
Abstract
Early efforts in cystic fibrosis (CF) gene therapy faced major challenges in delivery efficiency and sustained therapeutic gene expression. Recent advancements in engineered site-specific endonucleases such as clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 make permanent CF transmembrane conductance regulator (CFTR) gene correction possible. However, because of safety concerns of the CRISPR/Cas9 system and challenges in in vivo delivery to inflamed CF airway, CRISPR-based gene correction strategies need to be tested in proper animal models. In this study, we aimed at creating vectors for testing CFTR gene correction in pig models. We constructed helper-dependent adenoviral (HD-Ad) vectors to deliver CRISPR/Cas9 and a donor template (a 6 kb LacZ or 8.7 kb human CFTR expression cassette) into cultured pig cells. We demonstrated precise integration of each donor into the GGTA1 safe harbor through Cas9-induced homology directed repair with 3 kb homology arms. In addition, we showed that both LacZ and hCFTR were persistently expressed in transduced cells. Furthermore, we created a CFTR-deficient cell line for testing CFTR correction. We detected hCFTR mRNA and protein expression in cells transduced with the hCFTR vector. We also demonstrated CFTR function in the CF cells transduced with the HD-Ad delivering the CRISPR-Cas9 system and hCFTR donor at late cellular passages using the membrane potential sensitive dye-based assay (FLIPR®). Combined with our previous report on gene delivery to pig airway basal cells, these data provide the feasibility of testing CRISPR/Cas9-mediated permanent human CFTR correction through HD-Ad vector delivery in pigs.
Introduction
Cystic fibrosis (CF)
CF lung disease is traditionally treated with mucolytic drugs, antibiotics, physiotherapy, nonsteroidal anti-inflammatory drugs, and inhaled hypertonic saline. 5 However, these treatments only deal with symptoms, not the cause of the disease. In addition, the accumulative toxicity and drug-resistant pathogens as adverse outcomes from antibiotics are still inevitable. 6,7 Thus, recent effort has been directed to develop small-molecular drugs to target the CFTR protein. The first such molecule drug, ivacaftor, has been shown to sustain increased FEV1, improve weight gain, and reduce pulmonary exacerbations in patients with at least one G551D mutation. 8,9 A combination of ivacaftor with another such CFTR modulator drug, lumacaftor, has been shown to improve FEV1 of patients with homozygous F508 mutations. 10 Although present CFTR modulators do bring promising clinical outcomes, there are still limitations to be overcome in the health of the CF community. First, patients with a type I mutation do not respond to modulators because their cells do not produce CFTR protein. 11 Second, treatments with the new drugs must be combined with other prescribed medications with no reduction of the drug burden on patients. 8,10 Moreover, not all ivacaftor-treated patients experience improved lung function. 8 Finally, side effects such as abnormal liver function also make some patients intolerable during the lumacaftor–ivacaftor treatment. 10
Therefore, novel approaches to treat all types of CFTR mutations are needed. One of these methods is gene therapy, because CF is a monogenic and recessive disease and introducing one copy of the functional CFTR gene into targeted cells theoretically restores the CFTR channel function. Although previous in vivo lung gene therapy studies suggested the feasibility of CFTR gene delivery to the airway epithelia of patients, no sustained clinical benefits have been demonstrated. 12 –18 There are two major challenges in lung gene therapy. First, the physical barrier of lung epithelia and the strong immune defense prevent exogenous genes from being efficiently and safely delivered to airway cells. Second, airway epithelial cells that receive the therapeutic gene are lost eventually due to slow cell turnover, unless basal stem and progenitor cells are targeted. Thus, a permanent gene correction strategy is needed for lung gene therapy to overcome both categories of challenges.
The recent advancements in engineered site-specific endonucleases make it possible to overcome these challenges by permanently correcting CFTR-deficient cells. Programmable nucleases, such as zinc-finger nucleases (ZFNs), TAL effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR)-associated Cas9, are efficient gene editing tools in generating double-strand breaks at targeted genomic locus to facilitate transgene integration through homology-directed repair (HDR). 19 –21 Both ZFNs and TALENs require laborious work to generate a site-specific enzyme for each target site. On the contrary, the programmable CRISPR/Cas9 system is relatively simple and flexible, because changing guide RNA sequences allows multiple loci to be targeted. 22
CRISPRs are known to have off-target effects, and thus, animal studies in appropriate models are necessary to reveal the impact of these effects on the safety and efficiency of gene therapy approaches. CF pigs generated by the Welsh group are an ideal model for the test. 23 These CF pigs develop lung symptoms similar to those observed in CF patients, such as airway infection, airway obstruction, and bronchiectasis. 23,24 To examine our CRISPR-mediated gene therapy strategy in pigs, we plan to integrate a wild-type human CFTR gene expression cassette into the pig GGTA1 locus, potentially to correct all CFTR mutation types. The GGTA1 gene encodes α-1, 3-galactosyltransferase that is expressed in many mammals, however, it is a transcribed pseudogene in humans 25 and the pig gene product is a major xenoantigen that can be recognized by human antibodies. 26 The pig GGTA1 locus has been proved as a safe harbor for exogenous transgene integration and expression in both in vitro and in vivo studies. 27 –29 We hypothesized that integration of a transgene cassette into the genomic safe harbor of pig cells will achieve sustained and functional transgene expression.
To test this approach, we generated two helper-dependent adenoviral (HD-Ad) vectors, termed HD-Ad-Cas9-hCFTR and HD-Ad-Cas9-LacZ, to deliver the CRISPR/Cas9 gene editing system with LacZ or hCFTR donor in a single vector for integration at the GGTA1 locus. The HD-Ad vector has a large capacity of around 36 kb to package multiple transgenes and their regulatory elements. Moreover, deletion of all viral coding sequences minimizes viral protein synthesis, making HD-Ad less immunogenic. 30 By using human keratin 18 (K18) promoter to enhance transgene expression in epithelial cells, we showed that CFTR knockout mice gained resistance to pathogen infection in airways after transducing with an HD-Ad vector containing the K18-CFTR expression cassette. 31 Recently, we also demonstrated that HD-Ad vectors can deliver transgenes into airway basal progenitor cells by using mice and pigs in vivo, as well as primary human nasal cells in vitro. 32
In this study, we have delivered CRISPR/Cas9 and LacZ transgene into the porcine IPEC-J2 cell line, 33 and examined transgene integration and expression at different cellular passages. We have also generated a porcine CFTR knockout cell line based on IPEC-J2 cells and examined hCFTR gene targeting and functional correction in the knockout cell line.
