotype capsids. We performed an AAV2 peptide display library screening on primary human macrovascular EC in the absence of helper virus co-infection and in the presence of high selection pressure. In addition, production of sub-libraries was optimized through a novel next generation sequencing (NGS)-based amplification and cloning strategy that led to the identification of AAV-VEC. With >85% of transduced cells, AAV-VEC is far more efficient than the parental serotype or first-generation capsid-engineered vectors at transducing EC. Using AAV-VEC and the second most enriched variant AAV-NEC as tools, we identified distinct steps in the AAV infection pathway that limit transduction of EC. Finally, we demonstrated AAV-VEC's potency in modifying induced pluripotent stem cell (iPSC)-derived endothelial progenitor cells (EPC). As EPC home to damaged tissue or growing vessels, 15 genetic modification of such cells might be envisioned as a valuable addition to current “Trojan horse” concepts in regenerative medicine and antitumor therapy.
Materials and Methods
Cell lines and primary cells
The human embryonic kidney cell line HEK293 (ATCC number: CRL-1573) and human cervix carcinoma cell line HeLa (ATCC number: CCL-2) were maintained in Dulbecco's modified Eagle's medium (DMEM) with GlutaMAX-I (Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 100 IU/mL of penicillin (Invitrogen), and 100 μg/mL of streptomycin (Invitrogen). AAV receptor knock-out (AAVR KO) cell lines based on HEK293 and HeLa, respectively, were generated before 16 and cultivated as described earlier. Human umbilical vein EC (HUVEC) (PromoCell) was maintained in EC growth medium 2 (PromoCell) supplemented with 2% FBS, 5 ng/mL endothelial growth factor (EGF), 10 ng/mL basic fibroblast growth factor, 20 ng/mL insulin-like growth factor, 0.5 ng/mL vascular EGF (VEGF), 1 μg/mL ascorbic acid, 0.2 μg/mL hydrocortisone, 100 IU/mL of penicillin (Invitrogen), and 100 μg/mL of streptomycin (Invitrogen). Throughout the experiments, only low-passage HUVEC were used (passage 4–7). All cells were maintained in a humidified incubator with 5% CO2 at 37°C.
Cell surface expression of AAV2 receptors
Analyses of cell surface expression of AAV2 receptors on HUVEC (low passage) and HeLa cells were performed as described. 8,17 Briefly, the following primary antibodies were used in this work: mouse anti-human αvβ5 antibody (MAB1961; Millipore), mouse anti-human α5β1 antibody (MAB1999; Millipore), and mouse anti-human heparan sulfate delta antibody (USBiological). Goat anti-mouse IgG was used as secondary antibody (Southern Biotech). Cell surface expression was determined by flow cytometry (FACS Calibur; Becton Dickinson) after harvesting of cells by trypsin (HeLa; Invitrogen) or Accutase (HUVEC; Sigma) treatment. A minimum of 10,000 cells was acquired for each sample.
AAV peptide selection on HUVEC and AAV vector production
AAV2 peptide display library 11 was produced in HEK293 cells. Phenotype and genotype of variants was coupled. Low-passage HUVEC, 24 h post-seeding, were incubated with the AAV peptide library at a viral genome-per-cell ratio (GOI) of 1000 on ice for 1 h, followed by 3 h at 37°C. Particles that failed to enter the cells were removed by exchanging the medium. Cells were washed and then harvested by Accutase (Sigma) treatment. After a further washing step, total DNA was isolated (DNeasy Blood and Tissue kit; Qiagen). Amounts of particles that successfully entered cells were quantified by quantitative polymerase chain reaction (qPCR) using cap gene-specific primers. Cap sequences neighboring the insertion site were amplified by PCR (Supplementary Figure S1) and sub-cloned into pLG backbone with CLO primers (Supplementary Table S1). The sub-library was produced as described earlier. Meanwhile, one third of viral DNA was PCR amplified with NGS primers for sequencing. To raise the selection pressure, GOI was reduced in subsequent selection rounds (GOI 100 for 2nd and GOI 10 for 3rd round). The 3rd round of AAV peptide display selection was repeated. Cells were harvested, and DNA was isolated from cell nuclei after subcellular fractionation (Subcellular Protein Fractionation Kit for Tissues; Thermo Fischer Scientific).
NGS data obtained from that selection (Sub-Nuc) were compared with NGS data from round 1 to 3 of whole cell selection (Sub-Lib1–Sub-Lib3). Purity of fraction at the protein level was assayed by western blot analysis (Supplementary Fig. S2) using antibodies against AKT (cytosol; Cell Signaling Technologies, Inc.) and Lamin B (nucleus; Santa Cruz Biotechnology, Inc.).
Peptide sequences of the most prominent two variants were ordered as oligonucleotides and cloned into pRC’99 by using MluI/AscI sites 18 generating pRC’99-VEC and pRC’99-NEC, respectively. For production of AAV vectors, HEK293 cells were cotransfected with a total of 37.5 μg of pRC, 19 pRC‘99-VEC, pRC’99-NEC, pRC’99-SIG 18 or pRC’99-NDVRAYS, pscAAV/EGFP 20 or pGFP, 21 and pXX6. 22 Of note, the AAV helper plasmid pRC’99-NDVRAYS was cloned as described in Nicklin et al. 18 by using the peptide sequence reported by Muller et al. 12 Cells were harvested 48 h post-transfection, cell pellet was lysed, and vectors were purified by iodixanol step gradient centrifugation. 20 Genomic particle titers were determined by qPCR (LightCycler System; Roche Diagnostics) using green fluorescent protein (GFP)-specific primers, 20 whereas capsid titers were determined by A20-ELISA (Progen).
Quantification of cell entry and nuclear entry efficiency
For determining vector uptake, HUVEC were incubated with the indicated GOI for 30 min on ice, followed by a shift to 37°C and 5% CO2. Cells were harvested at the indicated time points post-transduction (p.t.) by Accutase or trypsin treatment and washing steps as described earlier. Total DNA was isolated (DNeasy Tissue Kit; Qiagen), and vector genomes were quantified by qPCR (Roche Diagnostics) using GFP-specific and plasminogen activator (PLAT)-specific primers. 23 After confirmation of target specificity by melting curve analysis, GFP values were normalized to PLAT levels by using the LightCycler 480 software 1.5 (Roche Diagnostics). For determining nuclear entry efficiency, subcellular fractionation was performed as described earlier. The nuclear fractions were subjected to DNA isolation and analyzed as described earlier.
Cell binding assay
To determine the cell surface binding, HUVEC were seeded 24 h before transduction in 12-well plates with a density of 2.5 × 105 cells per well. Cell number per well was determined before transduction and used to calculate the GOI. To synchronize cell transduction, cells were incubated on ice for 10 min, followed by a washing step with phosphate-buffered saline (PBS) (cold). Cold fresh media were added to cells, which were maintained on ice for a further 20 min. Then, indicated vectors (GOI 5 × 103) diluted in cold media were added to the cells. After a further 1-h incubation on ice, one sample for each vector—corresponding to the time point 0 h p.t.—was washed with PBS intensively (two times), harvested by trypsin treatment, and finally stored at −80°C. The remaining samples were incubated at 37°C until harvesting (4, 6, and 24 h p.t.). Again, supernatant was removed, and cells were washed with PBS. Trypsin treatment followed by two washing steps of the cell pellet was performed to remove membrane-bound particles. A part of the “24-h samples” was analyzed by FACS (CytoFLEX platform; Beckman Counter). All samples were then subjected to DNA isolation and qPCR analyses as described earlier.