Materials and Methods
CRISPR design
Online resource (chopchop) was used to design the 5′-N20NGG-3′ target site for CFTR −/− IPEC-J2 pig cell line generation. sgRNA1, 5′-GAA GCA AAT GAC ATC ACC GC-3′ and sgRNA2, 5′-TGG CAT TAG GAG CTC GAG CC-3′ were selected and cloned into pSpCas9(BB)-2A-Puro (Addgene, PX459). The single guide RNA (sgRNA) guide sequence 5′-GAG AAA ATA ATG AAT GTC AA-3′ was selected for HD-Ad-Cas9-hCFTR vector and HD-Ad-Cas9-LacZ vector.
HD-Ad plasmid construction and vector production
The pC4HSU-PN was used as backbone for viral vector cloning. The HD-Ad-Cas9-LacZ vector was constructed as inverted terminal repeats (ITR)-green fluorescent protein (GFP) and Cas9 under chicken β-actin (CBA) promoter-sgRNA under U6 promoter-left homology arm (LHA)-LacZ under polyubiquitin C gene (UbC) promoter-right homology arm (RHA)-ITR. The HD-Ad-Cas9-hCFTR vector carried ITR-sgRNA under U6 promoter-Cas9 and GFP under CBA promoter-RHA-hCFTR under human K18 promoter-LHA-ITR. The hCFTR expression cassette previously synthesized in our laboratory contained a 2.5 kb genomic sequence upstream of the human K18 gene, K18 promoter with its first intron, hCFTR cDNA (containing a silent mutation to eliminate cryptic splicing with the K18 intron), 3′ untranslated region of K18, and a polyadenylation signal. 34 The HD-Ad-LacZ control vector carried LacZ gene under UbC promoter. The HD-Ad-hCFTR control vector contained hCFTR under the control of K18 promoter. The HD-Ad-GFP carried GFP under Cytomegalovirus (CMV) promoter. The HD-Ad viral vectors were produced as previously described by Ng et al. 35
Cell culture and transduction
The nontransformed and nontumorigenic porcine IPEC-J2 cell line was isolated from the epithelium of jejunum of unsuckled piglets. 33 IPEC-J2 cells used in this study were kindly provided by Dr. Julang Li (the University of Guelph). All cells were cultured in Dulbecco's modified Eagle's medium and Ham's F-12 nutrient mixture (DMEM/F12/HAM; Gibco), 5% porcine serum (Gibco), 1% insulin–transferrin–selenium (ITS-G; Thermo Fisher Scientific), 5 ng/mL epidermal growth factor recombinant human protein (EGF, Thermo Fisher Scientific), and 1% penicillin–streptomycin (10,000 U/mL; Thermo Fisher Scientific), unless specified. On 80–90% confluency, cells were washed twice with 1 × phosphate-buffered saline (PBS, pH7.4), dispersed by 0.25% Trypsin-EDTA (Gibco) at 37°C for 5–8 min, and split at 1:7 ratio.
Cells were seeded into a six-well plate at a density of 0.3 × 106 and cultured until 70% before transduction. The cells were transduced with HD-Ad vectors in 0.5 mL of serum-free, antibiotic-free DMEM/F12 for 2 h. After 2 h, a serum-contained medium was added to a final volume of 2 mL per well.
Junction PCR
Genomic DNA was extracted by phenol–chloroform extraction. The quality and concentration of DNA were measured by OD readings with the SimpliNano spectrophotometer (GE healthcare Life Science). In LHA junction PCR, the forward primer annealed upstream of LHA in genome, while the reverse primer bound within the transgene cassette. In RHA junction PCR, the forward primer annealed in the transgene cassette, while the reverse primer bound downstream of RHA in genome. For cell line generation, the following primers were used: GFP integration, LHA, primer 1-F: 5′-CAA GTG CCC ACC AAC AAA TGA ATG-3′, primer 1-R: 5′-GCC CAT TGA TGT ACT GCC AAA ACC-3′; GFP integration, RHA, primer 2-F: 5′-TGT CAG TTT CAT AGC CTG AAG AAC G-3′, primer 2-R: 5′-AAA AGC AAG TCC AAG TGA TCA GTC C-3′; and heterozygosity, primer 3-F: 5′-TAT TTC TGC TAA GGG GTC CGA CTT G-3′, primer 3-R: 5′-ATT CCA CAA AAC CCT CAT TCT CGT C-3′. For LacZ and hCFTR integration, the sequences of primers are as follow: LacZ integration, LHA, primer 1-F: 5′-AAT GTG GAC TAA CAC TGA CCT TCC-3′, primer 1-R: 5′-AAG GCC GAG TCT TAT GAG CAG-3′; LacZ integration, RHA, primer 2-F: 5′-GCA TCG CCT TCT ATC GCC TTC-3′, primer 2-R: 5′-GCA GAG CAA AAT GCA GGT CTT ATC-3′; hCFTR integration, LHA, primer 3-F: 5′-AAT GTG GAC TAA CAC TGA CCT TCC-3′, primer 3-R: 5′-GTG GAG TCA ACA AGC TAT GTA CTG C-3′; and hCFTR integration, RHA, primer 4-F: 5′-GAG GAG GGA GCA GTA GAT GTG ATG-3′, primer 4-R: 5′-GCA GAG CAA AAT GCA GGT CTT ATC-3′. Sanger sequencing was performed by the Centre for Applied Genomics at the Hospital for Sick Children.
T7E1 assay
Cells transduced with Cas9-contained HD-Ad vector or non-Cas9-contained HD-Ad vector were collected and lysed in 50 μL of cell lysis buffer with 2 μL of protein degrader (GeneArt Genomic Cleavage Detection Kit; Life Technologies, Carlsbad, CA), 3 days post-transduction. The forward and reverse primers for PCR were 5′-ACA ACG GCA ACT CTC TGG AAT GC-3′ and 5′-GCA CTC CTT AGC GCT CGT TGA CTA-3, respectively. A fragment, ∼350 bp, containing target site for cleavage assay, was amplified by AmpliTaq Gold 360 Master Mix. Five microliters from each PCR product was mixed with 1 μL of 10 × NEB buffer 2.1 and 3 μL of distilled water. The mixtures were denatured at 95°C for 5 min and reannealed to room temperature. The mixture was then added with 1 μL of T7 endonuclease I (New England Biolabs) and incubated at 37°C for 1 h for cleaving. The intensity of PCR band was analyzed by ImageJ. The cleavage efficiency was calculated as follows:
Off-target analysis
Off-target frequencies were measured by online tool ICE. Genomic DNA was extracted 3 days after transduction with HD-Ad-Cas9-LacZ at 50 MOI. Unedited cells were used as negative control. Purified PCR samples were used for Sanger sequencing. The PCR primers to amplify off-target sites were as follows: off-target site #1, forward: 5′-GGA ACA TTT CTG AAT GGT GTA TCT C-3′, reverse: 5′-TAT CAG TCC TTG TCC AGT AGA GGT C-3′; off-target site #2, forward: 5′-AGT GAA ACC GGA AAC CAT TAA TAA G-3′, reverse: 5′-TGA GCG TAA TAA TCT CAT TTG ATC C-3′; off-target site #3, forward: 5′-ATG TTT TTG GAG GAC AAA TAC AGT C-3′, reverse: 5′-ATA TTT TAG GAC ATG GAA AAC ATG G-3′; and off-target site #4, forward: 5′-TCA GGA TCT AGT TGC TTT CTT TTT G-3′, reverse: 5′-ACT CAT TAG AAA CCA AAA CTT GCA G-3′. The internal primers for Sanger sequencing were as follows: off-target site #1, 5′-CAC GAA AAC CCT ATG ACA TGC-3′; off-target site #2, 5′-TGC AGC AAA AAC ATT ACT GA-3′; off-target site #3, 5′-TGG TAA CCA GAG TTG GTT TT-3′; and off-target site #4, 5′-AAG GAG CAC CTT TTC TGT CG-3′.
X-gal staining
Cells cultured in six-well plates were washed three times with PBS (pH 8.0). Cells on each well were then fixed with 1 mL of 0.5% glutaraldehyde in PBS for 15 min. The cells were washed an additional three times in PBS and stained with 1 mL of β-galactosidase staining solution (300 μL of 40 mg/mL X-gal, 16 μL of 1.5 M MgCl2, 600 μL of 100 mM K-ferricyanide, 600 μL of 100 mM K-ferrocyanide, and 10.5 mL of PBS for 12 mL in total). Cells were incubated at 37°C for 3 h. The staining was stopped by removing the β-galactosidase staining solution and washing with PBS.
To determine integration efficiency, nine images of β-galactosidase-stained cells from each condition were taken randomly at passage 8 under 20 × objective. The number of LacZ positively stained cells and total number of cells were recorded. The formula used for calculating integration efficiency was as follows:
Single-cell colony analysis
After transducing with HD-Ad-Cas9-LacZ or HD-Ad-LacZ control vector at 20 MOI, cells were harvested at passage 8 and diluted to 1 cell per 100 μL by growth media supplemented with 20% fetal bovine serum. The resuspended cells were then pipetted into 96-well plates at 1 cell/well and incubated at 37°C for 20 days with routine media replacement. All single-cell derived colonies were stained for β-galactosidase activity. The percentage of integration efficiency was calculated as follows:
Quantitative real-time-PCR
Quantitative real-time-PCR (qRT-PCR) was performed to analyze CFTR expression in the CFTR −/− cell line and in HD-Ad-transduced cells. Total RNA was extracted from corresponding cell samples by the PureLink RNA mini kit (Thermo Fisher Scientific). For transduced cells, RNA samples were collected at day 3 post-transduction, passage 10, and passage 20, and genomic DNA was eliminated by DNase treatment (PureLink DNase set; Invitrogen). One microgram of total RNA from each sample was reverse-transcribed using random hexamers by SuperScript IV VILO Master Mix kit (Thermo Fisher Scientific) following the manufacturer's protocol. Ten nanograms of cDNA was loaded on as template for qRT-PCR using SYBR Green PCR Master Mix (Thermo Fisher Scientific) on a QuantStudio 3 Real-Time PCR System (Applied Biosystems).
Specific primers to detect endogenous pig CFTR expression in the CFTR −/− cells were designed: 5′-GCCAGCATCTTCTCCAAAC-3′ and 5′-TTCCAGGCGCTGTCTATATC-3′. To measure human CFTR expression in transduced cells, the following primers were used: 5′-CCTGAGTCCTGTCCTTTCTC-3′ and 5′-CGCTGTCTGTATCCT TTCCTC-3′. Primers for pig glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA were used as internal control: 5′-GTTCGACAGACAGCCGTGTG-3′ and 5′-ATGGCGACAATGTCCACTTTGC-3′. In each reaction, 50 and 10 ng of cDNA was used as template for hCFTR and GAPDH, respectively.
The relative expression of each gene was calculated as follows:
Relative expression = 2−ΔΔCt, in which −ΔΔCt = [(Cttreatment − Ctreference)−(Ctstandard – Ctreference)]. Our CFTR −/− pig cell line did not express pig CFTR mRNA, and so, in this experiment, Ctstandard was regarded as P0 cells transduced with HD-Ad-hCFTR control vector at 10 MOI.