Transduction of HUVEC, AAVR KO, and iPSC-derived EPC
To determine transduction efficiency, HUVEC, HEK293-AAVR-KO, HEK293, HeLa-AAVR-KO, and HeLa cells, respectively, were incubated with indicated vectors and GOI for 24 h followed by flow cytometric analyses (FACS Calibur; Becton Dickinson). Background fluorescence was set to 1% by using nontreated cells as reference. Total RNA was isolated at indicated time points. Following cDNA synthesis, GFP expression was quantified by qPCR (Roche Diagnostics) using GFP-specific and GAPDH-specific primers. For transduction of quiescent EC, HUVEC were cultivated until they reached a confluency of ∼90%, followed by incubation for another 24 h at 37°C and 5% CO2 with EC Basal Medium (PromoCell), which was devoid of all supplements and FBS. Finally, the status of quiescence was confirmed by staining with 5-ethynyl-2′-deoxyuridine (EdU) according to the manufacturer's instruction (Invitrogen).
For transduction of iPSC-derived EPC (iEC), iPSC clone H2E6C 24 was differentiated into EC lineages by using a modified protocol based on a previously described procedure. 25 Briefly, iPSC were cultured as monolayers in murine embryonic fibroblasts (MEF)-conditioned medium on Geltrex (Thermo Fischer Scientific) and mesodermal induction was initiated by the GSK3-inhibitor CHIR (Axon Medchem). Cells were further differentiated into EPC in VEGF (Peprotech) containing medium for 6 days. On day 7 of differentiation, EC were harvested with StemPro Accutase (Thermo Fischer Scientific) and seeded on a gelatine (Sigma-Aldrich)-coated plate in EGM2 medium (Lonza). Transduction was performed at indicated days of differentiation with indicated vectors in EGM2 medium. GFP expression was analyzed 48 h p.t. by flow cytometry (FACS Calibur; Becton Dickinson).
Heparin competition assay
Overall, 2.5 × 104 HUVEC per well were seeded 24 h before transduction. This cell number was used for calculation of GOI. Cells were incubated for 10 min on ice; after a washing step (cold PBS), cold fresh media were added to the cells, which were maintained on ice for a minimum of 20 min. Viral vectors were diluted in heparin containing medium and added to the cells (final concentration indicated in Fig. 8). One hour post-vector application, samples were shifted to 37°C for 24 h. Percentage of transgene-expressing cells was determined by flow cytometry (CytoFLEX platform; Beckman Counter).
Heparin affinity chromatography
A HiTrap heparin affinity column (Amersham) was equilibrated with 1 × PBS/1 mM MgCl2/2.5 mM KCl. Gradient-purified AAV vector preparation diluted in 1 × PBS/1 mM MgCl2/2.5 mM KCl was loaded. Flow-through and 4 × 5 mL wash fraction (1 × PBS/1 mM MgCl2/2.5 mM KCl) were collected. Elution was performed with increasing ionic strength (1 × PBS/1 × mM MgCl2/2.5 × mM KCl plus 100 mM NaCl up to 1 × PBS/1 mM MgCl2/2.5 mM KCl plus 1 M NaCl). For each step, five column volumes were collected. Samples were analyzed by qPCR using transgene (GFP)-specific primers.
Capsid thermal stability assay
We performed the capsid thermal stability assay as described. 26 Briefly, wells of qPCR plates were loaded with 5 × 108 vector genome containing particles of indicated AAV vector preparations diluted in PBS. The temperature gradient was generated by a LightCycler® 96 System (Roche Life Science). Then, PBS was used to dilute samples. Samples were transferred to a nitrocellulose membrane by using a vacuum blotter at native dot blot conditions. Membrane was blocked and then incubated with primary (A20, or B1; Progen) and then secondary (peroxidase-conjugated antibodies) antibodies. Finally, membranes were incubated with an enhanced chemiluminescence reagent (West Dura; Pierce) and analyzed by autoradiography film exposure or FusionFX device (Peqlab).
Statistical analysis
Statistical analyses and significance tests were performed by applying unpaired, two-tailed t-tests. p-values of <0.05 were considered significant.
Results
Insufficient levels of heparan sulfate proteoglycan may limit EC transduction
At a vector GOI with which more than 80% of HeLa cells were transduced with a single-stranded AAV2 vector encoding for enhanced GFP, only 18.0 ± 4.95% of HUVEC expressed the reporter gene (Fig. 1A). This result is consistent with earlier reports showing that AAV2 and other AAV serotypes are inefficient in EC transduction. 12,18,27,28

Vascular endothelial cells are poor targets for AAV2 transduction.
Low permissiveness is frequently associated with inefficient provision of receptors; therefore, HUVEC were analyzed for the presence of AAV2's primary receptor, heparan sulfate proteoglycan (HSPG), 29 and the internalization receptors αvβ5 and α5β1 integrin, 30,31 respectively. As shown in Fig. 1B, HUVEC clearly stained positive for αvβ5 and α5β1, whereas HSPG was barely detectable. In contrast, HeLa cells showed considerable expression of both, HSPG and the two integrins (Fig. 1B, insert).
Given the dependency of AAV2 on HSPG for subsequent steps in cell transduction, such as priming the capsid for co-receptor binding, endosomal escape, and uncoating, 32 it is reasonable to postulate that the low HSPG expression level negatively impacts AAV2 transduction efficiency.
AAV peptide selection of AAV capsid variants on HUVEC
To develop an AAV variant that overcomes this pre- and maybe further post-entry barriers, an AAV peptide display screening was performed. The library was based on AAV2, displayed random 7mer peptides at position 587, and was screened on HUVEC by applying the following criteria: (1) selections were performed in the absence of helper virus co-infection to avoid any assistance in cell infection caused either directly by helper virus proteins or indirectly by changing the intracellular milieu, (2) time of library exposure was limited to three hours, and (3) selection pressure was constantly increased by decreasing the GOI to select for highly infectious variants.
In total, three rounds of selection were performed on low-passage HUVEC. Each time, viral genomes were isolated from whole cell extracts. Genomes served as the template for sub-library production by using a novel NGS-based protocol (Supplementary Fig. S1). In parallel, NGS was performed to monitor the selection procedure (Fig. 2). Cluster analysis revealed a strong selection of two viral variants, AAV-VSSSTPR and AAV-NNPLPQR (Fig. 2A). Both variants were recovered with a frequency of 0.03% from the 1st round of selection. Frequency of occurrence increased to 0.42% (VSSSTPR) and 0.88% (NNPLPQR) in the 2nd and finally to 2.93% (VSSSTPR) and 3.23% (NNPLPQR) in the 3rd round of selection.

Heatmap of variants determined by NGS.
Isolation of viral variants from whole cell lysate indicates successful viral cell entry; however, it does not indicate the ability of the mutants to reach the nucleus. Consequently, the 3rd round of selection was repeated. When cells were harvested this time, nuclei were isolated (Supplementary Fig. S2) and then subjected to NGS sequencing to identify viral variants that accumulated in this cell compartment (Fig. 2B). Comparing the sequence pattern of this selection with those obtained for the whole cell lysate suggests that indeed not all variants reached the nuclear area with the same efficiency (Fig. 2A vs. B). VSSSTPR and NNPLPQR, however, remained the most frequently selected variants, although the ranking changed (3.27% for VSSSTPR vs. 2.62% for NNPLPQR). Analyzing genomes encoding for VSSSTPR and NNPLPQR, respectively, revealed at least three independent selection events, that is, selection of three independent clones for each of the two peptide motifs (Table 1).