Western blot
Protein expression levels were measured by an automated capillary-based immunoassay (Jess, Simple Western; ProteinSimple). Cells were lysed in RIPA buffer with proteinase inhibitor for protein extraction. Cell lysates were mixed with an equal volume of 4 × Laemmli sample buffer (Bio-Rad). β-mercaptoethanol (Sigma-Aldrich) was added to protein sample buffer mix to a final concentration of 5%. Protein concentrations were measured by BCA assay (Pierce bicinchoninic acid assay [BCA] Protein Assay kit; Thermo Fisher Scientific). Samples were diluted to 3 μg/μL with supplied sample buffer. Immunoprobing process for CFTR was first incubated with a primary antibody (596, 1:400 dilution), followed by a secondary antibody (HRP-conjugated anti-mouse IgG, 1:2000). The chemiluminescent reaction was captured by CCD camera and analyzed by Compass software (ProteinSimple). As a positive control for CFTR protein expression, the doxycycline-inducible IB3-8-3-7 cells were treated with 1 μg/mL doxycycline for 5 days before protein extraction. 36
Membrane potential assay
Cells were grown at 37°C on black 96-well plates to reach 100% confluency and were washed by PBS before incubation in 0.5 mg/mL of blue membrane potential dye (FLIPR® Membrane Potential Assay Kit; Molecular Devices) for 30 min at 37°C. The membrane potential dye was dissolved in chloride-free buffer, which contained 10 mM glucose, 20 mM HEPES, 136 mM sodium gluconate, 3 mM potassium gluconate, pH 7.35, 300 mOsm. The plates were then transferred and read in a fluorescent microplate reader (SpectraMax i3X Multi-Mode microplate reader; Molecular Devices) at 37°C. The baseline fluorescence (excitation: 530 nm, emission: 560 nm) was scanned 11 times before addition of drugs. After adding forskolin (FSK) (10 μM in dimethyl sulfoxide [DMSO]; Sigma-Aldrich) and CFTRinh-172 (10 μM in DMSO; Sigma-Aldrich), the fluorescence was scanned for 31 times and 21 times, respectively. DMSO vehicle was used as a negative control.
Digital droplet PCR
To measure HDR, TaqMan copy number probe (Life Technologies) and ZEN Double-Quenched Probes (IDT, IA) were used for estimating the copy number of CF11–12 in QX200 Droplet Digital PCR system (Bio-Rad Laboratories, Inc., Hercules, CA). To measure non-homologous end joining (NHEJ), HEX-labeled probe was designed to bind to unedited target locus. An FAM-labeled reference probe could bind to site distant from expected target locus, and was used to count total copy numbers of amplicons. The digital droplet PCR (ddPCR) copy number reaction mixture was prepared as following: 1 μL of copy number target assay (labeled with FAM), 1 μL of copy number reference assay (Pig Tfrc, labeled with HEX), 10 μL of 2 × ddPCR SuperMix for probes (no dUTP; Bio-Rad Laboratories), 225 ng of digested genomic DNA, and 5 μL of water. Droplets were generated on a QX200 droplet generator (Bio-Rad Laboratories) and transferred into a 96-well PCR plate. PCR was performed on a thermal cycler (Veriti; Life Technologies) using the following program: 95°C for 10 min, 94°C for 30 s, and 58°C for 1 min; repeat 45 cycles: 98°C for 10 min and hold at 10°C. Data obtained were analyzed by QuantaSoft v1.4 (Bio-Rad Laboratories).
Statistical analysis
Statistical significance was determined by one-way ANOVA using Prism 6 software. A p-value <0.05 was considered statistically significant.
Results
Integration of LacZ reporter at GGTA1
We generated HD-Ad-Cas9-LacZ and a control vector HD-Ad-LacZ without CRISPR/Cas9. The HD-Ad-Cas9-LacZ carried sgRNA targeting exon 3 of porcine GGTA1 gene, a LacZ donor gene under control of the UbC promoter, flanked by ∼3 kb homology arms (Fig. 1a). In both vectors, a nuclear localization signal (NLS) was added to the LacZ reporter.

Integration of LacZ reporter at the GGTA1 target site.
We transduced IPEC-J2 cells with HD-Ad-Cas9-LacZ at 50, 20, and 5 MOI (infectious vector particle per cell), or HD-Ad-LacZ control vector at 20 MOI. Transduced cells were cultured for multiple passages to dilute episomal viral vectors. After eight passages, X-gal staining was performed and the percentage of X-gal-stained cells, which represent the LacZ expression cells, was calculated by cell counting manually (Fig. 1b).
As shown in Fig. 1c, when cells were transduced with 50 MOI of HD-Ad-Cas9-LacZ, we observed 6.98% LacZ-positive cells. We detected 6.24% and 1.83% LacZ-positive cells in those transduced at 20 and 5 MOI, respectively.
Junction PCR performed on cells transduced with HD-Ad-Cas9-LacZ after passage 20 indicated successful integration of the LacZ gene at the GGTA1 locus (Fig. 1d). Sanger sequencing results of junction PCR products further confirmed on-target transgene integration (Fig. 1d). No PCR product was detected from HD-Ad-LacZ control vector-infected cells.
Single-cell culturing was performed to further confirm if LacZ transgene inserted could be stably expressed. When the LacZ transgene is integrated into the genome of a specific cell, then, the whole colony generated from this single cell would express the LacZ gene. Wild-type IPEC-J2 cells were transduced with HD-Ad-Cas9-LacZ or HD-Ad-LacZ control vector at 20 MOI. As shown in Supplementary Fig. S1, at 20 MOI, 12 of 247 (5.95%) single-cell colonies from the transduced cells had a positive LacZ signal, while none of the 132 single-cell colonies from cells transduced with HD-Ad-LacZ control vector had LacZ signal.
Several studies showed that Scr7, a DNA ligase inhibitor, enhances HDR-mediated transgene integration by inhibiting the NHEJ pathway. 37,38 As shown in Supplementary Fig. S2, the LacZ integration frequency was 5.35% in HD-Ad-Cas9-LacZ-infected cells at 20 MOI without Scr7. The frequencies increased significantly to 9.2% and 12.5% in 0.1 and 1 μM of Scr7-treated cells, respectively. This result indicates a dose-dependent effect of Scr7 on LacZ integration.
One concern of CRISPR/Cas9-mediated gene editing is the off-target cleavage. We selected the top four loci for off-target analysis (Supplementary Table S1). The IPEC cells were transduced with HD-Ad-Cas9-LacZ at 50 MOI. As shown in Supplementary Fig. S3, the off-target frequencies from the top four loci were 0.33%, 1%, 1%, and 0.33%, respectively.
Generation of CFTR−/− IPEC-J2 porcine cell line via CRISPR/Cas9 gene editing
There was no commercially available porcine CFTR knockout cell line; so, we generated a CFTR-deficient porcine cell line to measure transgenic hCFTR protein function. This was carried out in two steps by knocking in GFP and mCherry fluorescent protein (mCherry) maker genes sequentially into each of the porcine CFTR alleles with CRISPR/Cas9 (Fig. 2a). Two sgRNAs, sgRNA1 and sgRNA2, were designed to target the sites 39 bases and 109 bases upstream of the CFTR start codon, respectively (Fig. 2a). Two donor plasmids, pdonor-GFP and pdonor-mCherry, were constructed (Fig. 2a), and each flanked by two ∼3 kb homology arms complementary to target sites. Fluorescent markers were driven by a CMV promoter and followed by an SV40 polyA tail.
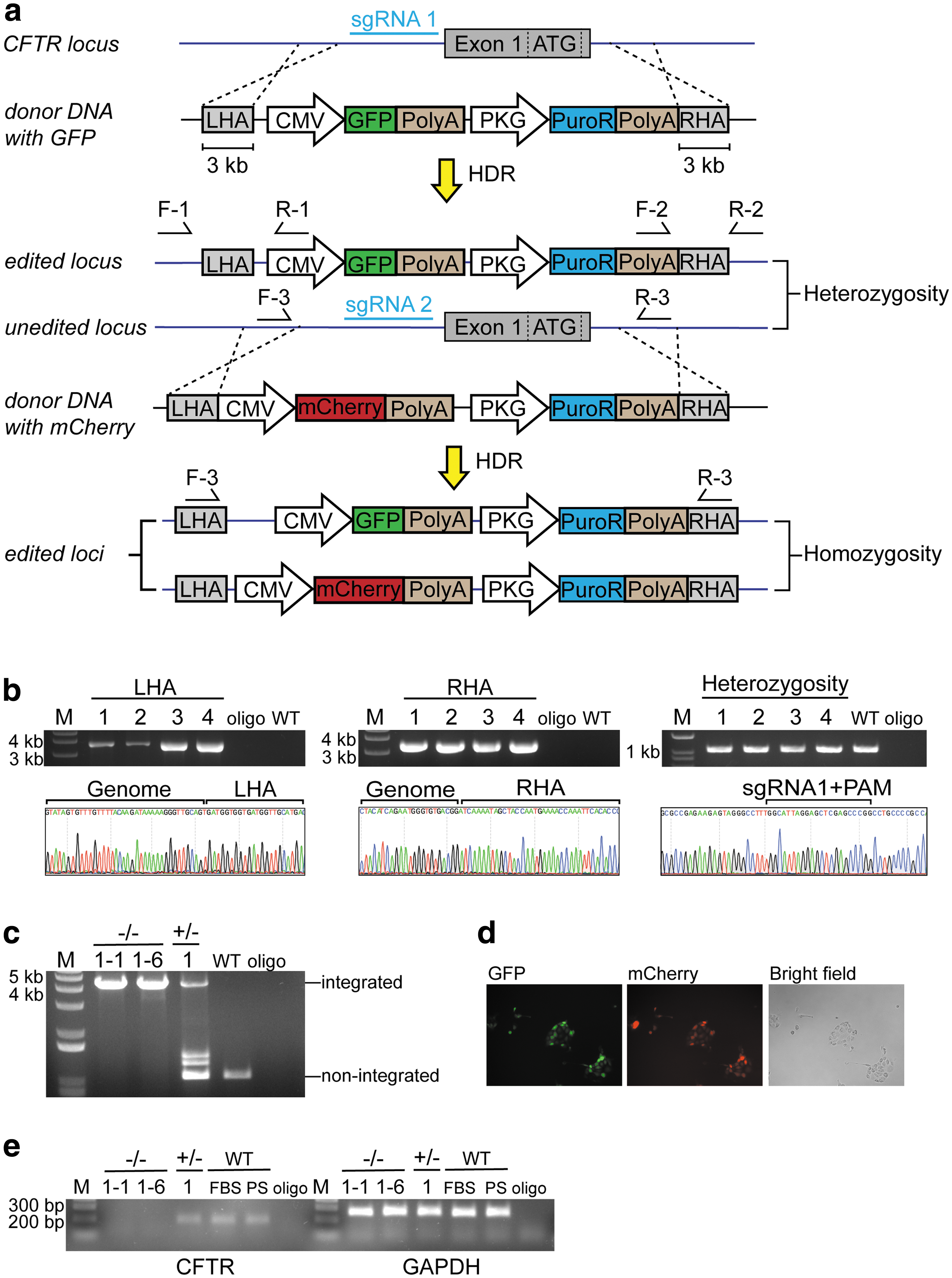
Generation of CFTR
−/− IPEC-J2 porcine cell line via CRISPR/Cas9 gene editing.
Wild-type IPEC-J2 cells were cotransfected with psgRNA1/Cas9 and pdonor-GFP by lipofection. Single GFP-positive cells were sorted into 96-well plates by fluorescence-activated cell sorting. However, due to low postsorting survival rate, only four single-cell clones, #1–#4, survived. Targeted GFP insertion was verified by LHA and RHA junction PCR and Sanger sequencing (Fig. 2b). In addition, a pair of primers was designed to amplify unintegrated locus to check heterozygosity (Fig. 2b). Clones #1–#4 were heterozygous CFTR+/ − (Fig. 2b).