Viral genomes encoding selected capsid-modified adeno-associated viral variants
Next generation sequencing revealed selection of the same peptide motif by different viral clones. The percentage represents the portion of the individual sequence coding for the corresponding motif.
AAV targeting vectors transduce HUVEC with higher efficiency
To characterize the tropism of AAV-VSSSTPR and AAV-NNPLPQR, variants were produced as vectors (AAV-VEC and AAV-NEC) displaying the respective peptide at position 587 in each of the 60 capsid subunits. Vectors again delivered GFP as reporter gene in a single-stranded vector genome conformation. AAV2 was prepared in parallel, to act as a control. The parental serotype and the newly developed variants showed a comparable packaging efficiency, revealing that ligand insertion did not interfere with vector production or with vector genome packaging (Supplementary Table S2).
Vectors were then tested on low-passage HUVEC (Fig. 3). Of the two mutants, AAV-VEC showed the highest transduction efficiency at all vector GOIs. At the lowest GOI (GOI 3600), AAV-VEC transduced 28.1 ± 6.8% of HUVEC cells. Transduction rate increased to up to 68.2 ± 6.9% at a GOI of 30,000, representing an up to 2.7-fold increase in transduction efficiency over AAV2. In contrast, although selected with a comparable efficiency as AAV-VEC, AAV-NEC was significantly less efficient in transducing HUVEC. However, AAV-NEC demonstrated a significantly improved EC transduction rate as compared with AAV2 at the highest vector dose tested.
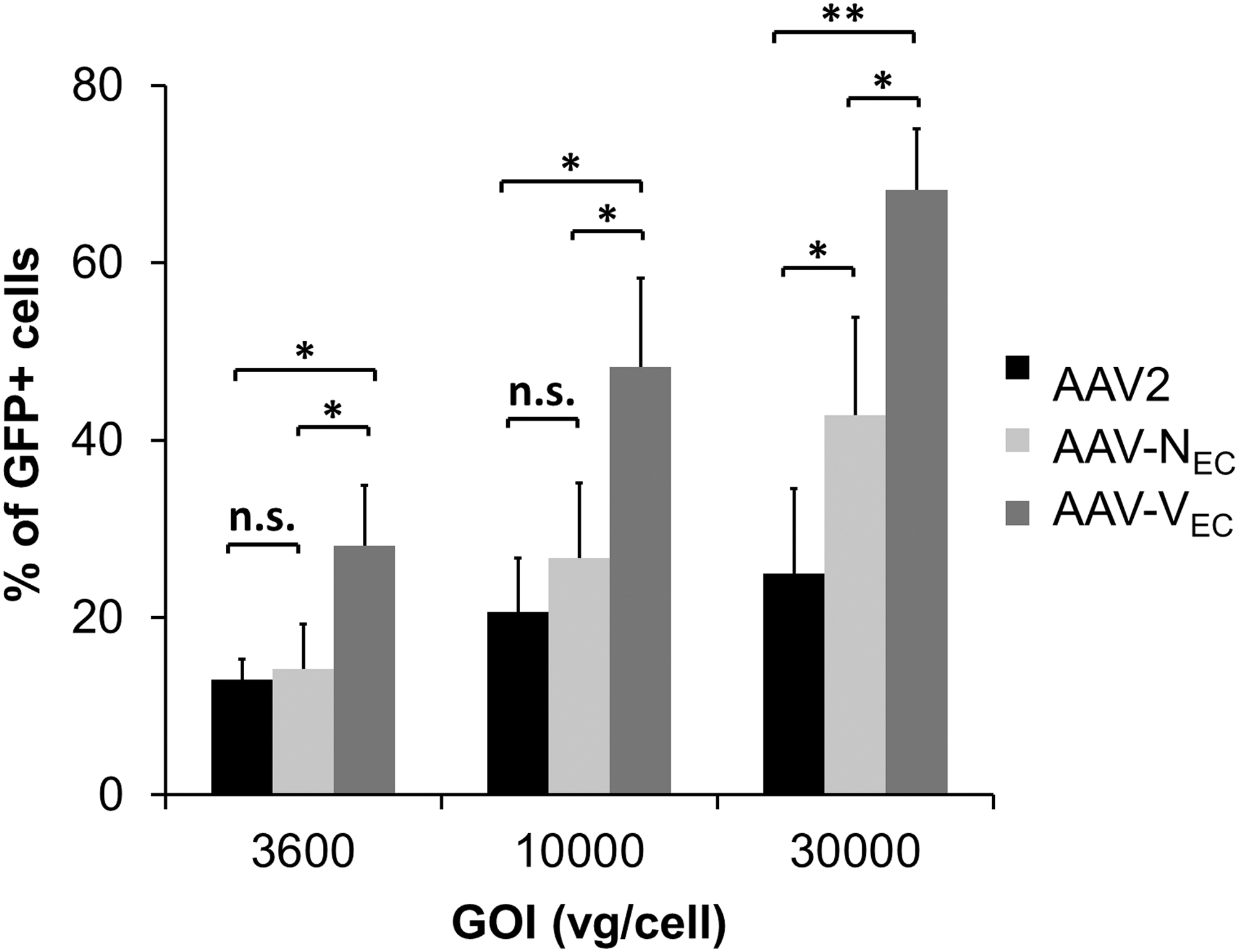
Transduction efficiency of AAV variants compared with AAV2. Low-passage HUVEC were incubated with a serial dilution of AAV2 (black), AAV-NEC (light gray), or AAV-VEC (dark gray). Vectors encode for GFP controlled by CMV promoter in a single-stranded vector genome conformation. Numbers on X-axis indicate genome (vg)-per-cell ratio. The percentage of transgene (GFP)-expressing cells was determined by flow cytometry 24 h p.t. Experiments were performed three times independently. Error bars indicate SD. **p < 0.001; *p < 0.05. n.s., nonsignificant; vg, vector genome.
Particularly in primary cells and in vivo, conversion of single-stranded AAV (ssAAV) vector genomes into a transcriptionally active double-stranded conformation has been identified as a post-entry barrier that can be overcome by usage of a self-complementary genome conformation. 33 To evaluate whether EC transduction efficiency can be further improved by switching to a self-complementary genome design, we performed a side-by-side comparison of conventional single-stranded and self-complementary vector genomes delivered by AAV-VEC, our best performing variant (Fig. 4). Especially at lower GOI, self-complementary AAV (scAAV)-VEC showed a higher transduction efficiency compared with ssAAV-VEC. Of note, scAAV-VEC at a GOI of 2500 was already sufficient to achieve a transduction efficiency of 50%.

Self-complementary vector genome conformation improves transduction efficiency. Low-passage HUVEC were incubated with a serial dilution of AAV-VEC delivering a single-stranded (ss; black) or self-complementary (sc, dark gray) vector genome encoding for GFP controlled by a CMV promoter. Numbers on X-axis indicate GOI. Percentage of GFP-expressing cells
AAV-VEC is superior to previously developed capsid variants
AAV targeting vectors for EC transduction were previously developed. 12,18,28,34 –39 The first example—already described in 2001—is AAV-SIG, which displays the SIGYPLP peptide at position 587. 18 Direct comparison of AAV-SIG with AAV-VEC by incubating low-passage HUVEC at a GOI of 10,000, followed by quantification of GFP-expressing cells 24 h later, revealed AAV-VEC to have a 13-fold greater transduction efficiency (Fig. 5A).