Then, the CFTR+/ − clone #1 was cotransfected by psgRNA2/Cas9 and pdonor-mCherry to generate CFTR −/−cells. Single cell expressing mCherry was sorted, and PCR was performed to check the homozygosity of the fluorescent makers. As shown in Fig. 2c, CFTR −/− clones #1–#1 and #1–#6 carried both markers. Fluorescence detection further confirmed GFP and mCherry expression in the two clones (Fig. 2d). The qRT-PCR results demonstrated that biallelic knockout of CFTR gene at targeted porcine CFTR locus resulted in no detectable CFTR transcript (Fig. 2e). The CFTR+/ − and wild-type IPEC-J2 cells had low but still detectable levels of CFTR transcript.
Quantification of Cas9-induced NHEJ in cells transduced with HD-Ad-Cas9 CFTR
To examine if hCFTR transgene could be integrated and stably expressed at this safe harbor, we constructed an HD-Ad-Cas9-hCFTR vector (Fig. 3a) for integration of the K18-hCFTR gene expression cassette into the GGTA1 locus. The CFTR −/− IPEC-J2 cells were transduced with HD-Ad-Cas9-hCFTR at 50, 25, and 10 MOI or HD-Ad-hCFTR control vector at 50 MOI. The hCFTR gene in the control vector was also regulated by a K18 promoter, and the vector did not contain CRISPR/Cas9. We quantified NHEJ event of the transduced cells by ddPCR to obtain data on the efficiency of Cas9 cleavage. The primer and probe designs of NHEJ are shown in Fig. 3b. As shown in Fig. 3c, the cleavage efficiency was ∼56% in cells infected with HD-Ad-Cas9-hCFTR at 50 MOI, and was ∼50% and ∼45% at 25 and 10 MOI, respectively. In addition, no cleavage event was detected from HD-Ad-hCFTR control vector-transduced cells. The Cas9-induced cleavage efficiency was also detected by T7E1 assay. As shown in Supplementary Fig. S4, the cleavage efficiency was ∼66.01% in cells infected with Cas9-contained HD-Ad vector at 50 MOI, and was ∼49.81% at 20 MOI.
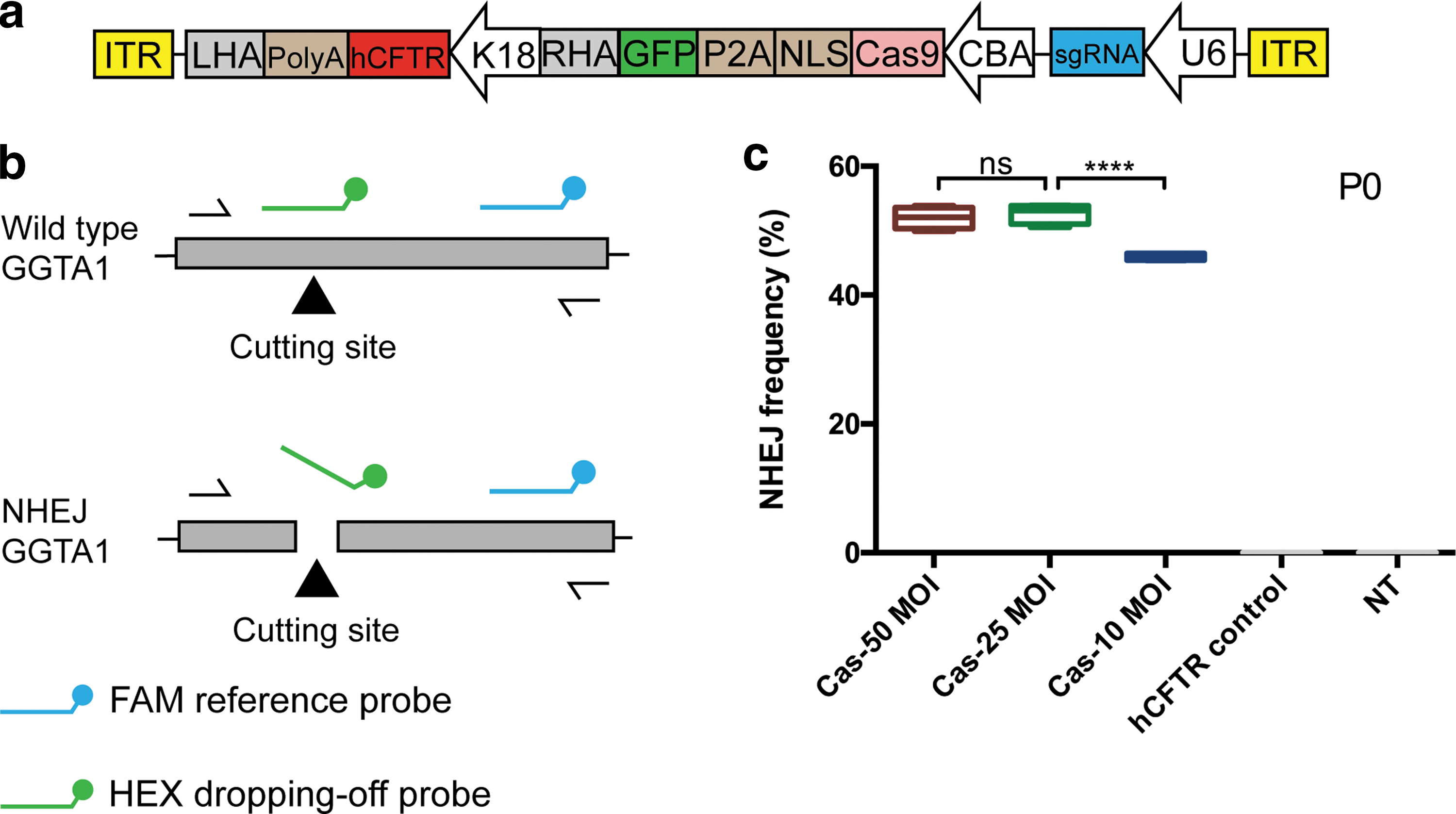
Analysis of CRISPR/Cas9-induced on-target cleavage efficiency in porcine cells following delivery of HD-Ad-Cas9-hCFTR.