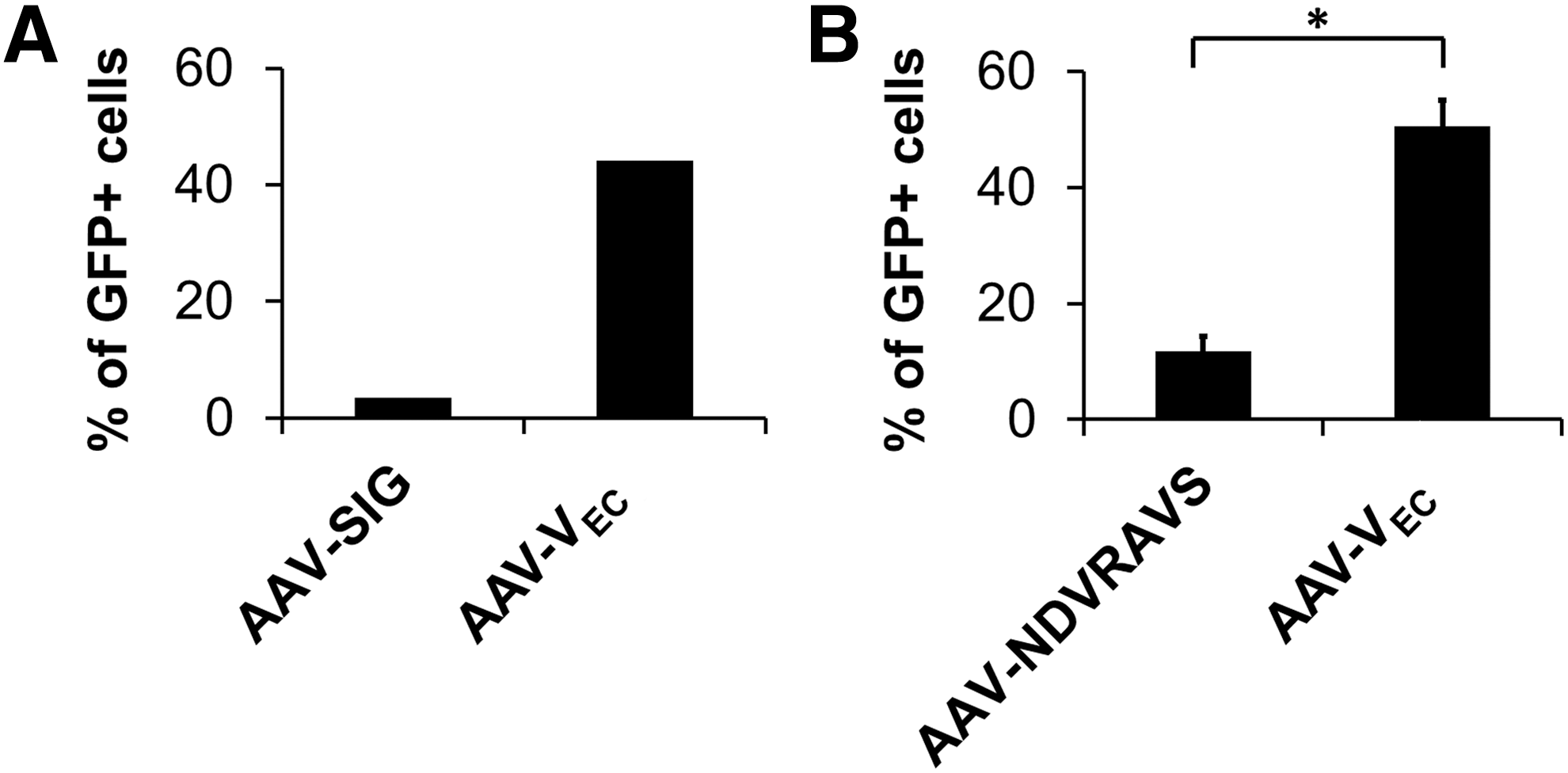
Improved transduction efficiency compared with previously developed capsid-engineered AAV vectors. Low-passage HUVEC were incubated with indicated capsid-modified vectors encoding for GFP in a single-stranded vector genome conformation at GOI of 10,000. The percentage of GFP-expressing cells was determined by flow cytometry 24 h p.t.
While the SIGYPLP peptide was selected on HUVEC, the same cell type as AAV-VEC, but by phage display and not by AAV peptide display, Muller et al. performed the first AAV2 peptide display on EC and identified AAV-NDVRAVS as an efficient variant for transducing human coronary artery EC. 12 Comparison of AAV-NDVRAVS with AAV-VEC again revealed the superiority of AAV-VEC, which transduced HUVEC with a fourfold higher efficiency (Fig. 5B).
Barriers toward AAV2-mediated transduction of vascular EC
AAV peptide display selects from a pool of capsid variants those that comply best with the selection criteria. Our results argue that, in particular, not only AAV-VEC but also AAV-NEC possess features that are beneficial for EC transduction that are not only superior to other variants in our library but also superior to AAV2 (Fig. 3). Based on this, we characterized the vector-EC interaction in more detail.
The kinetics and efficiency of cell entry as an indicator of successful uptake were determined by quantifying intracellular vector genomes at indicated time points by qPCR (Fig. 6). Highest vector copy numbers (VCN) were detected for AAV-VEC at all time points. AAV-VEC and the parental serotype AAV2 reached a plateau at 4 h p.t., whereas VCN continuously increased with time in the case of AAV-NEC, reaching a VCN comparable to AAV-VEC at 8 h p.t. To discriminate whether improved binding or internalization is responsible for the improved entry efficiency, we measured cell binding in the cold and again intracellular vector particles on shifting samples to 37°C (Supplementary Fig. S3). Although AAV-VEC and AAV2 did not differ in cell binding, we again observed significantly higher intracellular VCN for AAV-VEC compared with AAV2. Overall, the results indicate that AAV-VEC is internalized into HUVEC with a significantly higher efficiency than AAV2.

AAV targeting vectors differ in pre-entry and intracellular steps. Low-passage HUVEC were incubated with AAV2 (black), AAV-NEC (light gray), or AAV-VEC (dark gray), at a GOI of 10,000. Cells were harvested at the indicated time points (p.t.), and unbound and membrane-bound particles were removed.
Determination of intranuclear vector genomes at 8 h p.t. by isolating cell nuclei of vector-treated cells followed by qPCR measurements revealed that AAV-VEC and AAV-NEC showed a comparable VCN, which again was significantly higher than the VCN of AAV2 (Fig. 6). Measuring VCN does not allow to discriminate between vector genomes that have been released from the capsid (uncoating) and those that are still contained in the capsid and are thus not available for transcription. As an indirect measure for uncoating efficiency, we therefore determined the onset and level of transgene expression. Interestingly, our two targeting vectors that reached the nuclear area with comparable efficiency differed with regard to the onset of transcriptional activity and transgene expression level, arguing for an improved uncoating efficiency of AAV-VEC compared with AAV-NEC (Fig. 7). Not only AAV-NEC but also AAV2 demonstrated a significantly later onset of transcriptional activity (Fig. 7A), a difference confirmed by flow cytometry (Fig. 7B). However, in case of AAV2, it remains to be shown whether the lower transcriptional activity is, indeed, due to a lower uncoating efficiency or caused by the lower number of particles since significantly fewer AAV2 particles were internalized and accumulated in the nuclear area. An argument for the former, however, is the lower (thermal) stability of AAV-VEC compared with AAV2 (Supplementary Fig. S4).
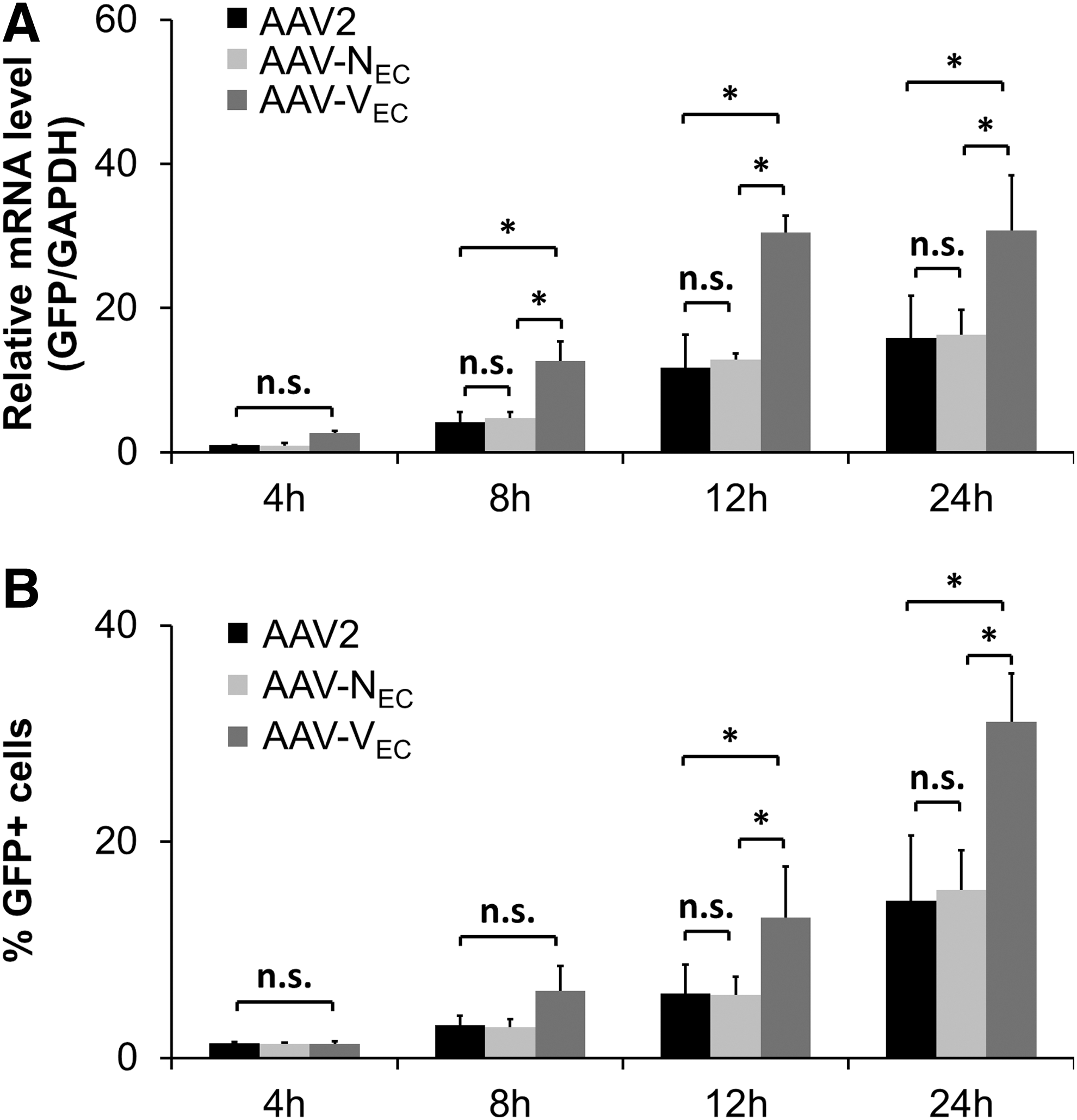
AAV-VEC does show a fast onset of transgene expression. HUVEC were incubated with AAV2 (black), AAV-NEC (light gray), or AAV-VEC (dark gray) encoding for GFP in a single-stranded vector genome conformation at a GOI of 10,000.
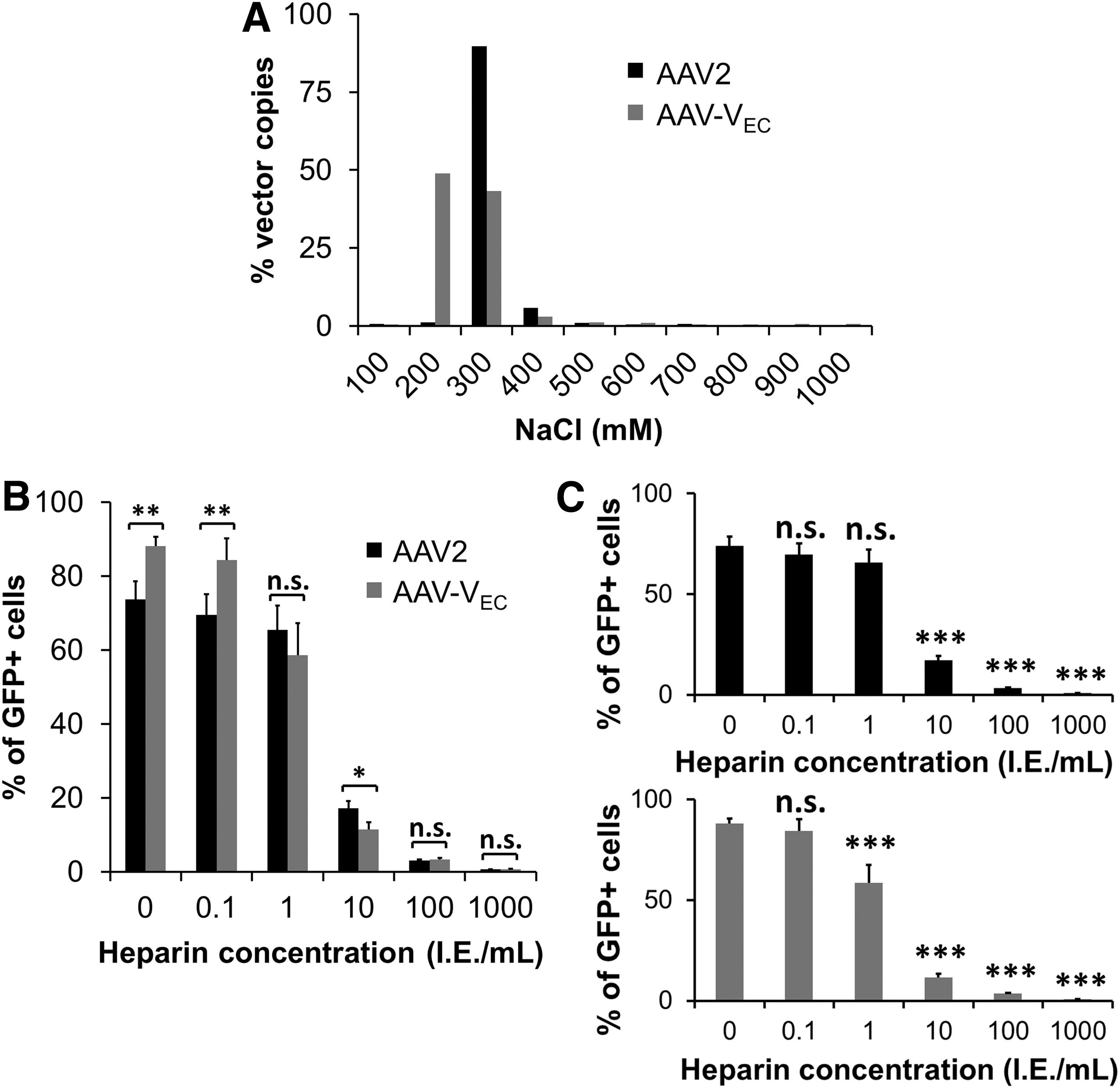
AAV-VEC does bind heparin and thus HSPG.