Transgene hCFTR was integrated at the GGTA1 locus
To validate the specificity of gene targeting, we designed primers to assess the site-specific integration of hCFTR at the GGTA1 locus, based on the strategy shown in Fig. 4a. The site of integration in cells transduced with HD-Ad-Cas9-hCFTR at 50, 25, and 10 MOI was confirmed by left and right junction PCR and Sanger sequencing (Fig. 4b). Cells transduced with HD-Ad-hCFTR control vector had no detectable integration (Fig. 4b). We also quantified HDR event of the transduced cells by ddPCR at P20. The primer and probe designs of HDR are shown in Fig. 4c. As shown in Fig. 4d, the integration efficiency from HD-Ad-Cas9-hCFTR-transduced group was 10% at 50 MOI, 4.2% at 25 MOI, and 3.1% at 10 MOI.

Integration of hCFTR at GGTA1 target site.
Transgene hCFTR was expressed at the GGTA1 locus
We measured hCFTR mRNA levels in transduced cells by RT-qPCR. The hCFTR mRNA expression level at P0 was set arbitrarily at 100%. As shown in Fig. 5a, the percentages of hCFTR mRNA expression relative to P0 were 0.65% and 0.5% at P10 and P20, respectively, in cells transduced with 50 MOI of HD-Ad-Cas9-hCFTR. At 25 MOI, the percentages were 1.95% and 0.84% at P10 and P20, respectively. At 10 MOI, the percentages of hCFTR mRNA expression were 7.94% and 2.53% in HD-Ad-Cas9-hCFTR-transduced cells. We also quantified copy numbers of HD-Ad vector at P0, P10, and P20 (Fig. 5b). It showed that viral genome copies were largely diminished at P10 and P20.

Expression of hCFTR at GGTA1 target site.
The hCFTR protein expression was measured by Western blot (Fig. 5c). We detected dose-dependent hCFTR protein expression by Western blot at P0 in cells transduced with HD-Ad-Cas9-hCFTR. Cells infected by the control vector at 50 MOI also had hCFTR protein expression, but the expression level was lower than the CRISPR-Cas9 group at the same viral dose. However, the Western blot was not sensitive enough to detect hCFTR protein expression from any group at P10 and P20.
Transduced and integrated hCFTR was functionally expressed
To assess hCFTR channel function, the membrane potential-sensitive dye-based FLIPR assay was performed. The CFTR −/− cells were transduced with HD-Ad-Cas9-hCFTR or HD-Ad-hCFTR control vector at 50 MOI. A GFP vector, named HD-Ad-GFP, was transduced at 50 MOI as a negative control.
Figure 6a shows elevated hCFTR channel activity on addition of FSK, detected as membrane depolarization from HD-Ad-Cas9-hCFTR or HD-Ad-hCFTR control vector-transduced cells at P0. The hCFTR channel-mediated membrane depolarization was also confirmed by adding CFTRinh-172 (Fig. 6a). The peak hCFTR channel activities in both groups were significantly higher than that in HD-Ad-GFP-transduced cells (Fig. 6d). At P10 and P20, the HD-Ad-Cas9-hCFTR-targeted cells still showed significant and stable responses to FSK and CFTRinh-172 (Fig. 6b–d). However, no obvious response was observed in the control group (Fig. 6b–d). In addition, the hCFTR peak response was stable at P10 and P20 from HD-Ad-Cas9-hCFTR-infected cells (Fig. 6d). The HD-Ad-GFP-transduced cells did not show detectable response to FSK or CFTRinh-172 at any time point (Fig. 6a–c).

Measurement of hCFTR channel activity in transduced cells by FLIPR assay.
Discussion
A major goal of CF gene therapy is to achieve sustained expression of therapeutic CFTR transgene. The advent of nuclease-based gene editing tools makes transgene integration feasible. A recent study showed the possibility of using CRISPR/Cas9 to functionally correct CFTR defect in cultured CF patient-derived intestinal organoids. 39 The mutated CFTR locus was replaced by a fragment of nonmutated template through CRISPR/Cas9-induced HDR. Although the gene targeting efficiency was not specified, the organoid expressed CFTR and had functional response to FSK stimulation. 39
In this study, we used HD-Ad vectors carrying LacZ reporter/hCFTR combined with CRISPR/Cas9 to examine our gene targeting strategy in vitro. In our donor template, the homology arms were about 3 kb. Longer homologous sequences were correlated with higher recombination efficiency. 40 –42 CRISPR/Cas9 can induce deletions that may damage homologous sequences on the target locus 43 ; long homology arms also guarantee sufficient sequences for recombination. We performed junction PCR and sequenced the PCR products to verify that the 6 kb LacZ reporter expression cassette was precisely incorporated into the targeted genomic site. We also demonstrated the long-term expression of integrated LacZ transgene by LacZ staining and single-cell culture. Our results indicate the feasibility of HD-Ad vector-mediated transgene integration.
Many studies have been carried out to enhance nuclease-mediated transgene knockin by regulating DNA repair mechanisms. Small molecules RS-1, NU7441, and KU-0060648 have been reported to increase transgene knockin by enhancing HDR. 44,45 DNA ligase inhibitor, Scr7, also increased HDR-mediated transgene integration by inhibiting NHEJ repair. 37,38 We observed increased LacZ integration frequencies after HD-Ad delivery when combined with Scr7.
We demonstrated that the 8.7 kb hCFTR expression cassette can be precisely integrated at GGTA1 locus through CRISPR/Cas9-induced HDR. This was again validated by junction PCR and Sanger sequencing of the PCR products. At P10 and P20, low but detectable hCFTR mRNA levels were present in cells transduced with HD-Ad-Cas9-hCFTR, while almost no hCFTR mRNA expression was detectable in cells transduced with the control vector, indicating that the mRNA expression was from the integrated hCFTR transgene. Protein expression in transduced cells was detected at P0 by capillary-based Western blot system. However, the level of expression was too low to be detectable at either P10 or P20, which was probably due to sensitivity of the Western blot system, considering detectable mRNA expression in same cellular passages.