AAV-VEC binds heparin, a soluble analogue of HSPG
Insertion of peptides in position 587 of the AAV capsid proteins destroys the HSPG-binding motif of wild-type AAV2, 40 which hampers the natural infection process. This is an advantage for cell surface targeting approaches aiming at re-directing AAV tropism. 40 However, on insertion of overall positively charged peptides or of peptides that form an HSPG-binding motif, targeting vectors regain the ability to bind to HSPG although affinity might differ. 41,42
Interestingly, our selection on HUVEC, which were performed without any selection pressure regarding specificity of cell transduction, yielded exclusively variants that present an arginine (R) residue at position 7 of the peptide insertion (Fig. 2). This arginine might form together with the two linker amino acids (AA in our case) and R588 of the wild-type AAV2 sequence a heparin binding motif (RXXR) 41,42 that contributes to cell transduction. To test whether AAV-VEC is able to bind to HSPG, we performed a heparin affinity chromatography and eluted viral particles with increasing salt concentration (Fig. 8A). For comparison, AAV2 was subjected to the same test. AAV2 was eluted as a single peak when adding 300 mM NaCl. In contrast, lower salt concentrations were sufficient to compete with the binding of AAV-VEC. In line, the addition of increasing concentrations of heparin to AAV2 and AAV-VEC before cell transduction revealed that a lower concentration of heparin is sufficient to interfere with AAV-VEC-mediated transduction of HUVEC (Fig. 8B, C).
AAVR is an essential receptor for the large majority of AAV serotypes to transduce human cells and mouse tissues. 16,43 Recently, two groups mapped the AAVR binding site for AAV2 to the spike region adjacent to the threefold axis of symmetry. 44,45 Since our capsid modification is located at the tip of the second highest spike, we were interested in determining whether AAV-VEC differs from AAV2 regarding the dependency on AAVR for cell transduction. In the absence of an EC-specific AAVR knockout cell line, we used HEK293-AAVR-KO and HeLa-AAVR-KO as model systems. These cells, and as control parental AAVR wild-type cells, were transduced with increasing doses of AAV-VEC and AAV2, respectively (Supplementary Fig. S5). As expected, in the absence of AAVR, the transduction efficiency of AAV2 was reduced to background. AAV-VEC also transduced both parental cell types, but not HEK293-AAVR-KO or HeLa-AAVR-KO cells, revealing that AAVR is an essential factor for AAV2 as well as for AAV-VEC at least for these otherwise highly permissive cell lines.
Overall, these results indicate that AAV-VEC binds heparin/HSPG, although with a lower affinity compared with AAV2. Based on the strong selection for R in position 7 of inserted peptides, we might argue that binding to HSPG or other negatively charged cell surface molecules is an advantage for transducing EC. However, binding to HSPG is also correlated with a broad tropism (Uhrig et al. 42 and Supplementary Fig. S5) and accumulation in liver tissue when vectors are applied intravenously. 41 Thus, AAV-VEC might be best suited for local in vivo application or ex vivo transduction of EPC that are subsequently infused in case an EC-selective transduction is desired.
AAV-VEC transduces quiescent EC with higher efficiency than AAV2 or AAV-NEC
Proliferating EC as used in this study are located at the tip of sprouting vessels under physiological conditions (i.e., wound healing) and during tumor-induced angiogenesis, whereas the majority of EC lining the inner part of vessels are nonproliferating. To mimic these conditions, quiescence was induced by depleting supplements and FBS from culture medium as indicated by reduced incorporation of EdU (Fig. 9A vs. B). Side-by-side comparison of AAV2, AAV-VEC, and AAV-NEC demonstrated that AAV-VEC was the most efficient vector with exception of transduction at GOI 2000 (Fig. 9C). For AAV-NEC, an advantage compared with AAV2 was again only observed at the highest vector dose. However, transduction efficiency for AAV2, AAV-VEC, and AAV-NEC on quiescent cells was lower compared with proliferating cells.
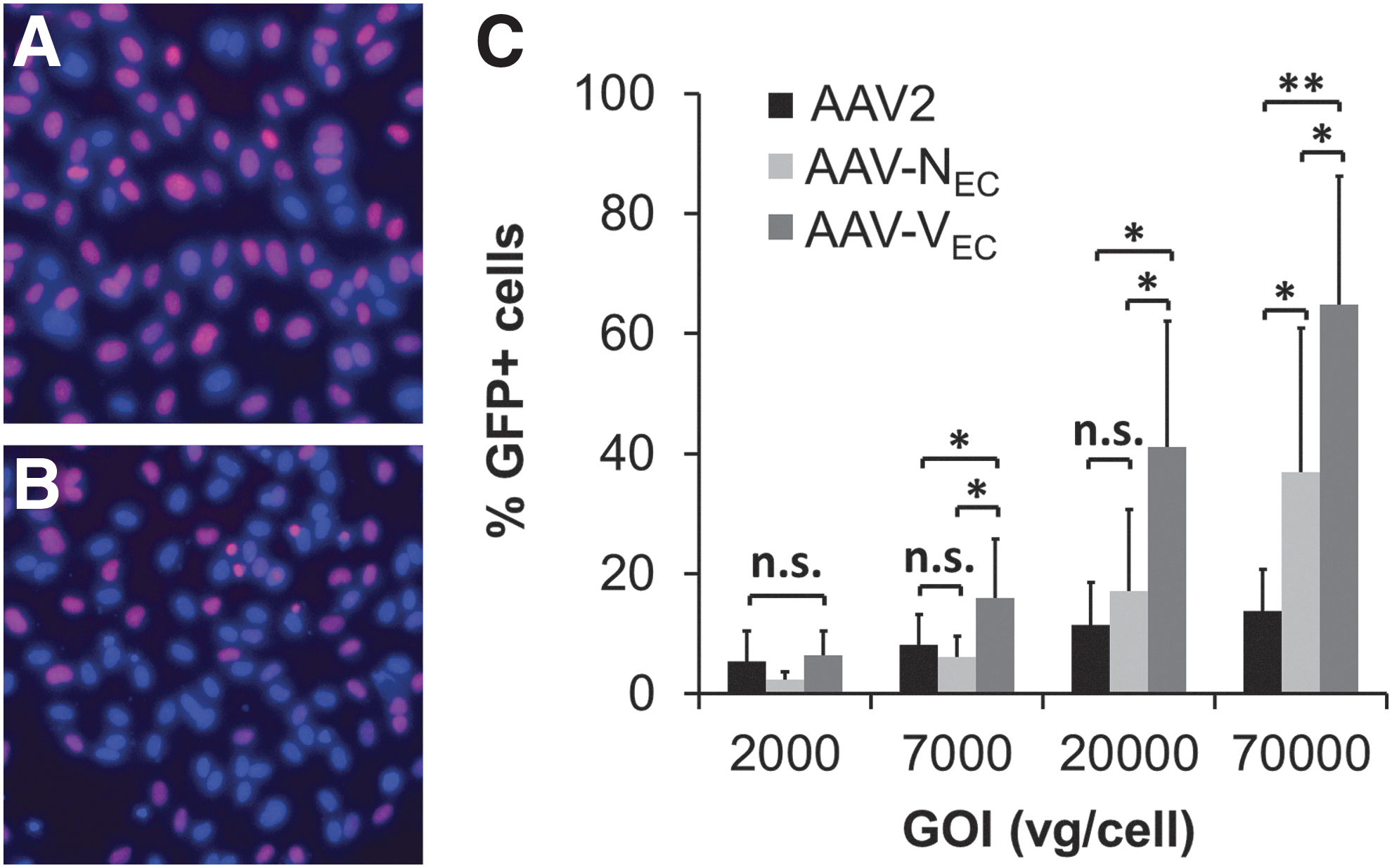
Quiescent HUVEC are permissive, in particular, for AAV-VEC.
iPSC-derived EPC are highly susceptible toward AAV-VEC transduction
Ex vivo genetic modification of EPC followed by re-infusion might represent a promising alternative to direct in vivo modification, because EPC home to sites of tissue damage or tumor growth. 46 –48 Consequently, we tested iPSC-derived EPC (iEC), which are generated in a patient-specific manner, but they also hold promise as a future source for off-the-shelf EPC, regarding their transducibility with AAV vectors. Specifically, a human fibroblast-derived iPSC clone was differentiated into iEC by using a forward reprogramming protocol. 24 At days 8 and 14 of differentiation, iEC were incubated with AAV-VEC and AAV2 vectors at a GOI of 200, followed by transduction efficiency analyses 48 h later (Fig. 10).
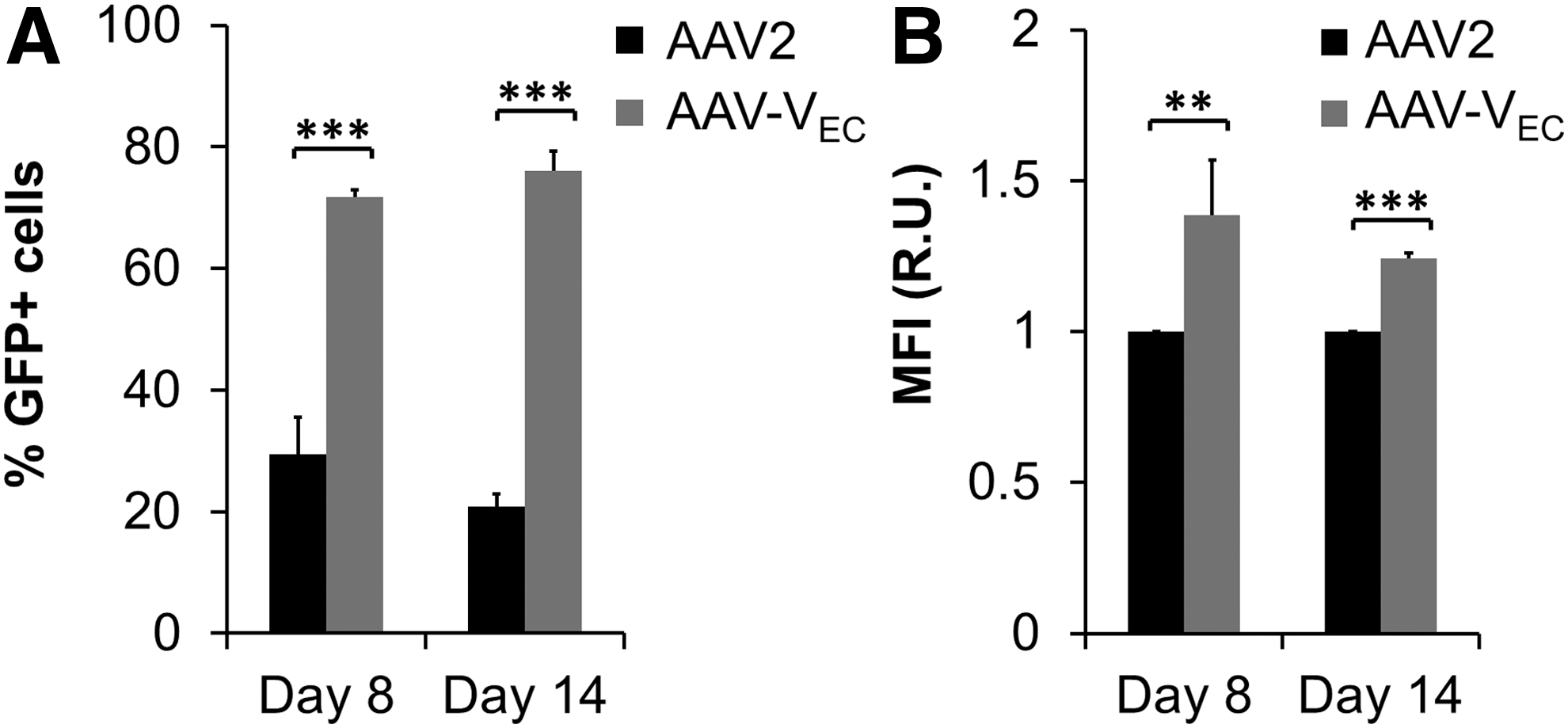
Transduction of iEC on days 8 and 14 of differentiation. The human fibroblast-derived iPSC clone H2E6C
24
was differentiated into iEC. On days 8 and 14 of differentiation, iEC were incubated with AAV2 (black) or AAV-VEC (dark gray). Vectors delivered a self-complementary vector genome encoding for GFP controlled by a CMV promoter. Transduction efficiency and transgene expression level was determined by flow cytometry 48 h p.t.
At both stages of differentiation, iEC were susceptible toward AAV2 and AAV-VEC. AAV-VEC, however, was clearly superior in transducing iEC compared to AAV2 (e.g., d14: e.g., 76.1% ± 1.9% for AAV-VEC versus 20.8% ± 1.3% for AAV2) (Supplementary Fig. S6). Surprisingly, with a GOI as low as 50, >40% of AAV-VEC treated cells showed reporter gene expression clearly above background. Transduction efficiency of AAV-VEC could be increased to nearly 100% with a GOI of only 1000 (Supplementary Fig. S6).
Discussion
EC form the inner layer of vessels controlling vasomotor tone, transport, and permeability. 49,50 In addition, EC contribute to the innate immune response, possess key functions in vascular homeostasis, and secrete proteinous factors into the blood stream. 49 –51 Consequently, disruption of the EC layer or of vascular homeostasis triggers disease processes, making EC attractive targets for therapeutic interventions. 50 The latter include genetic modification in gene therapy or regenerative medicine settings employing the homing capability of EC to repair damaged tissue or to target growing (tumor) vessels. 15 EC are, however, only weakly susceptible to the most common viral vectors. 27 Here, we focused on the AAV vector system and developed an AAV variant, AAV-VEC, to improve transduction of proliferating, quiescent, and iPSC-derived EC. As this variant clearly outperformed the parental serotype, we used this variant (and AAV-NEC) to identify barriers that limit transduction of EC.