Porcine is an ideal animal model to study gene therapy for CF lung diseases due to the similarity in lung anatomy, histology, and biology with human. 46,47 However, there is at present no porcine airway cell line available for our study. Instead, we used a porcine intestine epithelial cell line. A limitation of IPEC-J2 was a very low level of endogenous CFTR expression in the wild type. This natural characteristic of the cell line may restrict hCFTR transgene expression. Therefore, other pig cell lines, including porcine airway primary cells, should be tested to further verify our gene targeting strategy.
We found that both NHEJ and integration frequencies induced by HD-Ad-Cas9-hCFTR were dose dependent. In HD-Ad-Cas9-hCFTR-infected cells, the NHEJ frequencies were ∼56%, ∼44.2%, and ∼38.1% from 50, 25, and 10 MOI-infected cells, respectively. The hCFTR integration frequencies were ∼10% at 50 MOI, ∼4.2% at 25 MOI, and ∼3.1% at 10 MOI. By adding HDR frequencies together, the final CRSPR/Cas9-induced cleavage frequencies were ∼66%, ∼54.2%, and ∼48.1% from cells infected by 50, 25, and 10 MOI, respectively. Some studies have been done to identify the minimal amount of CFTR correction required to achieve a phenotypic normal channel function. A study used human bronchial epithelial cells and showed that close to normal CFTR channel function was detected when coculturing 10% of CF cells with 90% of non-CF cells. 48 Another study indicated that around 6–10% of corrected cells were sufficient to maintain almost normal chloride channel function. 49
One limitation of our study was that we could not directly measure HDR-mediated hCFTR integration, because our homology arms were too long to be amplified by ddPCR. Therefore, we measured copy numbers of hCFTR and viral backbone separately, and we calculated the frequencies of integrated hCFTR by the difference between hCFTR and viral backbone at P20. This was because viral vectors were diluted and mostly lost during continuous culture for 20 passages. By this method, however, a low level of random integration could not be excluded. Another limitation was that we could not directly measure the cleavage efficiency, because there was no HD-Ad vector that only contains CRISPR/Cas9, but without hCFTR donor template. Thus, the cleavage efficiency in our study could be slightly underestimated.
In CF airway, the defective CFTR channel is related to depleted airway surface liquid, impaired mucus clearance, and biofilm formation. Viral vector-based CFTR delivery could phenotypically restore CFTR channel function. 50,51 In the FLIPR membrane potential assay, 52,53 we detected agonist-induced channel opening as membrane depolarization and antagonist-mediated CFTR inhibition as membrane hyperpolarization in the transduced cells. We demonstrated that transduced hCFTR was functionally expressed in the tested cell line. At P10 and P20, HD-Ad-Cas9-hCFTR-infected cells showed a similar response to FSK (CFTR activator) and CFTR-172 (CFTR inhibitor), while the control vector-infected cells had no detectable response. These findings, take together with junction PCR and mRNA expression, suggest that the steady hCFTR function at P10 and P20 is a result of integrated hCFTR gene.
The delivery efficiency of therapeutic transgene is the main obstacle affecting CF lung gene therapy. Many factors, such as clogged mucus, pathogen biofilm, and hyperinflammatory environment, can restrict transferring of therapeutic agents to the inside of cells. 54 Viral vector-based gene delivery is one of the approaches being developed to improve the delivery efficacy. 50,55 –57 A recent study showed that CFTR channel defect was partially complemented by lentivirus-based delivery of a CFTR cassette in CF pig. 50 Steines et al. developed an adeno-associated virus vector with high tropism for pig airway, and showed enhanced CFTR minigene delivery efficiency and phenotypic correction in CF pig. 51 Our previous studies proved the feasibility of HD-Ad vector-based hCFTR transgene delivery. We demonstrated that HD-Ad vectors could deliver transgenes efficiently in human primary nasal cells, porcine primary airway cells in vitro, and porcine animal model in vivo. 32 Moreover, in primary nasal cells of CF patients, defected CFTR channel function was restored by HD-Ad vector-based CFTR delivery. 32
The turnover of well-differentiated airway epithelial cells puts limits on long-term transgene expression. Basal cells are multipotent, self-renewal stem cells that can differentiate into major CFTR-expressing ionocytes. 58,59 Therefore, our future work is to target basal progenitor cells for permanent transgene expression in airway, and our general strategy is shown in Fig. 7. In our previous study, we successfully delivered transgenes into basal progenitor cells of mouse and pig airway in vivo and human primary nasal cell in vitro with HD-Ad vectors. 32 We also showed that the anion channel function of transduced basal cells was corrected by hCFTR gene delivery. 32 However, it is more challenging to edit basal progenitor cells in vivo than the cell line in vitro at the present targeting efficiency. Further studies need to be carefully designed and performed to test if gene editing in basal progenitor cells in vivo is feasible.

Overview of the in vivo CFTR correction strategy in CF pig model. HD-Ad vectors will be constructed for delivery of both the CRISPR-Cas9 system and donor DNA to airway epithelial cells. The vector transduced cells will produce Cas9 protein and guide RNA for generation of a double-stranded DNA break at GGTA1 locus. When homology-directed DNA repair takes place, the donor DNA will be integrated into the target locus, while the rest of the compromised vector genome would be degraded.
In conclusion, the results have shown precise integration of HD-Ad vector-delivered hCFTR at GGTA1 safe harbor locus through CRISPR/Cas9 induced HDR; long-term, functional in vitro expression was achieved in transduced cells. Because pig GGTA1 locus was demonstrated as a safe harbor, it may be possible to target human GGTA1 locus as a new safe harbor since it is a pseudogene. 25 The results that we have obtained so far, together with those from our future studies, will provide important information for us to design CFTR gene targeting therapy to treat CF patients.
Footnotes
Acknowledgments
We thank Dr. Tara Paton and Mr. Guillermo Casallo for designing and performing the ddPCR analyses in this study. We thank Ms. Sara Chowns for reading and commenting on the article. This study was funded by Canadian Institute for Health Research (CIHR) grants (MOP 125882), Cystic Fibrosis Foundation Therapeutics, Inc. grant (HU15XX0), and Cystic Fibrosis Canada grant (#3032) to Jim Hu. Zhichang Peter Zhou held an Ontario Trillium Graduate Scholarship.
Author Disclosure
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.