AAV vector transduction is a multistep process that is initiated by binding to cell surface glycans such as sialic acids in case of AAV1, -4, -5, and -6, or HSPG in case of AAV2, the serotype we used as the basis for our study. 52 This attachment primes vector internalization mediated by co-receptors such as αvβ5 or α5β1 integrins. 30,31 Antibody staining confirmed the presence of both integrins, whereas HSPG was barely detectable (Fig. 1B). This condition did not impair vector uptake since AAV2 vector particles were detected intracellularly, but seem to limit efficacy of AAV2 internalization as significantly higher VCN were detected with AAV-VEC, a vector that uses its inserted peptide for cell entry (Supplementary Fig. S3). AAV2, AAV-VEC, and AAV-NEC were trafficked to the nucleus, although significantly more capsid-engineered AAV than AAV2 particles reached the nucleus (Fig. 6). Interestingly, nuclear accumulation did not correlate with transduction efficiency. Specifically, AAV-VEC and AAV-NEC differed remarkably regarding the onset of transgene expression as well as the level of transgene expression, although both vectors exhibited a comparable extent of nuclear trafficking (Figs. 3, 6, and 7). Transgene expression requires availability of vector genomes for transcription. As vectors delivered identical vector genomes, AAV-VEC seems to be more efficient in releasing vector genomes. This feature is may be due to the specific peptide insert, VSSSTPR, or the receptor and respective downstream signaling pathways activated on binding of this engineered capsid to the EC surface or a combination of both mechanisms. Once released from the capsid, single-stranded AAV vector genomes need to be converted into a double-stranded DNA for initiation of transcription. Since AAV-VEC applied in the expression kinetics experiment (Fig. 7) delivered a single-stranded AAV vector genome from which transcribed mRNA is detectable by qPCR already at early time points p.t., EC—at least HUVEC—possess all necessary components to assist AAV in second-stranded synthesis. Nevertheless, using a self-complementary instead of the natural single-stranded vector genome conformation is advantageous, particularly when only a few vector genomes are present, that is, in conditions of low GOI (Fig. 4).
Thus, regarding the main steps in AAV infection—cell surface attachment, internalization, trafficking toward the nuclear area, release from the endosomal compartment, nuclear delivery, uncoating, and second-strand synthesis—vector uptake and uncoating appear as the most critical steps that impact EC transduction. Furthermore, use of a self-complementary vector genome conformation can further improve transduction efficiency.
AAV-VEC, selected by an AAV peptide display screen on HUVEC, differs from AAV2 in the 7mer peptide VSSSTPR inserted at position 587 of each of the 60 subunits of the AAV capsid. Peptides inserted at that position were demonstrated to mediate receptor binding and determine vector tropism. 5 The receptor for AAV-VEC is currently unknown, a general caveat of directed evolution approaches in which variants are selected for tropism for a predefined target cell population without the option to predefine the target receptor. The latter—if required—needs to be determined afterward, for example, by comparative gene analyses. 8 Despite this drawback, high-throughput selection screens of AAV capsid libraries have—since their first application more than 15 years ago 11,12 –demonstrated their potency to tailor the AAV-target cell interaction without the need to know barriers that limit respective target cell transduction. 1,13,53
As mentioned earlier, the adaptation in AAV-EC interaction regarding AAV-VEC concerned vector uptake (compared with AAV2) and uncoating (compared with AAV-NEC). Comparison of AAV-VEC with AAV-SIG, a first-generation capsid-engineered vector that displayed a peptide identified by phage display selection on HUVEC, revealed a 13-fold higher transduction efficiency for AAV-VEC (Fig. 5A). This is likely due to the fact that selection of the SIGYPLP peptide was based on overcoming only one barrier, that is, the uptake process, since the SIGYPLP peptide was selected for improved binding to EC without tackling further downstream barriers. Our capsid variant selected here also demonstrated superior transduction efficiency compared with AAV-NADARVS (Fig. 5B).
Since we did not perform a side-by-side comparison of AAV-VEC and AAV-NADARVS for the various steps of EC transduction, we can only speculate on the reason(s) for this observation. It is possible that the NADARVS peptide is underestimated in our case, as we cloned this peptide into position 587, whereas Muller et al. selected the peptide from an AAV peptide display library displaying peptides at position 588, the neighboring position. 12 This peptide, however, mediated EC targeting with a comparable efficiency when displayed on an AAV9 capsid, which argues against a positional effect. 28 Alternatively, improved efficiency by AAV-NADARVS may be restricted to human coronary EC, the EC subtype this variant has been selected on, and/or requires co-factors since selection of AAV-NADARVS was performed in the presence of adenovirus, whereas we decided against adenovirus superinfection.
We did not perform extensive screenings for EC subtypes that were susceptible to AAV-VEC, but we demonstrated the ability of AAV-VEC to transduce HUVEC independent of their proliferation state as well as iPSC-derived EPC (Figs. 3, 9, and 10). The latter are currently entering the arena of drug screening as they allow patient-specific disease modeling. 50 In addition, EPC home to damaged vasculature, making them promising targets for genetic modification to develop novel therapeutic strategies in the context of regenerative medicine, gene therapy, organ transplantation, and cardiovascular disease 54 as well as antitumor therapy. 55 Given the recent advancement in iPSC technology, in particular, with regard to “off the shelf cell products,” iPSC-derived EPC will likely replace conventional EPC in the clinical setting. With its impressive efficacy in transducing iPSC-derived EPC, AAV-VEC may represent a highly potent delivery system also in this setting.
In summary, we here report on the development of AAV-VEC, a capsid-engineered AAV vector, that is internalized and consequently transported to the nucleus with significantly higher efficacy than the parental serotype. AAV-VEC showed efficient transduction of conventional EC at different states of differentiation and also demonstrated an impressive efficacy in iPSC-derived EPC. Of note, efficient and fast onset in transgene expression was observed even when delivering the vector genome in its natural single-stranded DNA conformation. This option is particularly promising for clinical application, as the full coding capacity of the AAV vector system is thereby available for delivery of transgene expression cassettes of ∼5 instead of 2.5 kb, the upper limit of AAV vectors with a self-complementary vector genome design.
Footnotes
Acknowledgments
The authors thank Jude Samulski (University of North Carolina at Chapel Hill, NC) for providing plasmid pXX6. They also thank Hanna Janicki (University of Cologne, Germany), Claudia Vollbrecht (University of Cologne, Germany), Nicki Lenort (Hannover Medical School), and Elke Barczak (Hannover Medical School) for their excellent technical assistance.
Author Disclosure
No competing financial interests exist.
Funding Information
This work was supported by grants from University Hospital Cologne (Köln Fortune) to H.B. and M.O., the Center for Molecular Medicine Cologne (CMMC) to H.B., BMBF and MWK Lower Saxony-funded Professorinnenprogramm Niedersachsen to H.B., the DFG-funded KFO286 to H.B., the DFG-funded cluster of excellence REBIRTH EXC62/2 to A.S. and H.B., and the DFG-funded SFB738 to A.S. L.Z. held a scholarship from the German Academic Exchange Service (DAAD).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.